

Abstract
MEK1 phosphorylates ERK1/2 and regulates T cell generation, differentiation and function. MEK1 has recently been shown to translocate to the nucleus. Its nuclear function is largely unknown. By studying human CD4 T cells we demonstrate that a low level of MEK1 is present in the nucleus of CD4 T cells under basal conditions. T cell activation further increases the nuclear translocation of MEK1. MEK1 interacts with the nuclear receptor co-repressor SMRT. MEK1 reduces the nuclear level of SMRT in an activation-dependent manner. MEK1 is recruited to the promoter of c-Fos upon TCR stimulation. Conversely, SMRT is bound to the c-Fos promoter under basal conditions and is removed upon TCR stimulation. We examined the role of SMRT in regulation of T cell function. siRNA-mediated knockdown of SMRT results in a biphasic effect on cytokine production. The production of the cytokines—IL2, IL4, IL10 and IFNγ increases in the early phase (8 hr) and then decreases in the late phase (48 hr). The late phase decrease is associated with inhibition of T cell proliferation. The late phase inhibition of T cell activation is, in part, mediated by IL10 that is produced in the early phase, and in part, by β-catenin signaling. Thus, we have identified a novel nuclear function of MEK1. MEK1 triggers a complex pattern of early T cell activation followed by a late inhibition through its interaction with SMRT. This biphasic dual effect likely reflects a homeostatic regulation of T cell function by MEK1.
Keywords: Nuclear MEK1, SMRT, ChIP, T cell activation, cytokine production
Introduction
Mitogen-activated protein kinases (MAPKs) play an essential role in many fundamental cellular functions including cell proliferation, differentiation, survival, locomotion and secretion (1). ERK1 and ERK2 represent a major subfamily of MAPKs. They are activated through unique threonine-tyrosine phosphorylation. MEK1 and MEK2 specifically phosphorylate the TEY motif of ERK1 and ERK2. MEK1 knockout is embryonic lethal (2, 3). Pharmacological inhibitors of MEK1/2 potently inhibit ERK1/2 activation. This approach allowed extensive characterization of the role of the MEK-ERK1/2 pathway in cellular function. The MEK-ERK1/2 signaling pathway plays an important role in different stages of thymic differentiation of CD4 and CD8 T cells (4–6). It is also important for mature T cell activation (7) and differentiation (8).
MEK1 has previously been localized to the cytosol (9) and late endosome (10). Recent studies have identified a novel and non-canonical nuclear localization motif (11). Phosphorylation of this motif leads to the nuclear translocation of MEK1 (12, 13). MEK1 also has an N-terminal nuclear export signal (NES: ALQKKLEELELDE, residues 32–44). The presence of the nuclear localization motif and an export signal allows MEK1 to shuttle between the nucleus and cytosol (14). The exact function of MEK1 in the nucleus is unclear. MEK1 but not MEK2 was reported to cause nuclear translocation of ERK2 (15). In addition to activating ERK1 and ERK2 MEK1 phosphorylates STAT5 (16) and MyoD (17). The phosphorylation of these transcription factors, especially MyoD is likely to occur in the nucleus. MEK1 also interacts with the nuclear receptor PPARγ and the nuclear co-repressor SMRT (silencing mediator of retinoid and thyroid hormone receptor, also known as NCoR2) and trigger their nuclear export (14, 18, 19). MEK1-mediated phosphorylation of SMRT prevents its interaction with the nuclear receptors. The interaction with SMRT was studied in an overexpression model with fusion proteins (16, 17). The direct interaction of endogenous MEK1 and SMRT in primary cells remains unknown.
SMRT is an NCoR (nuclear receptor co-repressor)-related transcriptional co-repressor (18, 20–22) and a component of a multi-molecular repressor complex that includes mSin3, TBL1, TBLR1, GPS2, and HDAC3 (23). The presence of HDACs in the complex prevents gene transcription. The SMRT targets two major groups of molecules in the nucleus. The first group includes the nuclear receptors—retinoic acid receptor (RAR), RXR, liver X receptor (LXR), vitamin D receptor (VDR), and thyroid hormone receptors (21, 22, 24, 25). The second group represents the transcription factors: AP1, NFkB, SRF, MEF2C, FoxP1, ETO1/2 and Ets family members (26–28). SMRT represses the histone 3 K27 methylase JMJD3, which de-represses many polycomb group silenced genes (29). SMRT knockout is embryonic lethal due to malformation of heart and palate (27). The function of SMRT in T cells is unknown.
In this manuscript we examined nuclear translocation of MEK1 and its consequences following activation of human CD4 T cells. We specifically examined the interaction of MEK1 with SMRT and the effect of SMRT inhibition on T cell function. We show that MEK1 interacts with SMRT in the nucleus. Both MEK1 and SMRT bind to the c-Fos promoter and regulate its transcription. SMRT knockdown results in an early phase stimulation followed by a late phase inhibition of T cell activation. IL10 and beta-catenin signaling, induced in the early stimulation phase, play an important role in the late phase negative feedback inhibition of T cell activation.
Material and Methods
Human subjects
The protocol for human blood draw and T cell signaling studies was approved by the Institutional Review Board of National Jewish Health (Denver, CO). Blood was drawn from healthy subjects upon written consent. Blood was anticoagulated with EDTA. In some experiments buffy coat (red cell-depleted leukocyte pack) was obtained from the blood bank donor through the Bonfils Blood Center.
Reagents
The mouse monoclonal anti- human CD3 (clone OKT3), and mouse monoclonal anti-human CD28 (clone CD28.2) were obtained from eBioscience (San Diego, CA). A concentration of 1–5μg/ml of each antibody was used for stimulation. Anti–human IL-2, IL-4, IL10 and IFNgamma ELISA kits were obtained from BD Biosciences. The dilution of each antibody was performed according to manufacture instructions. Rabbit monoclonal anti-p-ERK1/2, anti-pp38, and anti-MEK1 (clone 47E6, catalog number 9126) antibodies were obtained from Cell Signaling Technology (Danvers, MA), and a dilution of 1:1000 was used for each antibody as instructed by each antibody data sheet for Western Blotting purpose. For immunostaining purpose, the same anti-MEK1 antibody was used with a dilution of 1:50. Fluorescently labeled secondary antibodies are purchased from Molecular Probes (Invitrogen). Phycoerythrin (PE) and fluorescein isothiocyanate (FITC) labeled labeled mouse monoclonal anti-FoxP3 antibodies were purchased from BD Biosciences (San Jose, CA). Rabbit anti-SMRT antibody was purchased from Affinity Bioreagents/Thermo Scientific/Pierce Antibodies, Rockford, IL. For Western Blotting purpose, a concentration of 4ug/ml was used. All other antibodies were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA), and a final concentration of 0.5–2μg/ml was used for western blotting purpose. The PPARγ inhibitor T0070907 and the β-catenin/PPARγ inhibitor FH535 are from EMD/Millipore USA.
