

Abstract
A high throughput screen of the NIH’s MLSMR collection of ~340,000 compounds was undertaken to identify compounds that inhibit Plasmodium falciparum glucose-6-phosphate dehydrogenase (PfG6PD). PfG6PD is essential for proliferating and propagating P. falciparum and differs structurally and mechanistically from the human ortholog. The reaction catalyzed by glucose-6-phosphate dehydrogenase (G6PD) is the first, rate-limiting step in the pentose phosphate pathway (PPP), a key metabolic pathway sustaining anabolic needs in reductive equivalents and synthetic materials in fastgrowing cells. In P. falciparum the bifunctional enzyme glucose-6-phosphate dehydrogenase-6- phosphogluconolactonase (PfGluPho) catalyzes the first two steps of the PPP. Because P. falciparum and infected host red blood cells rely on accelerated glucose flux, they depend on the G6PD activity of PfGluPho. The lead compound identified from this effort, (R,Z)-N-((1-ethylpyrrolidin-2-yl)methyl)-2- (2-fluorobenzylidene)-3-oxo-3,4-dihydro-2H-benzo[b][1,4]thiazine-6-carboxamide, 11, (ML276), is a submicromolar inhibitor of PfG6PD (IC50 = 889 nM). It is completely selective for the enzyme’s human isoform, displays micromolar potency (IC50 = 2.6 μM) against P. falciparum in culture, and has good drug-like properties, including high solubility and moderate microsomal stability. Studies testing the potential advantage of inhibiting PfG6PD in vivo are in progress.
Keywords: 6-phosphogluconolactonase, glucose-6-phosphate, glucose-6-phosphate dehydrogenase, highthroughput screening, malaria, pentose phosphate pathway, Plasmodium, redox metabolism, benzothiazinone
INTRODUCTION
In 2010, around 1.2 million people died from malaria infections.1 Severe forms of the disease are mainly caused by Plasmodium parasites of the species falciparum, which are transmitted to the human host by female Anopheles mosquitoes.2 P. falciparum parasites have infected humans for thousands to millions of years,3,4 and yet malaria eradication has not been possible due to the parasites’ ability to develop resistance to most antimalarial drugs.5 To date, no vaccine against malaria is available,6 and infections are treated with drugs commonly targeting the blood stage of the parasite.7 The most common clinically used antimalarial drugs belong to seven classes, namely 4-aminoquinolines such as chloroquine and amodiaquine, arylaminoalcohols such as quinine and mefloquine, 8-aminoquinolines such as primaquine, artemisinins such as artemether and artesunate as well as synthetic peroxides such as tri- and tetraoxanes8, antifolates such as sulfadoxine and pyrimethamine, inhibitors of the respiratory chain such as atovaquone, and antibiotics.9,10 The occurrence of drug-resistant P. falciparum strains11–13 as well as side effects from drug administration14,15 resulted in the current WHO recommendation to use artemisinin-based combination therapies to treat P. falciparum malaria.16 However, recent reports of artemisinin resistances17–19 again create an urgent need for new anti-malarial drugs. Based on several findings, the enzyme glucose-6-phosphate dehydrogenase 6-phosphogluconolactonase of P. falciparum (PfGluPho) has emerged as an intriguing new target for anti-malarial drug development.20 Deficiency in human glucose-6-phosphate dehydrogenase (hG6PD), which affects around 330 million people worldwide,21 is associated with protection from severe malaria infections.22–24 It is suggested that both hG6PD and PfGluPho play an important role in Plasmodium development and survival.20,22,25 PfGluPho is the key enzyme of the parasitic pentose phosphate pathway (PPP), which is supposed to be the major NADPH source (reviewed by Preuss et al. 26) for these parasites highly sensitive to oxidative stress.27 A previous study showed that P. falciparum-infected red blood cells (IRBC) present an increased PPP activity compared to non-infected ones, 80% of which is caused by Plasmodium’s PPP.25 Moreover, silencing of PfGluPho with RNA interference (RNAi) stopped parasite development at the trophozoite stage.28 Although Baum et al. reported in 2009 that P. falciparum lacks an RNAi machinery,29 Lopez- Barragan et al. recently reported that antisense RNA may be an important factor in P. falciparum’s regulation of gene expression,30 strengthening the results of the previous RNAi-based study. Additionally, PfGluPho presents considerable structural differences compared to hG6PD, potentially allowing selective targeting.31,32
Herein we describe the high-throughput screening (HTS) of 348,911 compounds in the NIH’s Molecular Libraries Small Molecule Repository (MLSMR) collection (http://mli.nih.gov/mli/compound-repository/mlsmr-compounds) against PfGluPho and a subsequent structure-activity relationship (SAR) study of a class of benzothiazinone hits obtained from the screen. Optimization of the screening hits resulted in the discovery of (R,Z)-N-((1-ethylpyrrolidin-2-yl)methyl)- 2-(2-fluorobenzylidene)-3-oxo-3,4-dihydro-2H-benzo[b][1,4]thiazine-6-carboxamide, 11, (ML276),33 which inhibited PfGluPho selectively over hG6PD and inhibited P. falciparum growth in vitro with an IC50 in the low micromolar range. Compound 11 represents the first reported selective PfGluPho inhibitor and therefore could provide a basis for future drug design of new antimalarial therapeutics.
RESULTS AND DISCUSSION
High Throughput Screen
In a primary screening approach, the MLSMR compound library (~350,000 compounds) was screened at a concentration of 20 μM against PfGluPho. The screening campaign resulted in the identification of 2,709 compounds that inhibited PfGluPho activity ≥ 59%, corresponding to a hit rate of 0.8%. Of these, 2,429 were available from the MLSMR collection and were retested in triplicates at a concentration of 20 μM, resulting in 817 compounds inhibiting PfGluPho ≥ 50%, about 34% of the samples. To exclude samples that interfere with the coupled resazurin/diaphorase system used in our study,34 active compounds were run against a diaphorase inhibition assay at a concentration of 20 μM. Of the compounds tested, 230 inhibited the diaphorase reaction and thus were discarded as artifacts. The remaining 587 active compounds were further tested in dose response in the PfGluPho assay and an hG6PD counter-screen. As a result, 488 compounds presented IC50 values ≤ 20 μM vs. PfGluPho, and only 39 of the compounds were active against hG6PD. For final confirmation, the compounds were tested in an orthogonal assay that did not rely on the resazurin and diaphorase coupled system. Based on the initial slope of the reaction, around 50% of the compounds were confirmed to inhibit PfGluPho with an IC50 ≤ 80 μM. Several series of compounds were identified, of which a group of benzothiazinones was most promising based on their potency and selectivity. Detailed information is available in the PubChem database (AIDs 504690, 504753, 504863, 504862, 504765, 504792, 540252, 540269).
Synthesis
The synthesis of the benzothiazinone-derived compounds described herein was accomplished in 4–5 steps as outlined in Scheme 1.35 Starting from 4-fluoro-3-nitrobenzoic acid or the esters (1a–c), nucleophilic substitution of the fluorine substituent with ethyl thioglycolate under basic conditions provided thioethers (2a–c). Reductive cyclization mediated by iron in acetic acid provided benzothiazinones (3a–c). 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDCI)-mediated coupling of 3a with 2-aminomethyl-1-ethylpyrrolidine afforded amide 3d. The first point of diversity was introduced by condensation of benzothiazinones (3a–c) with a range of aromatic aldehydes resulting in intermediates (4a–j). Coupling of intermediates (4a–j) with 2-aminomethyl-1-ethylpyrrolidine resulted in the corresponding amides 5a–j (Scheme 1). The styryl moiety in the intermediate 4a was reduced with sodium borohydride in the presence of cobalt chloride, and the resulting acid was converted to amide 10 (Scheme 2). A second point of diversity resulted from amide synthesis starting from 4a using diverse amines to provide the amides 10–15, 17, 22, 29, 33, 34, 39–42 (Scheme 2). All final compounds were obtained in moderate yields and high purity needed to support SAR studies.
