

Abstract
Interactions between the aromatic amino acid residues have a significant influence on the protein structures and protein-DNA complexes. These interactions individually provide little stability to the structure; however, together they contribute significantly to the conformational stability of the protein structure. In this study, we focus on the four aromatic amino acid residues and their interactions with one another and their individual interactions with the four nucleotide bases. These are analyzed in order to determine the extent to which their orientation and the number of interactions contribute to the protein and protein-DNA complex structures.
Background
Aromatic compounds are unsaturated cyclic and planar molecules that contain an aromatic ring. They possess additional stability as a result of the arrangement of the π - electrons situated above and below the plane of the aromatic ring. These electrons give rise to what is known as a π-electron cloud over the ring. Aromaticity is a chemical property associated with such cyclic and planar compounds and is attributed to these π-electrons which are free to cycle around the circular arrangements of atoms found in the aromatic moieties. It can be considered as a manifestation of cyclic delocalization and resonance which is found in planar ring systems such as benzene [1, 2]. The flat face of an aromatic ring has a partial negative charge owing to these π electrons. Out of the 20 amino acids found in protein structures, four are aromatic. They are phenylalanine, tyrosine, tryptophan and histidine [3]. The interactions that take place between the sidechains of the aromatic amino acid residues are referred to as aromatic-aromatic interactions. Formally, aromatic-aromatic interactions are defined as pairs of interacting aromatic residues which satisfy the following criteria: (i) the centers of the aromatic rings of the two interacting residues are separated by a distance between 4.5 Å to 7 Å, (ii) the dihedral angle must fall between 30° to 90° and (iii) free energies of formation when such interactions take place should be between -0.6 and -1.3 kcal/mole [4]. The aromatic interactions are relatively nonpolar in nature. They have been found to play an important role in maintaining the overall structure of the protein molecules and protein-DNA complexes. The interactions between the aromatic residues within a protein and in protein and DNA complexes are integral part for the proper functioning of the protein molecule. This in turn influences various biological processes that take place within an organism in which the protein is involved [5].
The non-covalent interactions that take place within a protein contribute to its structural integrity and thereby influence its function. These non-covalent interactions include hydrogen bonds, electrostatic and van der Waals interactions. Studies on the aromatic interactions provided a new insight on the nature of these biologically important non-covalent interactions in terms of their driving force, stability and selectivity. Analysis of the binding patterns of the aromatic residues to nucleic acids provides some insight into the origin and nature of interactions that can take place between the amino acids and nucleic acid bases. The π-system of the aromatic rings gives rise to three types of interactions involving aromatic moieties: (i) π-π, (ii) cation-π and (iii) X-H-π. Cationic moieties that are within 6.0 Å of the face of an aromatic ring may engage in polar cation-π interactions [6]. These interactions give rise to three different types of geometries, namely, edge-to-face or T-shaped, face-toface and parallel displaced or offset stacked interactions (Figure 1). Among these, it has been found that most of the attractive orientations are usually T-shaped. The face-to-face orientation is rarely observed as it leads to an unfavorable electrostatic repulsion force between the two planar faces of the aromatic rings [7]. Apart from these three main types of interactions, there are also various sub-types present under offset stacked interactions [8].
Figure 1.
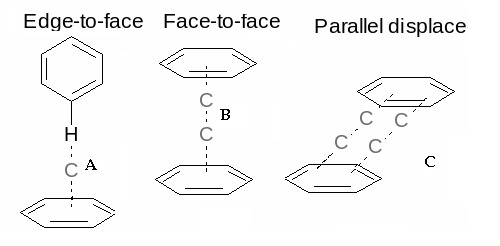
Schematic representation of the three types of orientations found in the aromatic-aromatic interactions.