CD4+ T cell isolation and transfection
Human CD4 T cells were purified according to the method described previously (30). Briefly, PBMCs were prepared by Ficoll-Hypaque (Histopaque) gradient centrifugation (Sigma-Aldrich, St. Louis, MO). Untouched CD4+ T cells were isolated by CD4+ T cell isolation kit-II (Miltenyi Biotec, Auburn, CA) according to the manufacturer instruction. CD4+ T cells were transfected with 30–300nM of siRNA against SMRT (siGENOME SMARTpool, Dharmacon siRNA technologies) or non-targeting control siRNA (siGENOME Non-Targeting Pool #2, Dharmacon siRNA technologies). Electroporation reagents were purchased from Lonza Allendale, NJ). Electroporation was performed according to the Amaxa protocol. After electroporation, cells were rested overnight before setting them up for experiments. Small hairpin RNAs (shRNA) for human SMRT and luciferase (control) were cloned into the retroviral vector Banshee-GFP as described previously (30). The SMRT- and luciferase-targeting shRNA sequences were: GTTCACAACACAGGCATGAACTC and AACGTACGCGGAATACTTC, respectively. Production of retroviruses, T cell infection, and sorting were done as described (31, 32).
CD4+ T cells activation and proliferation
CD4+ T cells transfected with siRNA were stimulated with plate bound anti-CD3 and anti-CD28 for various time. Cell culture supernatant was harvested for ELISA. For proliferation, cells were labeled with 5-(and-6)-carboxyfluorescein diacetate, succinimidyl ester (5(6)-CFDA, SE; CFSE) (Invitrogen) at a concentration of 500 nM according to manufacturer instruction, then stimulated for 72 hours. 3H-thymidine uptake assay was performed as described previously (31, 33).
Flow cytometry
Flow cytometry was performed as described previously (33). Cells were fixed with 2% paraformaldehyde for 10 minutes at room temperature. For intracellular staining, cells were further permeabilized in ice cold 90% methanol for 30 minutes before staining with antibodies. Primary and fluorescence conjugated secondary antibodies were diluted in 0.1% bovine serum albumin dissolved in PBS at desired concentration according to the supplier’s recommendation and incubated for 30 min. Cells were incubated with primary and secondary antibody plus 10% goat serum for 30 minutes at room temperature. Fluorescence intensity was detected by CYAN (Beckman-Coulter, Fullerton, CA) unless otherwise stated. Data was analyzed by the software FlowJo (Tree Star, Ashland, OR).
Immunofluorescence staining
Immunofluorescence staining was performed as described previously (34, 35). CD4 T cells were fixed in 2% paraformaldehyde, then placed and dried on slides. Slides were blocked for 1 h in 5% serum, 0.03% Triton X-100, and then incubated at 4°C overnight with primary Ab diluted at the supplier’s recommendation or isotype control Ab. Alexa Fluor fluorochrome-conjugated secondary Ab was used at 1 μg/ml. Cells were analyzed by a Nikon TE 2000 inverted microscope using a 100x objective. The microscope is equipped with a z-motor(Prior Scientific, Rockland, MA), excitation and emission filterwheels, and a CoolSnap HQ camera (Roper Scientific-Photometrics, Tuscon, AZ). The data acquisition and analysis was done by Metamorph, version 7.1 (Molecular Devices, Downingtown, PA). Images of experimental groups to be compared were acquired using the same software setting (e.g. same exposure time). The pixel level of the images was raised to an arbitrary level (a process called thresholding) for quantitative analyses in order to eliminate the input from the background fluorescence. The same threshold was applied to all images in comparison studies. Thresholded areas within the selected regions were used for integrated fluorescence intensity measurement. The integrated intensity is defined as a sum of intensities of all selected pixels.
Real-time PCR
Real-time PCR was done as described previously (35). RNA purified from Trizol (Invitrogen) lysates was reverse transcribed using ImProm-II Reverse Transcription System (Promega) according to the manufacturer’s protocol. Absolute SYBR Green ROX Mix (Thermo Fisher Scientific) was used for DNA amplification. Real-time PCR was performed with the ABI Prism 7000 Sequence Detection System (Applied Biosystems).
Chromatin Immunoprecipitation (ChIP) Assay
The ChIP assay was performed using the ChIP-IT® Express Enzymatic kit from Active Motif, Inc. We used 5–10 × 106 CD4 T cells per ChIP assay as described (36). We stimulated cells with PMA (50 ng/ml) and ionomycin (1 μM) for 15 min and then crosslinked with formaldehyde for 10 min. Cells were treated immediately with Glycine to stop crosslinking, washed three times and then frozen at −80 °C. The thawed cells were subjected to the enzymatic shearing step as per the kit instruction. Immunoprecipitation was performed with ChIP-grade mouse anti-SMRT (Thermo Fisher Scientific), anti-MEK1 (Santa Cruz Biotechnology (Santa Cruz, CA)) and anti-phosphohistone H3 antibodies (Millipore (Upstate, NY), and isotype control IgGs (Jackson Lab) (2 μg each), and protein A/G magnetic beads overnight at 4°C. Next day the beads were washed 3x with the provided buffer. DNA was eluted after reversal of crosslinking and used as a template for subsequent PCR. We designed 5 sequential primer sets spanning the proximal c-Fos promoter from +3 to −889 base-pair position and used them in the PCR. The primer sets were numbered as follows: #1:−889 to −665; #2:−680 to −444; #3:−463 to −210; #4:−348 to −141; and #5:−155 to +3.
Data analysis
Each experiment was done 3–10 times using blood lymphocytes from different donors. The statistical significance of difference between two samples was calculated by paired t test. Results are shown as mean ± standard error of means (SEM). P<0.05 was considered statistically significant.
Results
Nuclear translocation of MEK1
MEK1 and MEK2 are two threonine-tyrosine protein kinases that play a crucial role in T cell activation by activating ERK1/2. MEK1 but not MEK2 has a non-canonical nuclear localization motif—TPT (residues 386–388) (11). Phosphorylation of the threonine residues in this motif causes its translocation to the nucleus. MEK1 shuttles between the nucleus and cytosol at a low level under basal conditions. The nuclear translocation increases upon stimulation (12). We examined the expression of MEK1 and MEK2 in human CD4 T cells using an anti-MEK1 and an anti-MEK1/2 antibody. The anti-MEK1 antibody detected a major band with an approximate MW of 45kD (Figure 1A). There was a faint band below the 45 kD species. The anti-MEK1/2 antibody did not reveal any additional band. Based upon these findings we conclude that the 45 kD is the major MEK1 species in human CD4 T cells. The expression of MEK2 in this cell type is negligible. Next, we examined the nuclear translocation of MEK1 in resting and activated CD4 T cells by western blotting and immunofluorescence staining. Resting CD4 T cells from healthy human subjects had a low level of MEK1 in the nucleus (Figure 1B–D). Stimulation of CD4 T cells with anti-CD3 and anti-CD28 antibodies increased the overall expression of MEK1 and its nuclear translocation. Treatment of cells with leptomycin B, a known inhibitor of nuclear export, further increased the nuclear level of MEK1.
Figure 1.

A: Expression of MEK1 and MEK2 in human CD4 T cells. Purified human CD4 T cells from a healthy subject were western blotted using an anti-MEK1 and an anti-MEK1/2 antibody (N=3). Nuclear expression of MEK1. B: Western blotting of nuclear extract. Purified human CD4 T cells from a healthy subject were incubated in medium alone or with anti-CD3/CD28 antibodies for 1 hr and then were processed for nuclear and cytosolic extracts and western blotted for MEK1. Nuclear and cytosolic extracts were reprobed for JunD and tubulin, respectively (N=4). C&D: Immunoflurescence staining. Purified CD4 T cells were incubated in medium alone or with anti-CD3/CD28 antibodies +/− leptomycin B (LMB, 10 ng/ml) for 1 hr and then stained for MEK1 (green). Nuclei were stained blue with DAPI. All images were thresholded at the same upper and lower limits of the pixel count for comparison across the experimental groups. Integrated fluorescence intensity of nuclear (blue colored region) MEK1 from 25 cells per experiment and from 4 separate experiments was analyzed statistically (C). * P<0.05, paired t test.