Scheme 1.

Synthesis of 5a and analogs, conditions: i) ethyl thioglycolate, NaOAc, H2O, 90 °C; ii) ethyl thioglycolate, triethylamine, MeCN, 85 °C; iii) Fe, HOAc, 80 °C; iv) 2-aminomethyl- 1-ethylpyrrolidine, EDCI·HCl, HOBt, Et3N, CH2Cl2, 23 °C; v) aldehyde, Ac2O, Et3N, reflux; vi) aldehyde, Ac2O, Et3N, reflux; LiOH, MeOH, H2O, 23 °C.
Scheme 2.

Synthesis of compounds 10–15, 17, 22, 29, 33, 34, 39–42 including 11, conditions: i) CoCl2, NaBH4, THF, 23°C, 53%; 2-aminomethyl-1-ethylpyrrolidine, EDCI·HCl, HOBt, Et3N, CH2Cl2, 23°C, 41%; ii) RNH2, EDCI·HCl, HOBt, Et3N, CH2Cl2, 23 °C, 13–61%.
Structure Activity Relationships (SAR)
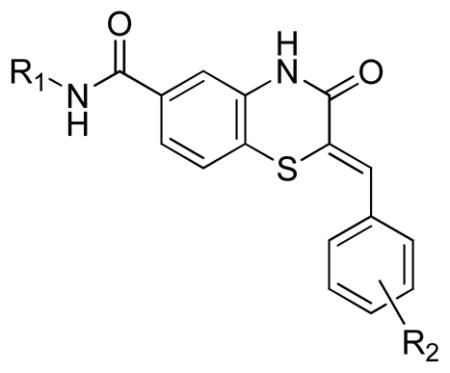
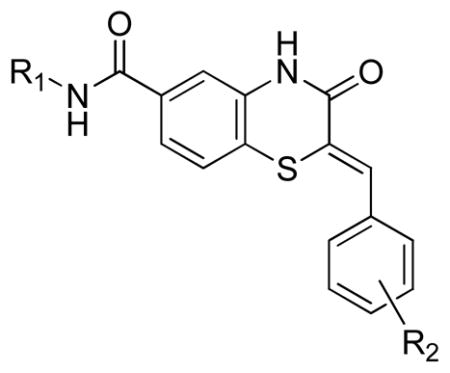
Three main assays were utilized to direct the SAR efforts. The primary SAR-driving assay was the dose response, resazurin/diaphorase-coupled PfGluPho assay, with the hG6PD assay serving as the counterscreen to ascertain specificity. None of the compounds from this series showed any activity in the hG6PD counterscreen (>80 μM) and thus were considered specific for the P. falciparum enzyme. The orthogonal PfGluPho assay, which relied on the direct detection of NADPH, was used to further confirm activity in the absence of the coupled resazurin/diaphorase system. Of the 156 substituted benzothiazinones tested in the high throughput screen, only five were confirmed active by dose response. All were inactive (IC50 > 80 μM) against hG6PD. These structures are represented in Table 1. The structural features of these active benzothiazinones that separate them from those that were either inactive or failed confirmation in dose response include: 1) the amide side chain contains a basic alkylamine; 2) the nitrogen in the benzothiazinone ring is not alkylated; and 3) the sulfur exists as a sulfide (Figure 1).
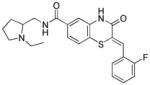
Table 1.
Structures of active benzothiazinone hits from HTS
| Entry | Structure | PfG6PD IC50 (μM) |
|---|---|---|
| 5a |
|
1.75 |
| 6 |
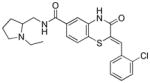
|
2.56 |
| 7 |
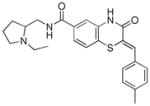
|
4.42 |
| 8 |
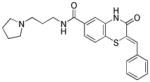
|
9.30 |
| 9 |
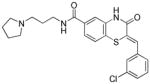
|
17.9 |
Figure 1.

Structural requirements for activity
From this preliminary SAR, we focused on further optimization of the scaffold. Specifically, we examined a wide range of amide substitutions as well as substitution around the pendant aryl ring. The general SAR strategy is depicted in Figure 2.
Figure 2.

General SAR strategy
Exploration of the styryl SAR was initiated by maintaining the preferred 1-ethyl-2-aminomethylpyrrolidine moiety from the HTS data and is summarized in Table 2. The primary hit 5a, identified during the HTS and subsequent hit confirmation, was resynthesized as described in Scheme 1. It was equipotent to the purchased compound (Entry 5a, Tables 1 and 2). 1H NMR data of the purchased sample and the synthesized sample of 5a were identical. The Z-geometry around the styryl portion was assigned based on comparison to previously reported NMR data.36
Table 2.
SAR of benzothiazinonesa inhibiting Plasmodium falciparum glucose-6-phosphate dehydrogenase
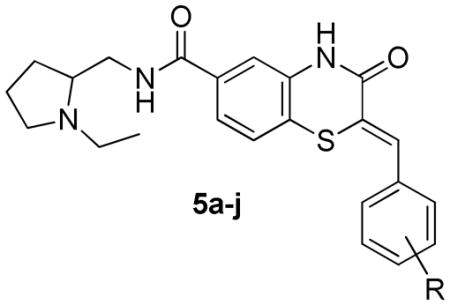
|
Potency (μM) Ave. ± S.E.M. (n= 4) | ||
|---|---|---|---|
| Compound | R | PfG6PD Primary Assay | PfG6PD NADPH Kinetic |
| 5a | 2-F | 1.73 ± 0.08 | 1.37 |
| 5b | H | 8.23 ± 2.11 | 19.7 |
| 6b | 2-Cl | 2.56 ± 0.19 | 3.3 |
| 5c | 2-Me | 5.93 ± 1.59 | 5.01 |
| 5d | 2-OMe | 9.79 ± 1.13 | 11.3 |
| 5e | 3-F | 2.35 ± 0.29 | 4.13 |
| 5f | 4-F | 1.41 ± 0.14 | 2.06 |
| 7b | 4-Me | 4.42 ± 0.71 | 11.6 |
| 5g | 2,3-difluoro | 5.72 ± 1.28 | 3.89 |
| 5h | 2,6-difluoro | 1.65 ± 0.08 | 2.61 |
| 5i | 1-napthyl | 4.28 ± 0.87 | 4.2 |
| 5j | 2-napthyl | 2.39 ± 0.28 | 2.88 |
all compounds represented in this table exhibited an IC50 > 80 μM against human G6PD
screen hit
A variety of substituted styryl analogs were synthesized; however, the potency range was only affected by 7-fold. The 2-fluoro substituent is important for activity, as the analog without any aryl substituent was ~ 4-fold less active (entry 5a vs. 5b, Table 2). Interestingly, neither the position nor the electronics of the substituent dramatically affected the observed potency. For example, analogs containing the 2-, 3-, or 4-fluoro substitution (entries 5a, 5e, and 5f, Table 2) were essentially equipotent. Replacement of the 2-fluoro substituent with 2-chloro did not significantly affect the potency, but replacement with electron-rich 2-methyl or 2-methoxy groups decreased potency by 3-fold and 5-fold respectively (Entries 5a, 6, 5c, and 5d, Table 2). Fused ring systems such as 1-naphthyl and 2-naphthyl (Entries 5i and 5j, Table 2) also slightly lowered potency. The unsubstituted phenyl and the 2-methoxy analogs (Entries 5b and 5d, Table 2) were the least preferred of the synthesized analogs.
While the SAR around the styryl moiety may appear to minimally affect potency, its presence was required for activity. When the styryl group was replaced with phenethyl or removed entirely, a complete loss of activity resulted (5a vs. 3d & 10, Table 3).
Table 3.