Aromatic interactions also play an important role in scaffolding in DNA and RNA binding [9, 10]. However, the role of the aromatic amino acids in protein-DNA interactions is still not fully understood. A seminal study was conducted by Luscombe and co-workers to identify the specificity with which the aromatic groups bind to nucleotide bases [11]. They found that the residue phenylalanine binds preferentially to adenine and thymine via a stacking interaction. They have speculated that tryptophan should also be capable of making such interactions. However, it occurs too infrequently in the DNA binding regions. They also found that histidine binds specifically to guanine via hydrogen bonding. They also observed that although tyrosine is commonly present at the interface between the protein and DNA, it did not show any particular binding specificity. Thus, the present study focuses on the aromaticaromatic interactions in proteins and in protein-DNA complexes.
Methodology
Dataset:
Structures of proteins and protein-DNA complexes resolved using X-ray diffraction and NMR were obtained from culled PDB [12]. X-ray structures having a crystallographic R-factor of 20% and with a resolution of 2.0Å were taken from 90% nonredundant protein chains. In the case of structures solved using NMR, the atomic coordinates of the first model were used. The number of resultant structures obtained was 12,026. The aromatic amino acids phenylalanine, tyrosine, histidine and tryptophan (along with its indole ring) were considered. The distance between the aromatic amino acid residues was calculated by taking into account the distance between the centroids of the aromatic rings.
Classification of the Dataset:
Luscombe and coworkers concluded that the protein–DNA interactions could be split into two classes, namely, those involving the DNA backbone and those involving the bases [11]. It was based on this that the interactions in the protein- DNA complexes were divided into two types: specific and nonspecific. Interactions between the aromatic residue side chains and nucleotide bases were defined as specific and those between the aromatic residue side chains and the DNA sugarphosphate backbone were treated as non-specific. Analysis was carried out keeping in mind that the distance range within which the aromatic interactions take place. The optimum distance (the distance at which the maximum number of interactions takes place) was calculated. The number of interactions observed for all possible 15 aromatics pairs is given in Table 1 (see supplementary material). The optimum interacting distance (the distance at which most number of interactions took place) was analyzed in six intervals as indicated in Table 2 (see supplementary material). The same method was used for calculating the distance at which the most number of specific and non-specific interactions took place. The distance at which the maximum number of interactions took place was found for specific and non-specific interactions are given in Table 3 (see supplementary material). The orientation in which the residues preferred to interact with each other was found by calculating the angle at which the residues made the maximum number of interactions. Perpendicular and parallel interactions were also calculated. The interactions where the angle between the residues was between 85°to 95° were categorized as perpendicular and those where the angle between the interacting residues was between 170° to 180° were designated as being parallel. Locally developed PERL scripts were used to perform all calculations. All diagrams were generated using Pymol [13] and ISIS Draw [14].
Discussion
Distance and angle based interactions:
Results obtained show that the aromatic pair phenylalaninetyrosine has more number of interactions than all other aromatic pairs (Table 1) and the interactions are most favorable in the distance range of 6.0Å to 6.5Å (Table 3). The maximum number of specific interactions is found to be between 6.5Å to 7.0Å and for non-specific, it is observed between 7.0Å to 7.5Å (Table 3). The number of interactions based on angle shows that a majority of the interactions between the residues fall in the range of 80°to 100° with the highest number of interactions occurring in the range 80° to 90° (Figure 2). From these results, it can be seen that the majority of interactions in the proteins fall into the T-shaped orientation. The T-shaped or edge-to-face interaction can make a significant contribution to the binding energy of the protein-DNA complex formation [5]. The π–π repulsion, in general, does not favor direct face-to-face interactions [7]. In specific interactions, the angle at which maximum number of interactions occurred is once again found to be in the range 80° to 90° (Figure 3). This may be due to the fact that the nucleotide base also interacts in a perpendicular Tshaped orientation with the residues in the protein. However, in the case of non-specific interactions, the maximum number of interactions between the sugar-phosphate backbone and the side chains of the aromatic residues are found to be between 130° to 140° indicating a more parallel orientation (Figure 3). Usually, it is the α-helices that engage in most of the basespecific interactions. Furthermore, these interactions are found mainly on the major groove of the DNA [11]. Analysis of the aromatic residues involved in DNA-protein complexes indicate that they play a significant role in binding to the bases and in strengthening such interactions [11].