MEK1 physically interacts with and regulates the transcriptional co-repressor SMRT
Next, we examined the nuclear function of MEK1. MEK1 has previously been shown to phosphorylate SMRT and prevents its interaction with the nuclear receptor-thyroid hormone receptor and the transcription factor PLZF (19). The foregoing studies were performed in overexpression models so their physiological implications remain unknown. We examined physical interaction of MEK1 with SMRT using nuclear extract from anti-CD3/CD28 antibody-stimulated CD4 T cells. MEK1 co-precipitated with SMRT from the nuclear extract (Figure 2A). SMRT exists in multiple molecular weight forms due to alternative splicing (19, 21, 22). We examined the presence of various molecular weight (MW) forms of SMRT in CD4 T cells by western blotting using a polyclonal antibody that has been used in other labs (28, 38). We observed a prominent high MW form around 170 kD and a low MW form around 120 KD (Figure 2B) in the nucleus and only the high MW form in the cytosol. There were two faint bands just below and above the 170 kD MW form. In most experiments we detected a doublet of the 170 kD form and its slightly higher MW form. The 170 kD doublet was consistently detected by antibodies obtained from three different vendors—Affinity Bioreagents/Thermo Fisher Scientific, Inc. (Waltham, MA), Santa Cruz Biotechnology, Inc. (Santa Cruz, CA) and Abcam, Inc., (Cambridge, MA). Its mRNA expression was reduced by SMRT siRNA (Figure 4D). For these reasons we will present data on the 170 kD doublet. Next, we studied the effect of TCR stimulation on SMRT mRNA expression by real-time PCR. Anti-CD3/CD28 stimulation decreased the expression of mRNA for SMRT in the first 4 hr and then increased its expression at 24 hr (Figure 2C). Immunofluorescence staining showed a predominant nuclear localization of SMRT under basal conditions (Figure 3). Anti-CD3/CD28 stimulation for 1 hr reduced the nuclear level and increased the cytosolic level of SMRT suggesting a nuclear export (Figure 3). Thus, TCR stimulation resulted in an immediate reduction in nuclear SMRT through two distinct mechanisms—inhibition at the transcriptional level and nuclear export at the protein level. The latter likely occurs due to the action of MEK1 as its inhibition with U0126 resulted in increased nuclear SMRT as shown by western blotting (Figure 2D) and immunofluorescence staining (Figure 3). We examined this issue further by overexpressing MEK1 using a bicistronic GFP-expressing retroviral vector (RV). Overexpression of MEK1 increased its level in the nucleus (Figure 2E). This was associated with a reduction in the nuclear level of SMRT.
Figure 2.

A: Co-precipitation of MEK1 with SMRT. We prepared nuclear extract from anti-CD3/CD28 stimulated CD4 T cells, immunoprecipitated with an anti-SMRT or control IgG antibody and then western blotted for MEK1. Because of disparate MWs of MEK1 (45kD) and SMRT (170 kD) the membrane was not reprobed for SMRT. Instead we show equal loading of IgH and IgL chains. The immunoprecipitate was separately western blotted for SMRT (bottom plan) (N=3). B: Identification of isoforms of SMRT in CD4 T cells. Nuclear and cytosolic extracts from CD4 T cells were western blotted for SMRT (N=6). C: Early inhibition and late induction of SMRT by CD3/CD28 stimulation. CD4 T cells were stimulated with anti-CD3/CD28 antibodies for the indicated periods of time and then used to isolate RNA and assay for mRNA for SMRT and beta-actin by real-time PCR. Data is presented as normalized ratio of SMRT to beta-actin (N=3, P<0.05, paired t test). D: MEK inhibition increases the level of nuclear SMRT. CD4 T cells were stimulated with anti-CD3/CD28 antibodies for 1 hr in the presence or absence of the MEK inhibitor U0126 (10 uM) and then western blotted for SMRT using the nuclear and cytosolic extracts. The membrane with the nuclear extract was reprobed for the nuclear repressor ZEB1 (N=3). The pixel density is shown above the bands. E: Effect of overexpression of MEK1 on SMRT expression. CD4 T cells were infected with a GFP-expressing bicistronic retrovirus containing cDNA for MEK1 (MEK1) or the empty retrovirus (RV). Cells were sorted for GFP and then western blotted for SMRT and MEK1 (N=3). A 50 kD non-specific bands in the MEK1 blot shows equal protein loading
Figure 4.

A&B: ChIP studies with c-Fos promoter. CD4 T cells were stimulated (stim) with and without (basal) PMA (50 ng/ml)/ionomycin (1 μM). DNA was cross-linked to proteins using formaldehyde and sheared by enzymatic processing using the ChIP-IT Express Enzymatic kit. ChIP grade rabbit anti-MEK1, anti-SMRT and anti-phospho-H3 antibodies and control rabbit IgG were used to immunoprecipitate the bound chromatin. Following reversal of cross-linking and digestion of the proteins the DNA was used for PCR. Five sets of primers (#1 through #5 where #5 is proximal to the start codon site) were designed from the proximal promoter sequence of c-Fos and used in PCR. An image of the electrophoresed amplicons is shown in Figure A. The input DNA was electrophoresed at 1:50 dilution. The density of the bands generated with the #1 primer set was analyzed and presented as enrichment over the input. Results from 3 separate experiments are shown in Figure 5B. C: Expression of SMRT mRNA following siRNA- and shRNA-mediated knockdown. Purified CD4 T cells were either transfected with the Smartpool siRNA or infected with the shRNA retrovirus for SMRT. After resting overnight cells were stimulated with anti-CD3/CD28 antibodies for 30 min. Purified RNA was used for real-time PCR (N=3). D&E: CD4 T cells were transfected with siSMRT or control non-targeting siRNA (siNT) and stimulated as above for 0.5, 2 and 4 hr. An aliquot of cells that were stimulated for 0.5 hr was western blotted for SMRT, c-Fos, MEK1 and beta-actin (control) (D). The other aliquots were used to isolate RNA and real-time PCR for c-Fos (E) and c-Jun (F). N=3, *P<0.05, paired t test.
Figure 3.

CD3/CD28 stimulation increases nuclear export of SMRT. Isolated CD4 T cells were incubated with medium and anti-CD3/CD28 (2 μg/ml each) antibodies +/− U0126 (10 μM) for 1 hr and then stained for SMRT. Nuclei were counterstained with DAPI. All images were thresholded at the same upper and lower limits of the pixel count for comparison across the experimental groups. Representative cells from one four separate experiments are shown.