Necessity of styryl moiety in SAR
| Entry | Structure | PfG6PD IC50 (μM) |
|---|---|---|
| 5a |
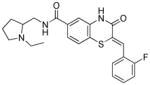
|
1.73 |
| 3d |
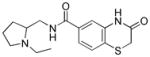
|
>80 |
| 10 |
|
>80 |
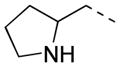
The SAR of the alkylamino side chain proved to be much more stringent than the styryl region, with a strong preference for 2-aminomethyl pyrrolidines. Potency was maintained within 5-fold whether the nitrogen of the pyrrolidine ring was substituted with ethyl, propyl, or was unsubstituted (Entries 5a, 13 and 15, Table 4). The t-butoxycarbonyl derivative proved inactive, consistent with observations from the HTS (Entry 14, Table 4). Less constrained examples such as dimethylaminoethyl, diethylaminoethyl, and dipropylaminoethyl demonstrated sharp decreases in potency (Entries 16–27, Table 5). Attachment of pyrrolidine through nitrogen via an aminoethyl linker led to a 5-fold loss of potency, which became more pronounced as the ring was expanded (Entries 8, 9, 28–39, Table 6). Examples of constrained tertiary or aromatic amides proved substantially less active (Entries 40–42, Table 6). All compounds represented in Tables 4, 5, and 6 exhibited an IC50 > 80 μM against hG6PD.
Table 4.
SAR of benzothiazinones inhibiting Plasmodium falciparum glucose-6-phosphate dehydrogenase
|
Potency (μM) Ave. ± S.E.M. | ||||
|---|---|---|---|---|---|
| Entry | R1 | R2 | Yield % | PfG6PD Primary Assay | PfG6PD NADPH Kinetic |
|
5a racemic |
|
2-F | 19 | 1.7 ± 0.1 | 1.4 |
|
11 R-enantiomer ML276 |
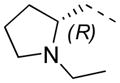
|
2-F | 32 | 0.9 ± 0.0 | 1.9 |
|
12 S-enantiomer |
|
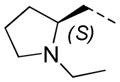
2-F | 28 | 6.6 ± 0.2 | 4.7 |
| 13 |

|
2-F | 48 | 3.1 ± 0.4 | 4.0 |
| 14 |
|
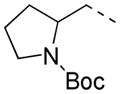
2-F | 61 | >80.0 | 24.4 |
| 15 |
|
2-F | 79 | 5.9 ± 0.6 | 5.0 |
Table 5.
SAR of benzothiazinones inhibiting Plasmodium falciparum glucose-6-phosphate dehydrogenase
|
Potency (μM) Ave. ± S.E.M. | ||||
|---|---|---|---|---|---|
| Entry | R1 | R2 | Yield %a | PfG6PD Primary Assay | PfG6PD NADPH Kinetic |
| 16 |
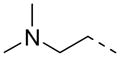
|
2-F | P | 16.7 ± 0.8 | 11.2 |
| 17 | 2-F | 13 | 17.5 ± 8.5 | 38.4 | |
| 18 | 3-Cl | P | 60.0 ± 8.5 | 43.6 | |
| 19 | 4-OMe | P | >80.0 | >80.0 | |
| 20 |
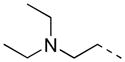
|
H | P | 8.8 ± 1.6 | 18.4 |
| 21 | 2-F | P | 7.4 ± 0.7 | 9.4 | |
| 22 | 2-F | 28 | 6.9 ± 0.3 | 4.8 | |
| 23 | 2-Cl | P | 16.2 ± 2.1 | 44.9 | |
| 24 | 3-Me | P | 14.5 ± 3.8 | 18.8 | |
| 25 | 3,4- (OMe)2 | P | 60.0 ± 10.0 | 24.5 | |
| 26 |
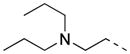
|
H | P | >80.0 | 27.2 |
| 27 | 4-Me | P | >80.0 | 45.9 | |
P: commercially purchased compounds
Table 6.
SAR of benzothiazinones inhibiting Plasmodium falciparum glucose-6-phosphate dehydrogenase.
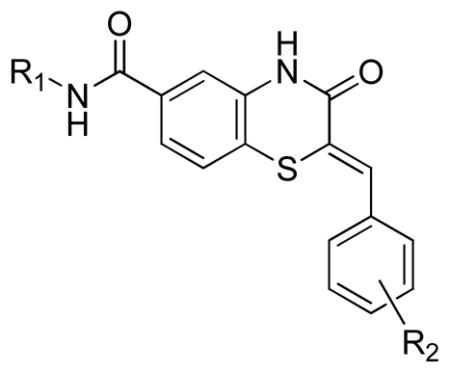
|
Potency (μM) Ave. ± S.E.M. | ||||
|---|---|---|---|---|---|
| Entry | R1 | R2 | Yield % a | PfG6PD Primary Assay | PfG6PD NADPH Kinetic |
| 28 |
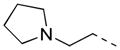
|
2-F | P | 9.5 ± 3.6 | 11.0 |
| 29 | 2-F | 18 | 16.2 ± 2.1 | 25.2 | |
| 30 | 4-OMe | P | 49.7 ± 13.7 | >80.0 | |
| 31 | 2-Cl | P | 11.1 ± 1.3 | >80.0 | |
| 32 | 3-Me | P | 11.8 ± 0.4 | 8.8 | |
| 33 |
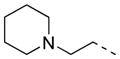
|
2-F | 6 | 21.6 ± 3.4 | 24.0 |
| 34 |
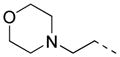
|
2-F | 15 | >80.0 | >80.0 |
| 35 |
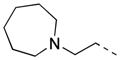
|
2-F | P | 46.6 ± 10.0 | 11.0 |
| 36 | 2-Cl | P | 40.9 ± 12.5 | >80.0 | |
| 37 | 3-OMe | P | >80.0 | 58.5 | |
| 38 | 4-OMe | P | >80.0 | >80.0 | |
| 8b |
|
H | P | 9.3 | 22.3 |
| 9b | 3-Cl | P | 17.9 | 34.2 | |
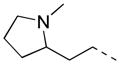
| 39 |
|
2-F | 27 | 22.7 ± 2.1 | 45.2 |
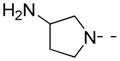
| 40 |
|
2-F | 36 | 45.0 ± 5.3 | 35.0 |
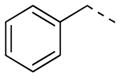
| 41 |
|
2-F | 36 | >80.0 | >80.0 |
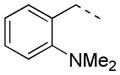
| 42 |
|
2-F | 46 | >80.0 | 67.8 |
P: commercially purchased compounds
screen hit
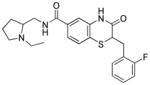
Stereochemical evaluation of the 2-aminomethyl pyrrolidines led us to synthesize each enantiomer of 5a from the corresponding commercially available chiral amines. An 8-fold preference for the Renantiomer versus the S-enantiomer was discovered (Entries 11 & 12, Table 4). Compound 11, (R,Z)-N- ((1-ethylpyrrolidin-2-yl)methyl)-2-(2-fluorobenzylidene)-3-oxo-3,4-dihydro-2H-benzo[b][1,4]thiazine- 6-carboxamide, was the most potent compound identified and was therefore declared a probe. Compound 11 was selective for PfGluPho ovr hG6PD (Figure 3).
Figure 3.

Dose response curve for 11 in the Plasmodium and human G6PD assays, indicating full selectivity for PfGluPho.
In vitro ADME/T profiling and chemical stability
Compound 11 was evaluated in a detailed in vitro pharmacology screen (absorption, distribution, metabolism, excretion, toxicity (ADME/T)) as shown in Table 7. Compound 11 is highly soluble especially at lower pH, which is likely due to the presence and protonation of the alkylamino moiety. Consistent with its solubility data, 11 exhibits high permeability with increased pH of the donor compartment in a standard parallel artificial membrane permeability assay (PAMPA). When incubated with an artificial membrane that models the blood-brain barrier (BBB), 11 was highly permeable. Compound 11 had good stability in plasma, thus increasing its exposure to blood-borne parasites such as P. falciparum, and it was highly plasma protein bound, which may contribute to its high plasma stability. Lastly, 11 shows moderate stability in human and modest stability in mouse liver homogenates and no toxicity (>50 OM) toward Fa2N-4 immortalized human hepatocytes.