Figure 2.
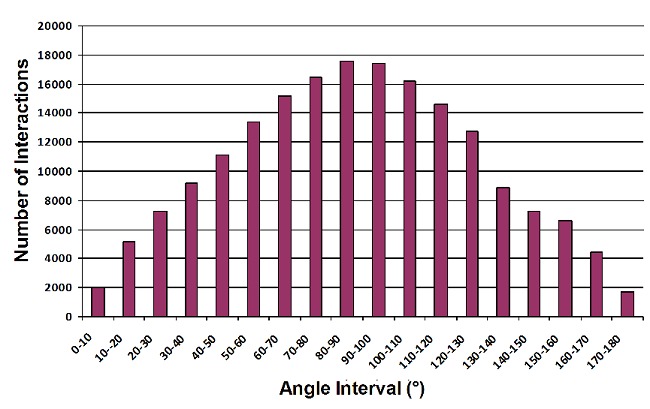
The number of interactions between the aromatic amino acids (based on angle).
Figure 3.
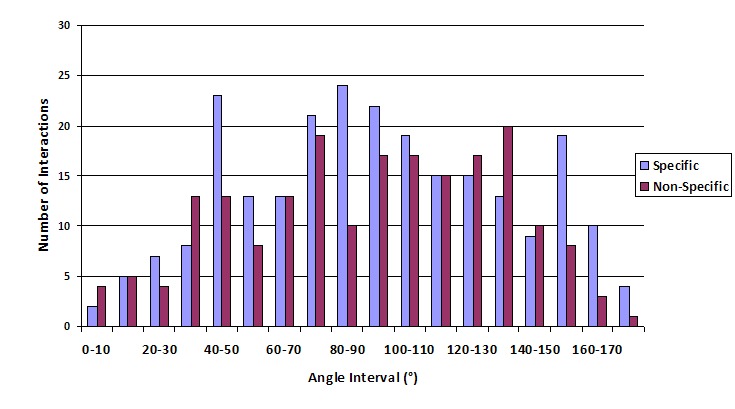
The number of specific and non-specific interactions (based on angle).
Promiscuity of Tyrosine:
Tyrosine binds strongly to both DNA and RNA and shows more promiscuous binding than other aromatic residues. The manner in which the residue tyrosine binds indicates strength rather than specificity. Tyrosine residues are also found frequently at DNA-protein interfaces. Tryptophan is found mostly in RNA rather than DNA indicating that it may aid in differentiation of these molecules. This suggests that these sorts of interactions play a major role in protein-nucleic acid recognition [5]. In the present study, tyrosine was frequently found at the interface between the protein and DNA. This may be due to the fact that the phenolic hydroxyl group is able to provide some amount of stability when exposed to solvent [15]. This could also be one of the reasons why tyrosine residues are involved in less favorable face-to-face interaction.
Energetics of Aromatic-Aromatic Interactions:
Energy calculations have shown that the T-shaped packing geometry is the most preferred and therefore most prevalent [4, 16]. Apart from this, parallel displaced interactions are also observed. In thermophilic proteins, the presence of the aromatic clusters increased thermal stability [15]. In DNA-protein interactions, the apolar surface of the sugar forms an attractive complementary surface for the aromatic rings [17]. As mentioned earlier, the aromatic interactions comprise of van der Waals interactions and electrostatic forces. It has been found that 60% of the aromatic residues in proteins are found in pairwise interactions and out of these, it has been found that the side-chains interacted in networks of three or more [4]. The orientation and packing of the aromatic moieties in a protein is driven by two main factors, namely, the need to remain in a hydrophobic environment and exclude water and to form a large number of weakly polar interactions which are believed to be electrostatic in nature. These enthalpically favorable interactions, albeit small, are large in number and are therefore capable of making a substantial contribution to the overall structural stability of the protein [4].