MEK1 and SMRT bind to the promoter of c-Fos and reciprocally regulate its expression
MEK1 along with ERK1/2 has been shown to bind the promoter of c-Fos and insulin in a ChIP assay (37). The exact role of MEK1 at the promoter site is unknown but could include phosphorylation of transcriptional activators and gene repressors, and the removal of the latter from the nucleus. We examined the binding of MEK1 and SMRT to the c-Fos promoter because of its importance in IL-2 production. We performed chromatin immunoprecipitation (ChIP) studies using 5 different primer sets spanning the promoter site from +3 to −889 base-pair position. We detected the presence of SMRT at the distal site (primer set #1) of the c-Fos promoter in non-stimulated CD4 T cells and its removal after stimulation of cells with PMA and ionomycin (Figure 4A & B). MEK1 was present at a low level under basal conditions and was recruited further upon stimulation (Figure 4B). As a positive control we used an anti-phospho-H3 antibody. As anticipated phospho-H3 was absent under basal conditions but became detectable at the distal promoter after stimulation.
Its dissociation from the c-Fos promoter upon T cell stimulation suggests that SMRT regulates the expression of c-Fos. To test this possibility we developed two approaches to knockdown SMRT in the cells. We used a “smartpool siRNA” for SMRT and a control non-targeting siRNA pool (siNT), and transfected cells with these siRNA using the Amaxa protocol. We also constructed bicistronic GFP-expressing retroviral vectors encoding shRNA against luciferase (negative control) and SMRT. siSMRT and shRNA for SMRT but not the non-targeting siNT and shRNA reduced the expression of SMRT by more than 65% (Figure 4C & D). The siSMRT-mediated knockdown was associated with an increase in c-Fos protein (Figure 4D) and mRNA (Figure 4E) within 30 min. SMRT knockdown did not affect the expression of mRNA for c-Jun (Figure 4F).
SMRT knockdown results in an early increase followed by a late decline in cytokine production
Next, we studied the effect of SMRT knockdown on T cell function. We measured cytokine production by ELISA following stimulation of CD4 T cells with anti-CD3/CD28 stimulation. Since we observed a biphasic (an early phase inhibition and a late phase upregulation) effect of TCR stimulation on SMRT expression (Figure 2C), in preliminary studies we examined T cell cytokine production at multiple time points following anti-CD3/CD28 stimulation. We observed an initial rise and a late fall in IL2 production in SMRT knockdown CD4 T cells (Figure 5A). In subsequent studies we studied cytokine production at an early (8 hr) and a late (48 hr) time points. We confirmed the biphasic profile of IL2 production in a larger study group. We also observed a similar early rise and a late fall in the production of IL4, IL10 and IFNγ in SMRT knockdown T cells as compared to the control cells (Figure 5B & C). SMRT knockdown was associated with an inhibition of T cell proliferation as demonstrated by reduced CFSE dilution (Figure 5D & E). The effect on proliferation likely results from the inhibition of IL2 production that commences by about 24 hr. The late decline in IL4 and IFNγ is secondary to the dramatic reduction in the level of IL2. Supplementation with IL2 reversed the inhibitory effect of SMRT knockdown on IL4 and IFNγ production (Figure 5F). The supplementation also reversed the inhibition of T cell proliferation with siSMRT (Figure 5G). In order to understand the phenotype of SMRT knockdown T cells we studied activation of three signaling pathways that are involved in T cell activation—ERK1/2, p38 and p65 NFκB. We also examined the expression of JunD, a negative regulator of T cell activation (38). There was no difference in ERK1/2 activation after TCR stimulation of SMRT knockdown T cells (Figure 6A). The phosphorylation of p38 was mildly elevated. There was no difference in the nuclear expression level of p65 NFκB and JunD (Figure 6B). We recognize that the p65 NFkB is just one of many members of the NFκB family and our result does not rule the involvement of other NFκB members in SMRT knockdown cells.
Figure 5.

A–C: Effect of SMRT knockdown on cytokine production. Purified CD4 T cells were transfected with siSMRT and siNT and then cultured on anti-CD3/CD28 antibody-coated plates for the indicated periods of time. Supernatant collected at different time points was assayed for cytokines by ELISA. IL2 production from 3 donors is shown in A. *P<0.04, paired t test. B & C show the production profile of IL2, IL4, IL10 and IFNγ at 8 and 48 hr. The numbers at the end of the line graphs represents P values for difference (N=6–8 donors per group). D &E: SMRT knockdown inhibits T cell proliferation. CD4 T cells, transfected as per A, were labeled with CFSE, cultured on an anti-CD3/CD28-coated plate and then analyzed by flow cytometry (N=3). F&G: IL2 prevents late decline in cytokine production and restores proliferation. CD4 T cells, transfected as per A, were cultured on an anti-CD3/CD28-coated plate with and without IL2 (10 ng/ml) for 48 hr. F: Supernatant was assayed for cytokines by ELISA (N=3). G: 3H-thymidine uptake was measured at 48 hr. *P<0.04, paired t test, N=3.
Figure 6.

Effect of SMRT knockdown on cytosolic and nuclear signaling. A: CD4 T cells were transfected with siSMRT and siNT Smartpool siRNA and then stimulated with and without (medium) anti-CD3/CD28 antibodies for 10 min and then western blotted for p-p38 and pERK1/2. The p-p38 membrane was reprobed for p38, and the pERK1/2 membrane was reprobed for beta-actin. (N=4). B: CD4 T cells were transfected with siSMRT and siNT siRNA and then stimulated with anti-CD3/CD28 antibodies for 8 and 48 hr. Nuclear extract was isolated and western blotted for p65 NFkB and JunD. The membrane was reprobed for HDAC3 (N=4).
IL10 establishes a negative feedback loop to inhibit T cell activation
Since SMRT knockdown CD4 T cells produced high quantities of IL10 at the early time point, we asked if this early rise in IL10 mediated the late inhibition of cytokine production. To this goal we infected CD4 T cells with the control shRNA- or SMRT shRNA-encoding bicistronic retrovirus. Infected cells were sorted for expression of GFP, rested overnight in the medium alone and then cultured on an anti-CD3/CD28-coated plate with a neutralizing anti-IL10 or an isotype control antibody. The anti-IL10 antibody partially reversed the inhibitory effect of SMRT knockdown on the late phase IL2 production (Figure 7A). We also checked Foxp3 expression in SMRT knockdown cells. There was no difference in FoxP3 expression following SMRT knockdown (Figure 7B). Next, we examined if IL10 exerted its inhibitory effect on CD4 T cells through the co-repressor SMRT. We cultured CD4 T cells with anti-CD4/CD28 antibodies for 48 hr in the presence and absence of IL10 and then examined the expression of SMRT by immunofluorescence staining and western blotting. Anti-CD3/CD28 stimulation significantly upregulated SMRT expression (Figure 7C–E). This expression was further amplified by the addition of IL10. Cells cultured with IL10 alone showed a negligible effect.
Figure 7.