Table 7.
Summary of in vitro ADME/T properties of P. falciparum G6PD inhibitor probe 11
| Aqueous Solubility in pION’s buffer (μg/mL) [μM]a pH 5.0/6.2/7.4 | >126/>126/29.4 [>296/>296/ 69] | |
| Aqueous Solubility in 1x PBS, pH 7.4 (μg/mL) [μM]a | 71.0 [166] | |
| PAMPA Permeability, Pe (x10−6 cm/s) Donor pH: 5.0 / 6.2 / 7.4 Acceptor pH: 7.4 | 29 / 737 / 806 | |
| BBB-PAMPA Permeability, Pe (x10−6 cm/s) Donor pH: 7.4 Acceptor pH: 7.4 | 233 | |
| Plasma Protein Binding (% Bound) | Human 1 μM /10 μM | 98.39 / 97.98 |
| Mouse 1 μM /10 μM | 98.95 / 98.34 | |
| Plasma Stability (% Remaining at 3 hrs) | Human/Mouse | 90.98 / 80.84 |
| Hepatic Microsome Stability (% Remaining at 1hr) Human/Mouse | 60.53 / 37.57 | |
| Toxicity Towards Fa2N-4 Immortalized Human Hepatocytes LC50 (OM) | >50 | |
Solubility also expressed in molar units (μM) as indicated in italicized [bracketed values], in addition to more traditional μg/mL units.
Compound 11 contains an α,β-unsaturated amide and is a potential Michael acceptor. To investigate this potential chemical liability, 11 (10 μM, 1% DMSO) was incubated with 5 equivalents of glutathione (GSH) in PBS buffer (pH 7.4) at 23 °C. Loss of compound was monitored at different times over 18 h using RP-LCMS (Figure 4). Ethacrynic acid, a diuretic containing an α,β-unsaturated ketone, was used as a positive control since it has been previously reported to form GSH-adduct.37 Loss of ethacrynic acid was observed as early as 30 minutes and was ~97% depleted in 24 h; whereas 11 remained intact for the duration of the study, indicating a lack of reactivity with nucleophiles (Figure 4). Thus, 11 represents a potential lead compound for the development of an in vivo probe.
Figure 4.

Investigation of chemical reactivity/stability of 11 via GSH adduct formation.
Activity against P. falciparum in culture
Compound 11 was included in P. falciparum growth assays to assess its effects on blood stage parasites in vitro. After incubation for 72 hours, 11 gave IC50 values of 2.3 ± 0.2 μM for the chloroquine sensitive 3D7 strain and 3.7 ± 0.9 for the chloroquine resistant Dd2 strain (Figure 5).
Figure 5.

P. falciparumgrowth inhibition by 11 of chloroquine-sensitive (A) and chloroquineresistant (B) strains. One representative data set out of two independent experiments is shown. Experiments were performed twice in triplicates, of which the average and standard deviation are given.
Mechanism of Inhibition (MOI) studies
The most likely type of inhibition was determined by titrating compound and substrate/co-substrate at the same time. G6P and 11 titration revealed increasing Km values and an α > 6.6 with increasing compound concentration, suggesting that 11 inhibits PfGluPho activity by competing with the substrate (Figure 6B). Increasing Km and decreasing Vmax values, and α between 1.4 and 4.7, were found when compound and co-substrate were titrated, indicating mixed-type inhibition of 11 vs. NADP+ (Figure 6A).
Figure 6.

Mechanistic characterization of 11 vs. NADP+ (A) and G6P (B) for PfGluPho. The signal (RFU/sec) was plotted against the compound and NADP+ or G6P concentration. One representative data set from two independent experiments is shown. Data points are given as averages of duplicates.
CONCLUSIONS
A series of benzothiazinones derived from an HTS of the NIH’s MLSMR collection that are potent, fully selective inhibitors of PfGluPho has been reported. The most potent compound in this series, 11, has antimalarial activity against both chloroquine-sensitive and chloroquine-resistant strains. Compound 11 has reasonable in vitro ADME/T properties, including excellent solubility, no toxicity, good permeability, and plasma stability and moderate microsomal stability. Further work in this series is in progress to optimize the potency and ADME/T properties, which may lead to validation in in vivo models.
EXPERIMENTAL SECTION
HTS/Biology
Materials
G6P, resazurin, diaphorase, Tween20, MgCl2, and BSA were obtained from Sigma, St. Louis, MO. NADP+ was purchased from Amresco, Solon, OH, and 1,536 well-plates from Aurora, Poway, CA.
Compound library
The Sanford-Burnham Center for Chemical Genomics’ copy of the NIH Molecular Libraries Small Molecule Repository (MLSMR) consisted of 348,911 compounds when the screen was run, which were present in 1,536-well plates at a concentration of 2 mM (in DMSO). The library was stored at −80°C until first use, after which it was kept at room temperature under desiccating conditions for a period not greater than six months.
HTS
Primary Screen
The previously developed HTS assay for PfGluPho34 was used in a 384-well plate format. This assay was slightly modified for use in 1,536-well plates and performed in the following way. A volume of 60 nL of the compounds (final 20 OM, 1% DMSO) was transferred to columns 5–48 of 1,536-well plates using the Echo Liquid Handler (LabCyte). Then 60 nL of DMSO were transferred to columns 1–4, which served as positive and negative controls. A volume of 3 μL of enzyme mix (50 mM Tris (pH 7.5), 0.005% Tween 20, 1 mg/mL BSA, 0.1 μg/mL PfGluPho) was added to all wells using the Kalypsys (Kalypsys Systems). To start the reaction, 3 μL substrate mix (50 mM Tris (pH 7.5), 40 μM G6P, 0.005% Tween 20, 1 mg/mL BSA, 6 μM NADP+, 6.6 mM MgCl2, 2 U/mL diaphorase, 0.05 mM resazurin) were added to columns 3–48 using the Kalypsys. Substrate mix without G6P was added to columns 1–2 for positive control. The plates were centrifuged at 1,500 rpm for 1 min and incubated in the dark for 2 hours. Fluorescence of resorufin was detected at excitation 530/emission 580 nm (ex530/em580) using a Viewlux (PerkinElmer) plate reader. Data was analyzed with CBIS (Chemical and Biological Information Systems, www.cheminnovation.com) and compounds inhibiting PfGluPho for at least 59% were retested as described before. Retested compounds inhibiting PfGluPho activity ≥ 50% were further investigated.
Diaphorase counter-screen
First 12 nL of 10 mM compounds in DMSO were transferred into columns 5–48 of 1,536-well plates using the Echo Liquid Handler. Next, 12 nL of DMSO were transferred to columns 1–4 for positive and negative controls. Subsequently, 3 μL of substrate mix (50 mM Tris (pH 7.5), 0.005% Tween20, 1 mg/mL BSA, 6.6 mM MgCl2, 0.06 mM/0.02 mM NADPH, 0.05 mM resazurin) were added to columns 1–48 using the Multidrop Combi reagent dispenser (Thermo Scientific). To initiate the reaction, 3 μL of enzyme solution (50 mM Tris (pH 7.5), 0.005% Tween 20, 1 mg/mL BSA, 6.6 mM MgCl2, 2 U/mL diaphorase) were added to columns 3–48 with the Multidrop Combi dispenser. For the negative control, 3 μL of the enzyme solution without diaphorase were added to columns 1–2. The plates were centrifuged at 1,500 rpm for 1 min, incubated in the dark for 2 hours, and read at ex530/em590 nm. Diaphorase inhibition was analyzed using CBIS and compounds exhibiting at least 50% enzyme inhibition were assigned active status and thus not included in further experiments.
Hit confirmation in dose response
Varying volumes of compounds in DMSO (starting final concentration of 20 μM) were transferred to columns 5–48 of 1,536-well plates using the Echo Liquid Handler. DMSO was added to control and test compound wells to equilibrate DMSO concentrations across plate (final concentration of 1.0%). The remaining assay procedures for the PfGluPho assay were performed as described in ‘Primary Screening.’ Compounds with an IC50 of ≤ 20 μM were further investigated.
hG6PD counter screen
Using the Echo Liquid Handler, varying volumes of compounds (starting final concentration of 20 μM) were transferred to wells of columns 5–48 of 1,536-well plates. DMSO was added to control and test compound wells to equilibrate DMSO concentrations across the plate (final concentration of 0.8%). Then 2.5 μL of enzyme solution (50 mM Tris (pH 7.5), 0.005% Tween 20, 1 mg/mL BSA, 0.05 μg/mL hG6PD) were transferred to all wells. Following enzyme addition, 2.5 μL substrate solution (0.64 mM G6P, 50 mM Tris (pH 7.5), 0.005% Tween 20, 1 mg/mL BSA, 12 μM NADP+, 6.6 mM MgCl2, 2 U/mL diaphorase, 0.28 mM resazurin) were added to columns 3–48 and solution without G6P to columns 1–2 for positive control. After 1 min of centrifugation at 1,500 rpm and 90 min incubation in the dark, fluorescence of resorufin was detected at ex530/em590 by using the Viewlux plate reader. IC50 values were calculated using CBIS.
Orthogonal assay
Using the Echo Liquid Handler, varying volumes of compounds were transferred to wells of columns 5–48 of 1,536-well plates. DMSO was added to control and test compound wells to equilibrate DMSO concentrations across the plate. Following compound and DMSO transfer, 2.5 μL of enzyme solution (50 mM Tris (pH 7.5), 0.005% Tween 20, 1 mg/mL BSA, 0.4 μg/mL PfGluPho) were added to all wells using a Multidrop Combi dispenser. Then 2.5 μL of substrate solution (40 μM G6P, 6 μM NADP+, 50 mM Tris (pH 7.5), 0.005% Tween 20, 1 mg/mL BSA, 6.6 mM MgCl2) were added to columns 5–48, and 2.5 OL of solution without G6P were added to columns 1–4 for positive control. The plates were centrifuged and either incubated for 45 min to determine endpoints of the reaction or kinetically read at ex 340nm /em 450 nm over 45 min. The reaction rate was calculated by dividing the relative fluorescence units (RFU) by time in minutes. CBIS was used to calculate IC50 values, and compounds with IC50s ≤ 80 μM were designated active.
Chemistry
All reactions involving air and moisture-sensitive reagents and solvents were performed under a nitrogen atmosphere using standard chemical techniques. Anhydrous solvents were purchased and freshly used from Sigma-Aldrich or EMD Biosciences. All organic reagents were used as purchased. (R)-(1-ethylpyrrolidin-2-yl)methanamine (Catalog no. 357000010, CAS 22795-97-7) and (S)-(1- ethylpyrrolidin-2-yl)methanamine (Catalog no. 367750010, CAS 22795-99-9) were purchased from Acros Organics and used without further purification. Analytical thin-layer chromatography was performed on Partisil K6F silica gel 60 A, 250 μm. Microwave-assisted reactions were performed using a CEM Discover system. 1H and 13C chemical shifts are reported in δ values in ppm in the corresponding solvent. All solvents used for chromatography on the synthetic materials were Fisher Scientific HPLC grade, and the water was Millipore Milli-Q PP filtered. All NMR spectra for the synthetic materials were recorded on a Bruker Avance II 400 or DRX-500 MHz instrument. High Resolution Mass Spectrometry (HRMS) spectra were carried out on an Agilent 6224A Accurate-Mass Time-of-Flight (TOF) LC/MS system with electron spray ionization (ESI). LCMS analysis of synthetic materials was completed on a Waters Autopurification system, which consists of a 2767 sample manager, a 2545 binary gradient module, a system fluidics organizer, a 2489 UV/vis detector, and a 3100 mass detector, all controlled with MassLynx software. A Sunfire Analytical C18 5 μm column (4.6 × 50 mm) and stepwise gradient {10% [(MeCN + 0.1% TFA) in (water + 0.1% TFA)] to 98% [(MeCN + 0.1% TFA) in (water + 0.1% TFA)] for 9 min.} was used for analytical LCMS of final compounds. The final compounds were purified by RP-HPLC on a SunFire preparative C18 column (5 μm; 19 × 50 mm) using a stepwise gradient {10% [(MeCN + 0.1% TFA) in (water + 0.1% TFA)] for 1 min.; 30% for 1 min.; 50% for 4 min.; 70% for 1.5 min. and 98% for 1.5 min.}. All synthesized final compounds 5a–j and 10–42 were determined to be ≥ 95% pure by LC-MS. Compound identity was verified by 1H NMR and HRMS, and additionally by 13C NMR for 11. The MestReNova program was used to interpret NMR spectra.
General procedure for the synthesis of compounds 5a-j & 10–42 as illustrated by synthesis of (R, Z)-N-((1-ethylpyrrolidin-2-yl)methyl)-2-(2-fluorobenzylidene)-3-oxo-3,4-dihydro-2Hbenzo[ b][1,4]thiazine-6-carboxamide (11)
4-(2-ethoxy-2-oxoethylthio)-3-nitrobenzoic acid (2a)
4-fluoro-3-nitrobenzoic acid (1a) (5.55 g, 30 mmol) and sodium acetate (2.71 g, 33 mmol) were combined in a round bottom flask. Ethyl mercaptoacetate (3.62 mL, 33 mmol) and water (30 mL) were then added, and the mixture was heated under nitrogen atmosphere at 90 °C for 24 h. The reaction mixture was cooled to 23 °C and then placed in an ice bath for 10–20 min. The precipitate formed was collected by filtration, washed with water, and dried to afford 2a as a tan solid (8.66 g, quant.) 1H NMR (500 MHz, CDCl3) δ 9.02 – 8.91 (m, 1H), 8.34 – 8.20 (m, 1H), 7.64 (d, J = 8.6 Hz, 1H), 4.26 (q, J = 7.1 Hz, 2H), 3.84 (s, 2H), 1.31 (t, J = 7.1 Hz, 3H); LRMS (ESI-ve): calculated for C11H11NO6S, [M-H] = 284.02, observed [M-H] = 283.99.
3-oxo-3,4-dihydro-2H-benzo[b][1,4]thiazine-6-carboxylic acid (3a)
2a (3.94 g, 13.8 mmol) and iron powder (3.08 g, 55 mmol) were suspended in glacial acetic acid (33 mL) in a round-bottom flask equipped with an air condenser under nitrogen atmosphere. The mixture was heated to 80 °C in an oil bath, during which the suspension turned reddish brown, and the temperature of the bath rose to 92 °C. After 30 minutes the temperature of the oil bath was lowered to 80 °C and maintained for 2 h. The mixture was then allowed to cool to 23 °C and stir for about 20 h and then was poured into 1N HCl solution (150 mL). After stirring the mixture for 30 minutes, the precipitate was filtered, washed extensively with water and diethyl ether, and dried to afford 3a as a light gray solid (2.56 g, 89%). 1H NMR (500 MHz, DMSO-d6) δ 12.99 (s, 1H), 10.72 (s, 1H), 7.55 (s, 1H), 7.50 (d, J = 7.9 Hz, 1H), 7.42 (d, J = 7.9 Hz, 1H), 3.31 (s, 2H); LRMS (ESI-ve): calculated for C9H6NO3S, [M-H] = 208.01, observed [M-H] = 207.95.