A: Anti-IL10 antibody partially prevents late decline in cytokine production. CD4 T cells were transfected with siSMRT and siNT siRNA and then stimulated with anti-CD3/CD28 antibodies for 48 hr in the presence of a neutralizing anti-IL10 antibody or IgG control (10 μg/ml each). IL2 in the supernatant was measured by ELISA. N=3, P=0.04, paired t test. B: SMRT knockdown does not affect FoxP3 expression. CD4 T cells were transfected with siSMRT and siNT siRNA and then stimulated with anti-CD3/CD28 antibodies for 48 hr. Cells were then fixed, stained with an anti-FoxP3 antibody and analyzed by flow cytometry (N=3). C–E: Effect of IL10 on SMRT expression. CD4 T cells were cultured with anti-CD3/CD28 antibodies +/−IL10. The expression of SMRT was examined by immunofluorescence staining (C) and western blotting (E, N=3) at 48 hr. Immunofluorescence staining (C) and statistical analyses of the morphometric data (D) are shown. *P<0.01, paired t test, N=4. Note that all images were thresholded at the same upper and lower limits of the pixel count for cross-comparison. F: Isolated CD4 T cells were transfected with siNT or siSMRT, then cultured in anti-CD3/CD28 antibody-coated plates for 48 hr in the presence of the PPARγ inhibitor T0070907 (T70907), β-catenin/PPARγ inhibitor FH535 or the vehicle (DMSO). IL2 in the culture supernatant was assayed by ELISA. One of three representative experiments done in triplicates is shown. Results were normalized to IL2 production in DMSO- and siNT-treated cells. *:P<0.05 compared to siNT and DMSO-treated cells (paired t test).
β-catenin signaling contributes to the late-phase inhibition of T cell IL2 production
SMRT antagonizes gene transcription induced by β-catenin (39, 40) and PPAR-γ (41). The latter molecules are known to regulate T cell function (42, 43). We applied two pharmacologic inhibitors to explore their involvement in inhibition of IL2 production in SMRT knockdown T cells. T0070907 is a specific inhibitor of PPAR-γ (44). FH535 inhibits both β-catenin and PPAR-γ (45). SMRT knockdown and control CD4 T cells were stimulated with anti-CD3/CD28 antibodies for 48 hr in the presence or absence of the inhibitors. In the presence of FH535 and T0070907 IL2 production changed to 153% and 56% of the baseline, respectively, in control T cells (Figure 7F). The result suggests that β-catenin and PPAR-γ inhibits and augments, respectively, IL2 production by human CD4 T cells. SMRT knockdown T cells without the inhibitors had an 89% inhibition of IL2 production. This inhibition was reduced to 84% and 34% of the baseline in the presence of T0070907 and FH535, respectively. Thus, FH535 increased IL2 production in SMRT knockdown cells by nearly 7-fold. The results suggest that SMRT antagonizes β-catenin signaling, which is lost in SMRT knockdown T cells. As a consequence, β-catenin-mediated inhibition dominates in the late phase of T cell activation.
Discussion
In this paper we have shown that MEK1 translocates to the nucleus following TCR stimulation and interacts with the nuclear co-repressor SMRT in CD4 T cells. MEK1 is bound to the c-Fos promoter at a low level under basal conditions and is further recruited following T cell activation. In contrast, SMRT is bound to the c-Fos promoter under basal condition and is removed following stimulation. These reciprocal changes in MEK1 and SMRT binding to the promoter site result in increased c-Fos expression and cytokine production in the early stage of T cell activation. The role of SMRT in T cell activation is far more complex than that would be expected from a simple co-repressor function. In the early stage of T cell activation SMRT functions as a co-repressor and its nuclear reduction plays a permissive role (Figure 8). In the late phase SMRT functions as an activator of T cells. SMRT antagonizes two inhibitory signals that arise in the late phase—IL10 and β-catenin. By antagonizing these two signals SMRT promotes sustained IL2 production and T cell proliferation.
Figure 8.
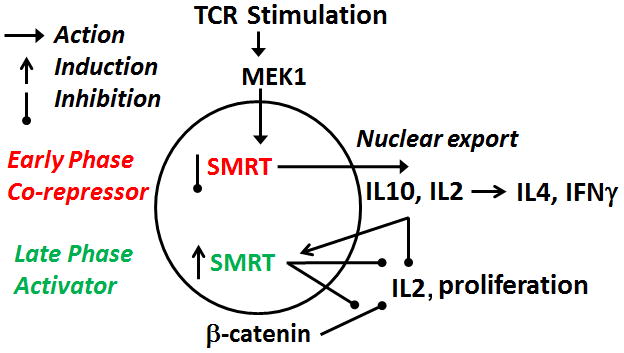
A diagrammatic presentation of the effect of nuclear MEK on SMRT and its early and late phase consequences on cytokine production and proliferation.
The translocation of MEK1 to the nucleus has previously demonstrated in other cells (10). MEK1 is translocated to the nucleus in an importin 7-dependent manner. Two distinct mechanisms have been identified for nuclear translocation (11). Translocation following a mitogenic stimulation requires MEK1 kinase activity. MEK1 also translocates to the nucleus at a low level under basal conditions. This is largely independent of MEK1 kinase activity. It requires phosphorylation of a unique TPT (residues 386–388) motif for translocation. ERK2 has been shown to phosphorylate the TPT motif. We have shown that MEK1 directly binds to the c-Fos promoter in T cells. This is agreement with a previous report where human pancreatic beta cells were studied (37). We have also shown that the recruitment of MEK1 to the promoter increases upon TCR stimulation. MEK1 has been shown to phosphorylate SMRT, which could cause its dissociation from HDAC3 and removal of this repressor complex from the promoter.
The N-terminal nuclear export signal allows MEK1 to exit the nucleus. MEK1 has previously been shown to cause nuclear export of SMRT (19) and PPARγ (14). We have demonstrated a direct interaction of endogenous MEK1 with SMRT in CD4 T cells in this study. Inhibition of MEK1 increases the nuclear content of SMRT suggesting that MEK1 regulates gene repression by controlling the nuclear level of the co-repressor SMRT. MEK1 is one of a number of molecules that regulate the nuclear level of SMRT. The others include MEKK1 (19), 14-3-3 and PIN1 (46). T cell stimulation results in activation and engagement of many of these molecules (47–49). The individual contribution of these molecules in regulating nuclear export of SMRT following T cell activation will require further studies.
SMRT has been shown to antagonize the transcriptional activity of β-catenin (39, 40). β-catenin is important for T cell development and function. β-catenin positively regulates thymopoiesis (50) and Th2 cell differentiation in the mouse model (51, 52). However, it inhibits differentiation of CD4 Th17 cells (53) and CD8 effector cells (54). β-catenin induces T cell anergy in mature T cells (55). In human T cells β-catenin signaling inhibits IL2 and IFNγ production, and blocks upregulation of the IL2 receptor (56). As a result, T cell proliferation is blocked. β-catenin impairs maturation of CD45RA naïve T cells into CD45RO memory cells. Our results are in agreement with the foregoing studies. β-catenin modestly but significantly inhibited IL2 production in human CD4 T cells (Figure 7F). This inhibition was remarkably pronounced in SMRT knockdown T cells as uncovered in our inhibitor studies with FH535. The results suggest that SMRT antagonizes β-catenin signaling in T cells. Although FH535 inhibits both β-catenin and PPARγ, we think PPARγ is unlikely to play any role in our model. The PPARγ specific inhibitor T0070907 actually inhibited IL2 production. IL2 production in SMRT knockdown T cells was unchanged in the presence of T0070907.
Previously SMRT was studied in Jurkat T cells (57). Through the use of reporter genes it was shown that CD3/CD28 ligation increased the association of SMRT with nuclear retinoic acid receptor (RAR), RXR and thyroid hormone receptor. This association resulted in silencing the target gene expression. The increase in SMRT-nuclear receptor association was PKC-theta dependent. RAR, RXR and LXR are negative regulators of T cells, although their effect on a specific T helper cell subtype may vary. A conducive T cell activation signaling requires silencing these receptors through SMRT. The late effect of SMRT knockdown in our study is in agreement with the foregoing study. We speculate that in the absence of SMRT gene transcription through RAR, RXR and LXR is uninhibited, which also contribute to the inhibition of cytokine production and T cell proliferation.
In addition to nuclear factors SMRT targets certain transcription factors and regulates the expression of their target genes. In macrophages SMRT regulates the expression of 87 LPS-inducible and LXR-regulated genes (58). Along with NCOR it co-regulates another 80 genes. We have observed SMRT recruitment to the c-Fos promoter. NFκB has been reported to be a target of SMRT (26). We have not observed any difference in the nuclear level of p65 NFκB in SMRT knockdown T cells. The results suggest that the nature of the gene targeted by SMRT varies depending upon the type of the cell. Our studies have revealed a biphasic effect of SMRT knockdown in human T cells. We believe that the early phase rise in cytokine production results from the absence of co-repressor activity of SMRT for c-Fos and possibly other SMRT-regulated genes. SMRT interacts with many histone deacetylases belonging to the class I and class II HDACs (59). Absence of SMRT could lead to the dissociation of HDACs from the gene promoters and initiation of gene transcription. The disruption of HDAC11 has been shown to induce IL10 production (60).
SMRT is a broad-spectrum co-repressor. It represses not only inhibitory but also stimulatory genes (58). Based upon this tenet we have anticipated a complex function of SMRT in T cells. Accordingly, we have observed an early phase repressive and a late phase stimulatory effects of SMRT in CD4 T cells. Our results suggest that a complete deficiency would be detrimental to CD4 T cells. However, a low level up- and down-regulation of SMRT would likely affect a wide spectrum of T cell functions.
Acknowledgments
The work was supported by NIH grants RO1 AI091614 and AI68088, PPG HL36577 and N01 HHSN272200700048C.
Abbreviation used
- MEK
Mek erk kinase
- SMRT
silencing mediator of retinoid and thyroid hormone receptor
- NCOR
nuclear receptor co-repressor
- HDAC
histone deacetylase
- PPAR
peroxisome proliferator-activated receptor
- RAR
retinoic acid receptor
Footnotes
Disclosures
The authors have no conflicting financial interests.
References
- 1.Roux PP, Blenis J. ERK and p38 MAPK-activated protein kinases: a family of protein kinases with diverse biological functions. Microbiology and molecular biology reviews: MMBR. 2004;68:320–344. doi: 10.1128/MMBR.68.2.320-344.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Giroux S, Tremblay M, Bernard D, Cardin-Girard JF, Aubry S, Larouche L, Rousseau S, Huot J, Landry J, Jeannotte L, Charron J. Embryonic death of Mek1-deficient mice reveals a role for this kinase in angiogenesis in the labyrinthine region of the placenta. Current biology: CB. 1999;9:369–372. doi: 10.1016/s0960-9822(99)80164-x. [DOI] [PubMed] [Google Scholar]
- 3.Scholl FA, Dumesic PA, Barragan DI, Harada K, Bissonauth V, Charron J, Khavari PA. Mek1/2 MAPK kinases are essential for Mammalian development, homeostasis, and Raf-induced hyperplasia. Developmental cell. 2007;12:615–629. doi: 10.1016/j.devcel.2007.03.009. [DOI] [PubMed] [Google Scholar]
- 4.Pages G, Guerin S, Grall D, Bonino F, Smith A, Anjuere F, Auberger P, Pouyssegur J. Defective thymocyte maturation in p44 MAP kinase (Erk 1) knockout mice. Science. 1999;286:1374–1377. doi: 10.1126/science.286.5443.1374. [DOI] [PubMed] [Google Scholar]
- 5.Fischer AM, Katayama CD, Pages G, Pouyssegur J, Hedrick SM. The role of erk1 and erk2 in multiple stages of T cell development. Immunity. 2005;23:431–443. doi: 10.1016/j.immuni.2005.08.013. [DOI] [PubMed] [Google Scholar]
- 6.D’Souza WN, Chang CF, Fischer AM, Li M, Hedrick SM. The Erk2 MAPK regulates CD8 T cell proliferation and survival. J Immunol. 2008;181:7617–7629. doi: 10.4049/jimmunol.181.11.7617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jorritsma PJ, Brogdon JL, Bottomly K. Role of TCR-induced extracellular signal-regulated kinase activation in the regulation of early IL-4 expression in naive CD4+ T cells. J Immunol. 2003;170:2427–2434. doi: 10.4049/jimmunol.170.5.2427. [DOI] [PubMed] [Google Scholar]
- 8.Goplen N, Karim Z, Guo L, Zhuang Y, Huang H, Gorska MM, Gelfand E, Pages G, Pouyssegur J, Alam R. ERK1 is important for Th2 differentiation and development of experimental asthma. The FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2012 doi: 10.1096/fj.11-196477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schipper HM, Agarwal-Mawal A, Paudel HK. The topography and subcellular distribution of mitogen-activated protein kinase kinase1 (MEK1) in adult rat brain and differentiating PC12 cells. Neuroscience. 1999;93:585–595. doi: 10.1016/s0306-4522(99)00120-7. [DOI] [PubMed] [Google Scholar]
- 10.Wunderlich W, Fialka I, Teis D, Alpi A, Pfeifer A, Parton RG, Lottspeich F, Huber LA. A novel 14-kilodalton protein interacts with the mitogen-activated protein kinase scaffold mp1 on a late endosomal/lysosomal compartment. The Journal of cell biology. 2001;152:765–776. doi: 10.1083/jcb.152.4.765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chuderland D, Konson A, Seger R. Identification and characterization of a general nuclear translocation signal in signaling proteins. Molecular cell. 2008;31:850–861. doi: 10.1016/j.molcel.2008.08.007. [DOI] [PubMed] [Google Scholar]
- 12.Jaaro H, Rubinfeld H, Hanoch T, Seger R. Nuclear translocation of mitogen-activated protein kinase kinase (MEK1) in response to mitogenic stimulation. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:3742–3747. doi: 10.1073/pnas.94.8.3742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wortzel I, Seger R. The ERK Cascade: Distinct Functions within Various Subcellular Organelles. Genes & cancer. 2011;2:195–209. doi: 10.1177/1947601911407328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Burgermeister E, Chuderland D, Hanoch T, Meyer M, Liscovitch M, Seger R. Interaction with MEK causes nuclear export and downregulation of peroxisome proliferator-activated receptor gamma. Molecular and cellular biology. 2007;27:803–817. doi: 10.1128/MCB.00601-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Skarpen E, Flinder LI, Rosseland CM, Orstavik S, Wierod L, Oksvold MP, Skalhegg BS, Huitfeldt HS. MEK1 and MEK2 regulate distinct functions by sorting ERK2 to different intracellular compartments. The FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2008;22:466–476. doi: 10.1096/fj.07-8650com. [DOI] [PubMed] [Google Scholar]
- 16.Maki K, Ikuta K. MEK1/2 induces STAT5-mediated germline transcription of the TCRgamma locus in response to IL-7R signaling. J Immunol. 2008;181:494–502. doi: 10.4049/jimmunol.181.1.494. [DOI] [PubMed] [Google Scholar]