(Z)-2-(2-fluorobenzylidene)-3-oxo-3,4-dihydro-2H-benzo[b][1,4]thiazine-6-carboxylic acid (4a)
3a (1.2 g, 5.7 mmol), was suspended in acetic anhydride (20 mL) in a 40 mL vial. Triethylamine (2.39 mL, 17.2 mmol) and 2-fluorobenzaldehyde (1.21 mL, 11.5 mmol) were added, and the mixture was heated at 145 °C for 15 h. The reaction mixture was cooled to 23 °C, poured into 100 mL of diethyl ether, and cooled at 4 °C for 20 min. The precipitate was collected by filtration, washed with ether (25 mL), and dried to afford 4a as a yellowish brown solid (1.28 g, 71%). 1H NMR (500 MHz, DMSO-d6) δ11.30 (s, 1H), 7.86 (s, 1H), 7.79 (t, 1H), 7.69 (d, J = 1.7 Hz, 1H), 7.55 (dd, J = 8.3, 1.7 Hz, 1H), 7.53 – 7.48 (m, 1H), 7.43 (d, J = 8.2 Hz, 1H), 7.40 – 7.33 (m, 2H); LRMS (ESI-ve): calculated for C16H10FNO3S, [M-H] = 314.03, observed [M-H] = 314.0.
(R,Z)-N-((1-ethylpyrrolidin-2-yl)methyl)-2-(2-fluorobenzylidene)-3-oxo-3,4-dihydro-2Hbenzo[ b][1,4]thiazine-6-carboxamide (11)
4a (300 mg, 0.95 mmol), HOBt (193 mg, 1.43 mmol) and EDCI·HCl (274 mg, 1.43 mmol) were combined in an 8 mL vial. Dry dichloromethane (4 mL) and triethylamine (0.4 mL, 2.85 mmol) were added, and the mixture was stirred at 23 °C for 5 minutes. (R)-(1-ethylpyrrolidin-2-yl)methanamine (0.2 mL, 1.43 mmol) was added, and within a few minutes the solution became homogenous and was allowed to stir for about 15 h. The reaction mixture was then diluted with 20% methanol/dichloromethane (5 mL) and extracted with water (4 mL, 3x). The organic layer was dried over anhydrous sodium sulfate and evaporated to afford a brown residue. The residue was purified by RP-HPLC on a SunFire preparative C18 column (5 μm; 19 × 50 mm) using a stepwise gradient {10% [(MeCN + 0.1% TFA) in (water + 0.1% TFA)] for 1 min.; 30% for 1 min.; 50% for 4 min.; 70% for 1.5 min. and 98% for 1.5 min.}. The fractions containing the expected molecular weight were collected and freeze-dried to afford a formate salt of the product as a pale yellow solid. The solid was dissolved in dichloromethane and extracted with 25% K2CO3 solution (3x). The organic layer was dried over anhydrous sodium sulfate and evaporated to afford 11 as a yellowish brown solid (0.13 g, 32%). Melting Point = 194–197 °C ; [α]D = (+)−12.9 (c = 0.001, MeOH, 23 °C); 1H NMR (500 MHz, acetic acid-d4) δ 8.05 (s, 1H), 7.83 (t, J = 7.3 Hz, 1H), 7.60 (dd, J = 8.3, 1.7 Hz, 1H), 7.52 (d, J = 1.6 Hz, 1H), 7.44 (dd, J = 13.3, 6.3 Hz, 1H), 7.36 (d, J = 8.3 Hz, 1H), 7.31 (t, J = 7.5 Hz, 1H), 7.20 (t, J = 7.5 Hz, 1H), 3.98 – 3.76 (m, 4H), 3.54 – 3.41 (m, 1H), 3.23 – 3.08 (m, 2H), 2.37 – 2.22 (m, 1H), 2.12 – 2.05 (m, 3H), 1.33 (t, J = 7.5 Hz, 3H). For 1H NMR in methanol-d4, see ancillary information. LRMS (ESI+ve): calculated for C23H24FN3O2S, [M+H] = 426.17, observed [M+H] = 426.26; HRMS (ESI+ve): calculated for C23H24FN3O2S, [M+H] = 426.1646, observed [M+H] = 426.1631.
Activity against P. falciparum in culture
The chloroquine-sensitive P. falciparum 3D7 and the resistant Dd2 strains were cultured as described elsewhere.38 Effects of PfGluPho inhibitors on Plasmodium parasites were tested using isotopic drug sensitivity assays,39,40 which are based on the incorporation of radioactive 3H-hypoxanthine. Hypoxanthine is taken up by Plasmodium parasites, since it is needed for nucleic acid synthesis. In brief, P. falciparum-infected RBCs were seeded in 96-well plates (Nunc) at a parasitemia of 0.25% (>70% ring forms) and 1.25% hematocrit in hypoxanthine-free medium. Varying concentrations of the test compounds and the control chloroquine (Sigma) were added to the wells. The parasites were incubated for 48 hours, after which 0.5 μCi 3H-hypoxanthine was added for a further 24 hours. The cells of each well were harvested on a glass fiber filter (Perkin-Elmer), washed, and dried. Finally, the radioactivity of each condition was detected, which is supposed to be proportional to the parasites’ growth. IC50 values were calculated using non-linear regression of the GraphPad Prism software.
MOI studies
PfGluPho inhibitors were mechanistically characterized as previously described.34 In brief, varying G6P or NADP+ concentrations were prepared in a substrate solution (final assay concentration (FAC) 3.3 mM MgCl2, 1 mM TCEP, 0.05 M Tris, for G6P titration: 5 μM NADP+/for NADP+ titration: 25 μM G6P) and transferred to 384-well plate. Varying compound concentrations (starting at around 8x IC50) titrated in DMSO were added to the wells using the Echo Liquid Handler. Addition of an enzyme mix (FAC 0.2 μg/mL PfGluPho, 0.005% Tween 20) started the reaction. Fluorescence of NADPH production was monitored over 10 min at ex340/em460 with a SpectraMax M5 (Molecular Devices) multiwell plate reader and the initial slope was determined with the SoftMax Pro 5.2 software (Molecular Devices). Based on the data, Lineweaver-Burk plots were created,41 and Km and Vmax values were calculated with the GraphPrad Prism software. Additionally, α from the following rate equation was calculated using SigmaPlot (Systat Software) (Eq. (1); v=velocity, S=substrate, I=inhibitor).
| Eq. (1) |
If Lineweaver-Burk plots intersected at the y-axis, Kmapp increased, and Vmaxapp was constant with increasing inhibitor concentrations, α = ∞, then compounds were assigned to inhibit competitively. Lineweaver-Burk plots intersecting at the x-axis, constant Kmapp and decreasing Vmaxapp values with increasing inhibitor concentrations, and α = 1 indicated non-competitive inhibition. Parallel lines in Lineweaver-Burk plots, decreasing Kmapp and Vmaxapp values with increasing inhibitor concentration, and α < 1 was characteristic for uncompetitive inhibition.42 Were parameters found to be between those for competitive and non-competitive inhibitors, compounds were assigned to follow mixed-type inhibition. Eq. (1)
Protocol for chemical stability (reactivity/GSH adduct formation)
Protocol: 10 μL of compound (1 mM stock in DMSO) was added to 985 μL PBS buffer (1x, pH =7.4) in a capped glass vial. 5 μL GSH solution (10 mM stock in PBS) was added to the solution, and the mixture was gently mixed on a Grant-Bio PS-M3D platform shaker at 23 °C. 20 mL aliquots were analyzed at 0, 0.5, 1, 2, 4, 6, 18, and 25h on a Waters HPLC-MS system using a SunFire analytical C18 column (5 μm; 4.6 × 50 mm) using a stepwise gradient [11: {10 % [(MeCN + 0.1% TFA) in (water + 0.1% TFA)] for 1 min.; 30 % for 1 min.; 50 % for 4 min.; 70 % for 1.5 min.; and 98 % for 1.5 min.}; Ethacrynic acid: {10 % [(MeCN + 0.1% TFA) in (water + 0.1% TFA)] for 1 min.; 30 % for 1 min.; 50 % for 4 min.; 70 % for 1.5 min.; and 98 % for 1.5 min.}]. The percentage remaining for each compound relative to the starting amount of compound at 0 h was calculated from the area under the curve (AUC) at different time points and plotted.