- 17.Jo C, Cho SJ, Jo SA. Mitogen-activated protein kinase kinase 1 (MEK1) stabilizes MyoD through direct phosphorylation at tyrosine 156 during myogenic differentiation. The Journal of biological chemistry. 2011;286:18903–18913. doi: 10.1074/jbc.M111.225128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen JD, Evans RM. A transcriptional co-repressor that interacts with nuclear hormone receptors. Nature. 1995;377:454–457. doi: 10.1038/377454a0. [DOI] [PubMed] [Google Scholar]
- 19.Hong SH, Privalsky ML. The SMRT corepressor is regulated by a MEK-1 kinase pathway: inhibition of corepressor function is associated with SMRT phosphorylation and nuclear export. Molecular and cellular biology. 2000;20:6612–6625. doi: 10.1128/mcb.20.17.6612-6625.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Perissi V, Jepsen K, Glass CK, Rosenfeld MG. Deconstructing repression: evolving models of co-repressor action. Nature reviews Genetics. 2010;11:109–123. doi: 10.1038/nrg2736. [DOI] [PubMed] [Google Scholar]
- 21.Goodson ML, Jonas BA, Privalsky ML. Alternative mRNA splicing of SMRT creates functional diversity by generating corepressor isoforms with different affinities for different nuclear receptors. The Journal of biological chemistry. 2005;280:7493–7503. doi: 10.1074/jbc.M411514200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen JD, Umesono K, Evans RM. SMRT isoforms mediate repression and anti-repression of nuclear receptor heterodimers. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:7567–7571. doi: 10.1073/pnas.93.15.7567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Perissi V, Scafoglio C, Zhang J, Ohgi KA, Rose DW, Glass CK, Rosenfeld MG. TBL1 and TBLR1 phosphorylation on regulated gene promoters overcomes dual CtBP and NCoR/SMRT transcriptional repression checkpoints. Molecular cell. 2008;29:755–766. doi: 10.1016/j.molcel.2008.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hu X, Li S, Wu J, Xia C, Lala DS. Liver X receptors interact with corepressors to regulate gene expression. Mol Endocrinol. 2003;17:1019–1026. doi: 10.1210/me.2002-0399. [DOI] [PubMed] [Google Scholar]
- 25.Ghisletti S, Huang W, Jepsen K, Benner C, Hardiman G, Rosenfeld MG, Glass CK. Cooperative NCoR/SMRT interactions establish a corepressor-based strategy for integration of inflammatory and anti-inflammatory signaling pathways. Genes & development. 2009;23:681–693. doi: 10.1101/gad.1773109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee SK, Kim JH, Lee YC, Cheong J, Lee JW. Silencing mediator of retinoic acid and thyroid hormone receptors, as a novel transcriptional corepressor molecule of activating protein-1, nuclear factor-kappaB, and serum response factor. The Journal of biological chemistry. 2000;275:12470–12474. doi: 10.1074/jbc.275.17.12470. [DOI] [PubMed] [Google Scholar]
- 27.Jepsen K, Gleiberman AS, Shi C, Simon DI, Rosenfeld MG. Cooperative regulation in development by SMRT and FOXP1. Genes & development. 2008;22:740–745. doi: 10.1101/gad.1637108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ogawa S, Lozach J, Jepsen K, Sawka-Verhelle D, Perissi V, Sasik R, Rose DW, Johnson RS, Rosenfeld MG, Glass CK. A nuclear receptor corepressor transcriptional checkpoint controlling activator protein 1-dependent gene networks required for macrophage activation. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:14461–14466. doi: 10.1073/pnas.0405786101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jepsen K, Solum D, Zhou T, McEvilly RJ, Kim HJ, Glass CK, Hermanson O, Rosenfeld MG. SMRT-mediated repression of an H3K27 demethylase in progression from neural stem cell to neuron. Nature. 2007;450:415–419. doi: 10.1038/nature06270. [DOI] [PubMed] [Google Scholar]
- 30.Hernández-Hoyos G, Anderson MK, Wang C, Rothenberg EV, Alberola-Ila J. GATA-3 expression is controlled by TCR signals and regulates CD4/CD8 differentiation. Immunity. 2003;19:83–94. doi: 10.1016/s1074-7613(03)00176-6. [DOI] [PubMed] [Google Scholar]
- 31.Gorska MM, Stafford SJ, Cen O, Sur S, Alam R. Unc119, a novel activator of Lck/Fyn, is essential for T cell activation. J Exp Med. 2004;199:369–379. doi: 10.1084/jem.20030589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Naviaux RK, Costanzi E, Haas M, Verma IM. The pCL vector system: rapid production of helper-free, high-titer, recombinant retroviruses. J Virol. 1996;70:5701–5705. doi: 10.1128/jvi.70.8.5701-5705.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liang Q, Guo L, Gogate S, Karim Z, Hanifi A, Leung DY, Gorska MM, Alam R. IL-2 and IL-4 stimulate MEK1 expression and contribute to T cell resistance against suppression by TGF-beta and IL-10 in asthma. J Immunol. 2010;185:5704–5713. doi: 10.4049/jimmunol.1000690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gorska MM, Liang Q, Karim Z, Alam R. Uncoordinated 119 protein controls trafficking of Lck via the Rab11 endosome and is critical for immunological synapse formation. J Immunol. 2009;183:1675–1684. doi: 10.4049/jimmunol.0900792. [DOI] [PubMed] [Google Scholar]
- 35.Liu W, Tundwal K, Liang Q, Goplen N, Rozario S, Quayum N, Gorska M, Wenzel S, Balzar S, Alam R. Establishment of extracellular signal-regulated kinase 1/2 bistability and sustained activation through Sprouty 2 and its relevance for epithelial function. Molecular and cellular biology. 2010;30:1783–1799. doi: 10.1128/MCB.01003-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yu M, Wan M, Zhang J, Wu J, Khatri R, Chi T. Nucleoprotein structure of the CD4 locus: implications for the mechanisms underlying CD4 regulation during T cell development. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:3873–3878. doi: 10.1073/pnas.0800810105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lawrence MC, McGlynn K, Shao C, Duan L, Naziruddin B, Levy MF, Cobb MH. Chromatin-bound mitogen-activated protein kinases transmit dynamic signals in transcription complexes in beta-cells. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:13315–13320. doi: 10.1073/pnas.0806465105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Meixner A, Karreth F, Kenner L, Wagner EF. JunD regulates lymphocyte proliferation and T helper cell cytokine expression. EMBO J. 2004;23:1325–35. doi: 10.1038/sj.emboj.7600133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Song LN, Gelmann EP. Silencing mediator for retinoid and thyroid hormone receptor and nuclear receptor corepressor attenuate transcriptional activation by the beta-catenin-TCF4 complex. J Biol Chem. 2008 Sep 19;283(38):25988–99. doi: 10.1074/jbc.M800325200. Epub 2008 Jul 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pendás-Franco N, García JM, Peña C, Valle N, Pálmer HG, Heinäniemi M, Carlberg C, Jiménez B, Bonilla F, Muñoz A, González-Sancho JM. DICKKOPF-4 is induced by TCF/beta-catenin and upregulated in human colon cancer, promotes tumour cell invasion and angiogenesis and is repressed by 1alpha,25-dihydroxyvitamin D3. Oncogene. 2008 Jul 24;27(32):4467–77. doi: 10.1038/onc.2008.88. Epub 2008 Apr 14. [DOI] [PubMed] [Google Scholar]