Supplementary Material
Acknowledgments
The authors would like to thank Michaela Stumpf, Beate Hecker, and Elisabeth Fischer for their excellent technical assistance. The study was supported by the NIH (1R21AI082434-01) to LB, the Deutsche Forschungsgemeinschaft (BE 1540/18-1) to KB, and an NIH Molecular Libraries grant (U54 HG005033-03) to the Conrad Prebys Center for Chemical Genomics at the Sanford Burnham Medical Research Institute, one of the comprehensive centers of the NIH Molecular Libraries Probe Production Centers Network (MLPCN).
Abbreviations Used
- ADME/T
absorption, distribution, metabolism, excretion, toxicity
- AUC
area under the curve
- BBB
blood-brain barrier
- CBIS
Chemical and Biological Information Systems
- EDCI
1-ethyl-3-(3- dimethylaminopropyl) carbodiimide
- ESI
electron spray ionization
- ex/em
excitation/emission
- FAC
final assay concentration
- G6P
glucose-6-phosphate
- G6PD
glucose-6-phosphate dehydrogenase
- hG6PD
human glucose-6-phosphate dehydrogenase
- HRMS
high resolution mass spectrometry
- HTS
high-throughput screening
- IRBC
infected red blood cells
- MLSMR
Molecular Libraries Small Molecule Repository
- MOI
mechanism of inhibition
- PAMPA
parallel artificial membrane permeability assay
- PfG6PD
Plasmodium falciparum glucose-6-phosphate dehydrogenase
- PfGluPho
glucose-6-phosphate dehydrogenase 6-phosphogluconolactonase
- PPP
pentose phosphate pathway
- RNAi
RNA interference
- SAR
structure-activity relationship
- TOF
time-of-flight
Footnotes
Supporting Information Available
Detailed experimental procedures for the synthesis of 1b, 1c, 2b, 2c, 3b, 3c, 3d, 4b, 4c, 4d, 4e, 4f, 4g, 4h, 4i, 4j, 5a, 5b, 5c, 5d, 5e, 5f, 5g, 5h, 5i, 5j, 10, 12, 13, 14, 15, 17, 22, 29, 33, 34, 39, 40, 41, 42 as well as NMR-spectra of 2a, 3a, 3d, 4a, 5a, 5b, 5c, 5d, 5e, 5f, 5g, 5h, 5i, 5j, 10, 11 (acetic acid-d4), 11 (methanol-d4), 12, 13, 14, 15, 17, 22, 29, 33, 34, 39, 40, 41, 42 are reported in the Supplementary Information. This material is available free of charge via the Internet at http://pubs.acs.org
References
- 1.Murray CJL, Rosenfeld LC, Lim SS, Andrews KG, Foreman KJ, Haring D, Fullman N, Naghavi M, Lozano R, Lopez AD. Global malaria mortality between 1980 and 2010: a systematic analysis. The Lancet. 2012;379:413–431. doi: 10.1016/S0140-6736(12)60034-8. [DOI] [PubMed] [Google Scholar]
- 2.WHO. World malaria report 2008. 2008 http://whqlibdoc.who.int/publications/2008/9789241563697_eng.pdf.
- 3.Baron JM, Higgins JM, Dzik WH. A revised timeline for the origin of Plasmodium falciparum as a human pathogen. J Mol Evol. 2011;73:297–304. doi: 10.1007/s00239-011-9476-x. [DOI] [PubMed] [Google Scholar]
- 4.Tanabe K, Mita T, Jombart T, Eriksson A, Horibe S, Palacpac N, Ranford-Cartwright L, Sawai H, Sakihama N, Ohmae H, Nakamura M, Ferreira MU, Escalante AA, Prugnolle F, Bjorkman A, Farnert A, Kaneko A, Horii T, Manica A, Kishino H, Balloux F. Plasmodium falciparum accompanied the human expansion out of Africa. Curr Biol. 2010;20:1283–1289. doi: 10.1016/j.cub.2010.05.053. [DOI] [PubMed] [Google Scholar]
- 5.Rodrigues T, Moreira R, Lopes F. New hope in the fight against malaria? Future Med Chem. 2011;3:1–3. doi: 10.4155/fmc.10.274. [DOI] [PubMed] [Google Scholar]
- 6.Hill AV. Vaccines against malaria. Philos Trans R Soc Lond B Biol Sci. 2011;366:2806–2814. doi: 10.1098/rstb.2011.0091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Murambiwa P, Masola B, Govender T, Mukaratirwa S, Musabayane CT. Anti-malarial drug formulations and novel delivery systems: a review. Acta Trop. 2011;118:71–79. doi: 10.1016/j.actatropica.2011.03.005. [DOI] [PubMed] [Google Scholar]
- 8.Bousejra-El Garah F, Wong MH, Amewu RK, Muangnoicharoen S, Maggs JL, Stigliani JL, Park BK, Chadwick J, Ward SA, O’Neill PM. Comparison of the reactivity of antimalarial 1,2,4,5-tetraoxanes with 1,2,4-trioxolanes in the presence of ferrous iron salts, heme, and ferrous iron salts/phosphatidylcholine. J Med Chem. 2011;54:6443–6455. doi: 10.1021/jm200768h. [DOI] [PubMed] [Google Scholar]
- 9.Schlitzer M. Antimalarial drugs - What is in use and what is in the pipeline. Arch Pharm. 2008;341:149–163. doi: 10.1002/ardp.200700184. [DOI] [PubMed] [Google Scholar]
- 10.Murambiwa P, Masola B, Govender T, Mukaratirwa S, Musabayane CT. Anti-malarial drug formulations and novel delivery systems: A review. Acta Trop. 2011;118:71–79. doi: 10.1016/j.actatropica.2011.03.005. [DOI] [PubMed] [Google Scholar]
- 11.Trape JF. The public health impact of chloroquine resistance in Africa. Am J Trop Med Hyg. 2001;64:12–17. doi: 10.4269/ajtmh.2001.64.12. [DOI] [PubMed] [Google Scholar]
- 12.Marks F, von Kalckreuth V, Kobbe R, Adjei S, Adjei O, Horstmann RD, Meyer CG, May J. Parasitological rebound effect and emergence of pyrimethamine resistance in Plasmodium falciparum after single-dose sulfadoxine-pyrimethamine. J Infect Dis. 2005;192:1962–1965. doi: 10.1086/497698. [DOI] [PubMed] [Google Scholar]
- 13.Egan TJ, Kaschula CH. Strategies to reverse drug resistance in malaria. Curr Opin Infect Dis. 2007;20:598–604. doi: 10.1097/QCO.0b013e3282f1673a. [DOI] [PubMed] [Google Scholar]
- 14.Gottschall JL, Elliot W, Lianos E, McFarland JG, Wolfmeyer K, Aster RH. Quinineinduced immune thrombocytopenia associated with hemolytic uremic syndrome: a new clinical entity. Blood. 1991;77:306–310. [PubMed] [Google Scholar]
- 15.Veinot JP, Mai KT, Zarychanski R. Chloroquine related cardiac toxicity. J Rheumatol. 1998;25:1221–1225. [PubMed] [Google Scholar]
- 16.WHO. Guidelines for the treatment of malaria. 2010 http://whqlibdoc.who.int/publications/2010/9789241547925_eng.pdf.