- 41.Yu C, Markan K, Temple KA, Deplewski D, Brady MJ, Cohen RN. The nuclear receptor corepressors NCoR and SMRT decrease peroxisome proliferator-activated receptor gamma transcriptional activity and repress 3T3-L1 adipogenesis. J Biol Chem. 2005 Apr 8;280(14):13600–5. doi: 10.1074/jbc.M409468200. Epub 2005 Feb 3. [DOI] [PubMed] [Google Scholar]
- 42.Xue HH, Zhao DM. Regulation of mature T cell responses by the Wnt signaling pathway. Ann N Y Acad Sci. 2012;1247:16–33. doi: 10.1111/j.1749-6632.2011.06302.x. [DOI] [PubMed] [Google Scholar]
- 43.Glass CK, Saijo K. Nuclear receptor transrepression pathways that regulate inflammation in macrophages and T cells. Nat Rev Immunol. 2010;10:365–76. doi: 10.1038/nri2748. [DOI] [PubMed] [Google Scholar]
- 44.Lee G, Elwood F, McNally J, Weiszmann J, Lindstrom M, Amaral K, Nakamura M, Miao S, Cao P, Learned RM, Chen JL, Li Y. T0070907, a selective ligand for peroxisome proliferator-activated receptor gamma, functions as an antagonist of biochemical and cellular activities. J Biol Chem. 2002 May 31;277(22):19649–57. doi: 10.1074/jbc.M200743200. Epub 2002 Mar 4. [DOI] [PubMed] [Google Scholar]
- 45.Handeli S, Simon JA. A small-molecule inhibitor of Tcf/beta-catenin signaling down-regulates PPARgamma and PPARdelta activities. Mol Cancer Ther. 2008 Mar;7(3):521–9. doi: 10.1158/1535-7163.MCT-07-2063. [DOI] [PubMed] [Google Scholar]
- 46.Rajendran P, Delage B, Dashwood WM, Yu TW, Wuth B, Williams DE, Ho E, Dashwood RH. Histone deacetylase turnover and recovery in sulforaphane-treated colon cancer cells: competing actions of 14-3-3 and Pin1 in HDAC3/SMRT corepressor complex dissociation/reassembly. Molecular cancer. 2011;10:68. doi: 10.1186/1476-4598-10-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Venuprasad K, Elly C, Gao M, Salek-Ardakani S, Harada Y, Luo JL, Yang C, Croft M, Inoue MK, Karin M, Liu YC. Convergence of Itch-induced ubiquitination with MEKK1-JNK signaling in Th2 tolerance and airway inflammation. J Clin Invest. 2006;116:1117–26. doi: 10.1172/JCI26858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Di Bartolo V, Montagne B, Salek M, Jungwirth B, Carrette F, Fourtane J, Sol-Foulon N, Michel F, Schwartz O, Lehmann WD, Acuto O. A novel pathway down-modulating T cell activation involves HPK-1-dependent recruitment of 14-3-3 proteins on SLP-76. J Exp Med. 2007;204:681–91. doi: 10.1084/jem.20062066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Esnault S, Shen ZJ, Whitesel E, Malter JS. The peptidyl-prolyl isomerase Pin1 regulates granulocyte-macrophage colony-stimulating factor mRNA stability in T lymphocytes. J Immunol. 2006;177:6999–7006. doi: 10.4049/jimmunol.177.10.6999. [DOI] [PubMed] [Google Scholar]
- 50.Ioannidis V, Beermann F, Clevers H, Held W. The beta-catenin--TCF-1 pathway ensures CD4(+)CD8(+) thymocyte survival. Nat Immunol. 2001 Aug;2(8):691–7. doi: 10.1038/90623. [DOI] [PubMed] [Google Scholar]
- 51.Yu Q, Sharma A, Oh SY, Moon HG, Hossain MZ, Salay TM, Leeds KE, Du H, Wu B, Waterman ML, Zhu Z, Sen JM. T cell factor 1 initiates the T helper type 2 fate by inducing the transcription factor GATA-3 and repressing interferon-gamma. Nat Immunol. 2009 Sep;10(9):992–9. doi: 10.1038/ni.1762. Epub 2009 Aug 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Notani D, Gottimukkala KP, Jayani RS, Limaye AS, Damle MV, Mehta S, Purbey PK, Joseph J, Galande S. Global regulator SATB1 recruits beta-catenin and regulates T(H)2 differentiation in Wnt-dependent manner. PLoS Biol. 2010 Jan 26;8(1):e1000296. doi: 10.1371/journal.pbio.1000296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yu Q, Sharma A, Ghosh A, Sen JM. T cell factor-1 negatively regulates expression of IL-17 family of cytokines and protects mice from experimental autoimmune encephalomyelitis. J Immunol. 2011 Apr 1;186(7):3946–52. doi: 10.4049/jimmunol.1003497. Epub 2011 Feb 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gattinoni L, Zhong XS, Palmer DC, Ji Y, Hinrichs CS, Yu Z, Wrzesinski C, Boni A, Cassard L, Garvin LM, Paulos CM, Muranski P, Restifo NP. Wnt signaling arrests effector T cell differentiation and generates CD8+ memory stem cells. Nat Med. 2009 Jul;15(7):808–13. doi: 10.1038/nm.1982. Epub 2009 Jun 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ding Y, Shen S, Lino AC, Curotto de Lafaille MA, Lafaille JJ. Beta-catenin stabilization extends regulatory T cell survival and induces anergy in nonregulatory T cells. Nat Med. 2008 Feb;14(2):162–9. doi: 10.1038/nm1707. Epub 2008 Feb 3. [DOI] [PubMed] [Google Scholar]
- 56.Muralidharan S, Hanley PJ, Liu E, Chakraborty R, Bollard C, Shpall E, Rooney C, Savoldo B, Rodgers J, Dotti G. Activation of Wnt signaling arrests effector differentiation in human peripheral and cord blood-derived T lymphocytes. J Immunol. 2011 Nov 15;187(10):5221–32. doi: 10.4049/jimmunol.1101585. Epub 2011 Oct 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ishaq M, DeGray G, Natarajan V. Protein kinase C theta modulates nuclear receptor-corepressor interaction during T cell activation. The Journal of biological chemistry. 2003;278:39296–39302. doi: 10.1074/jbc.M302767200. [DOI] [PubMed] [Google Scholar]
- 58.Ghisletti S, Huang W, Jepsen K, Benner C, Hardiman G, Rosenfeld MG, Glass CK. Cooperative NCoR/SMRT interactions establish a corepressor-based strategy for integration of inflammatory and anti-inflammatory signaling pathways. Genes & development. 2009;23:681–693. doi: 10.1101/gad.1773109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kao HY, Downes M, Ordentlich P, Evans RM. Isolation of a novel histone deacetylase reveals that class I and class II deacetylases promote SMRT-mediated repression. Genes & development. 2000;14:55–66. [PMC free article] [PubMed] [Google Scholar]
- 60.Villagra A, Cheng F, Wang HW, Suarez I, Glozak M, Maurin M, Nguyen D, Wright KL, Atadja PW, Bhalla K, Pinilla-Ibarz J, Seto E, Sotomayor EM. The histone deacetylase HDAC11 regulates the expression of interleukin 10 and immune tolerance. Nature immunology. 2009;10:92–100. doi: 10.1038/ni.1673. [DOI] [PMC free article] [PubMed] [Google Scholar]