- 17.Dondorp AM, Nosten F, Yi P, Das D, Phyo AP, Tarning J, Lwin KM, Ariey F, Hanpithakpong W, Lee SJ, Ringwald P, Silamut K, Imwong M, Chotivanich K, Lim P, Herdman T, An SS, Yeung S, Singhasivanon P, Day NP, Lindegardh N, Socheat D, White NJ. Artemisinin resistance in Plasmodium falciparum malaria. N Engl J Med. 2009;361:455–467. doi: 10.1056/NEJMoa0808859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Noedl H, Se Y, Sriwichai S, Schaecher K, Teja-Isavadharm P, Smith B, Rutvisuttinunt W, Bethell D, Surasri S, Fukuda MM, Socheat D, Chan Thap L. Artemisinin resistance in Cambodia: a clinical trial designed to address an emerging problem in Southeast Asia. Clin Infect Dis. 2010;51:e82–89. doi: 10.1086/657120. [DOI] [PubMed] [Google Scholar]
- 19.Carrara VI, Zwang J, Ashley EA, Price RN, Stepniewska K, Barends M, Brockman A, Anderson T, McGready R, Phaiphun L, Proux S, van Vugt M, Hutagalung R, Lwin KM, Phyo AP, Preechapornkul P, Imwong M, Pukrittayakamee S, Singhasivanon P, White NJ, Nosten F. Changes in the treatment responses to artesunate-mefloquine on the northwestern border of Thailand during 13 years of continuous deployment. PLoS One. 2009;4:e4551. doi: 10.1371/journal.pone.0004551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jortzik E, Mailu BM, Preuss J, Fischer M, Bode L, Rahlfs S, Becker K. Glucose-6- phosphate dehydrogenase-6-phosphogluconolactonase: a unique bifunctional enzyme from Plasmodium falciparum. Biochem J. 2011;436:641–650. doi: 10.1042/BJ20110170. [DOI] [PubMed] [Google Scholar]
- 21.Nkhoma ET, Poole C, Vannappagari V, Hall SA, Beutler E. The global prevalence of glucose-6-phosphate dehydrogenase deficiency: a systematic review and meta-analysis. Blood Cells Mol Dis. 2009;42:267–278. doi: 10.1016/j.bcmd.2008.12.005. [DOI] [PubMed] [Google Scholar]
- 22.Luzzatto L. Glucose 6-phosphate dehydrogenase deficiency: from genotype to phenotype. Haematologica. 2006;91:1303–1306. [PubMed] [Google Scholar]
- 23.Cappadoro M, Giribaldi G, O’Brien E, Turrini F, Mannu F, Ulliers D, Simula G, Luzzatto L, Arese P. Early phagocytosis of glucose-6-phosphate dehydrogenase (G6PD)- deficient erythrocytes parasitized by Plasmodium falciparum may explain malaria protection in G6PD deficiency. Blood. 1998;92:2527–2534. [PubMed] [Google Scholar]
- 24.Ruwende C, Khoo SC, Snow RW, Yates SN, Kwiatkowski D, Gupta S, Warn P, Allsopp CE, Gilbert SC, Peschu N, et al. Natural selection of hemi- and heterozygotes for G6PD deficiency in Africa by resistance to severe malaria. Nature. 1995;376:246–249. doi: 10.1038/376246a0. [DOI] [PubMed] [Google Scholar]
- 25.Atamna H, Pascarmona G, Ginsburg H. Hexose-monophosphate shunt activity in intact Plasmodium falciparum-infected erythrocytes and in free parasites. Mol Biochem Parasitol. 1994;67:79–89. doi: 10.1016/0166-6851(94)90098-1. [DOI] [PubMed] [Google Scholar]
- 26.Preuss J, Jortzik E, Becker K. Glucose-6-phosphate metabolism in Plasmodium falciparum. IUBMB Life. 2012;64:603–611. doi: 10.1002/iub.1047. [DOI] [PubMed] [Google Scholar]
- 27.Becker K, Koncarevic S, Hunt NH. Oxidative stress and antioxidant defense in malarial parasites. In: Sherman IW, editor. Molecular Approaches to Malaria. American Society of Microbiology Press; Washington DC: 2005. pp. 365–383. [Google Scholar]
- 28.Crooke A, Diez A, Mason PJ, Bautista JM. Transient silencing of Plasmodium falciparum bifunctional glucose-6-phosphate dehydrogenase- 6-phosphogluconolactonase. The FEBS J. 2006;273:1537–1546. doi: 10.1111/j.1742-4658.2006.05174.x. [DOI] [PubMed] [Google Scholar]
- 29.Baum J, Papenfuss AT, Mair GR, Janse CJ, Vlachou D, Waters AP, Cowman AF, Crabb BS, de Koning-Ward TF. Molecular genetics and comparative genomics reveal RNAi is not functional in malaria parasites. Nucleic Acids Res. 2009;37:3788–3798. doi: 10.1093/nar/gkp239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lopez-Barragan MJ, Lemieux J, Quinones M, Williamson KC, Molina-Cruz A, Cui K, Barillas-Mury C, Zhao K, Su XZ. Directional gene expression and antisense transcripts in sexual and asexual stages of Plasmodium falciparum. BMC Genomics. 2011;12:587. doi: 10.1186/1471-2164-12-587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Clarke JL, Scopes DA, Sodeinde O, Mason PJ. Glucose-6-phosphate dehydrogenase-6- phosphogluconolactonase. A novel bifunctional enzyme in malaria parasites. Eur J Biochem. 2001;268:2013–2019. doi: 10.1046/j.1432-1327.2001.02078.x. [DOI] [PubMed] [Google Scholar]
- 32.Clarke JL, Sodeinde O, Mason PJ. A unique insertion in Plasmodium berghei glucose-6- phosphate dehydrogenase-6-phosphogluconolactonase: evolutionary and functional studies. Mol Biochem Parasitol. 2003;127:1–8. doi: 10.1016/s0166-6851(02)00298-0. [DOI] [PubMed] [Google Scholar]
- 33.ML276 has been declared a probe via the Molecular Libraries Probe Production Centers Network (MLPCN) and is available through the network, see: http://mli.nih.gov/mli/
- 34.Preuss J, Hedrick M, Sergienko E, Pinkerton A, Mangravita-Novo A, Smith L, Marx C, Fischer E, Jortzik E, Rahlfs S, Becker K, Bode L. High-throughput screening for smallmolecule inhibitors of Plasmodium falciparum glucose-6-phosphate dehydrogenase 6- phosphogluconolactonase. J Biomol Screen. 2012;17:738–751. doi: 10.1177/1087057112442382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Trifilenkov AS, Kobak VV, Salina MA, Kusovkova JA, Ilyin AP, Khvat AV, Tkachenko SE, Ivachtchenko AV. Liquid-phase parallel synthesis of combinatorial libraries of substituted 6-carbamoyl-3,4-dihydro-2H-benzo[1,4]thiazines. J Comb Chem. 2006;8:469–479. doi: 10.1021/cc050132u. [DOI] [PubMed] [Google Scholar]
- 36.Worley JW, Ratts KW, Cammack KL. 2-Dialkylphosphonyl- and 2-Alkylidene-3,4- dihydro-3-oxo-2H-1, 4-benzothiazines. J Org Chem. 1975;40:1731–1734. [Google Scholar]
- 37.Ploemen JH, Van Schanke A, Van Ommen B, Van Bladeren PJ. Reversible conjugation of ethacrynic acid with glutathione and human glutathione S-transferase P1-1. Cancer Res. 1994;54:915–919. [PubMed] [Google Scholar]
- 38.Koncarevic S, Rohrbach P, Deponte M, Krohne G, Prieto JH, Yates J, 3rd, Rahlfs S, Becker K. The malarial parasite Plasmodium falciparum imports the human protein peroxiredoxin 2 for peroxide detoxification. Proc Natl Acad Sci U S A. 2009;106:13323–13328. doi: 10.1073/pnas.0905387106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Desjardins RE, Canfield CJ, Haynes JD, Chulay JD. Quantitative assessment of antimalarial activity in vitro by a semiautomated microdilution technique. Antimicrob Agents Chemother. 1979;16:710–718. doi: 10.1128/aac.16.6.710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fivelman QL, Adagu IS, Warhurst DC. Modified fixed-ratio isobologram method for studying in vitro interactions between atovaquone and proguanil or dihydroartemisinin against drug-resistant strains of Plasmodium falciparum. Antimicrob Agents Chemother. 2004;48:4097–4102. doi: 10.1128/AAC.48.11.4097-4102.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cornish-Bowden A. Introduction to enzyme kinetics. In: Cornish-Bowden A, editor. Fundamentals of enzyme kinetics. 3. Portland Press; London: 2004. [Google Scholar]
- 42.Cornish-Bowden A. Reversible inhibition and activation. In: Cornish-Bowden A, editor. Fundamentals of enzyme kinetics. 3. Portland Press; London: 2004. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.