Propensity of Phenylalanines to interact with each other:
As indicated earlier, a total of 15 different possible pairs of interactions between the aromatic residues are taken into consideration. Experimental evidences show that the interactions between the phenylalanines can stabilize aα-helix. It has also been observed that the interactions between the two phenylalanine residues can provide energy up to -3.3kJ/mol to stabilize aα-helix [12]. Also, in a separate study which conducted a comparison of the cross-strand interactions between the phenylalanines and cyclohexylamines in water, it is found that phenylalanines showed a preference for selfassociation. The phe-phe pair is also found to be highly enthalpically favourable [16]. This is in concordance with our results obtained for the aromatic residues with the second highest number of interactions found to be between two phenylalanines (Table 1). Serrano and co-workers had found that phenylalanine and tyrosine residues in proteins are frequently found to be in pair wise interactions. These phe-tyr interactions have been found to play important roles in protein folding and stability [18]. Since T-shaped interactions are preferred, it can be speculated that the phenolic hydroxyl of the tyrosine residue interacts in a XH-π type interaction with the phenylalanine.
Conclusion
Although, different types of aromatic interactions are not vast, they play a significant role in the overall structure and conformation of the protein molecules as well as protein-DNA complexes. The strong preference for the T-shaped interactions and various optimal distances and angles provide a better insight into how significant these interactions are to the protein structure. Aromatic interactions are made more complicated because the molecules involved are larger and more complex. This size-wise expansiveness also influences the solvent accessible surface area. This in turn brings van der Waals and desolvation parameters which must be taken into consideration. Thus, it is difficult to rationalize the behavior of the aromatic interactions in a straightforward manner and further investigations need to be performed to study how exactly they influence protein structure.
Supplementary material
Acknowledgments
The authors acknowledge the facilities offered by the Interactive Graphics Facility and SERC. One of the authors (KS) thanks the Department of Biotechnology, Govt. of India for financial support.
Footnotes
Citation:Anjana et al, Bioinformation 8(24): 1220-1224 (2012)
References
- 1.PvR Schleyer. Chemical Reviews. 2005;105:3433. [Google Scholar]
- 2.TA Balaban, et al. Chemical Reviews. 2005;105:3436. doi: 10.1021/cr0300946. [DOI] [PubMed] [Google Scholar]
- 3.L Brocchieri, S Karlin. Proc Natl Acad Sci. 1994;91:9297. [Google Scholar]
- 4.SK Burley, GA Petsko. FEBS Lett. 1986;203:139. doi: 10.1016/0014-5793(86)80730-x. [DOI] [PubMed] [Google Scholar]
- 5.EA Meyer, et al. Angew Chem Int Ed Engl. 2003;42:1210. [Google Scholar]
- 6.JC Ma, DA Dougherty. Dougherty. 1997;97:1303. doi: 10.1021/cr9603744. [DOI] [PubMed] [Google Scholar]
- 7.CA Hunter, et al. Chem Soc Perkin Trans. 2001;651 [Google Scholar]
- 8.LM Espinoza-Fonseca, et al. Mol Biosyst. 2012;8:237. [Google Scholar]
- 9.M Yarus, et al. J Mol Evol. 2009;69:406. doi: 10.1007/s00239-009-9270-1. [DOI] [PubMed] [Google Scholar]
- 10.S Aravinda, et al. J Am Chem Soc. 2003;125:5308. [Google Scholar]
- 11.NM Luscombe, et al. Nucleic Acids Res. 2001;29:2860. doi: 10.1093/nar/29.13.2860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.G Wang, RL Jr Dunbrack. Bioinformatics. 2003;19:1589. [Google Scholar]
- 13. The PyMOL Molecular Graphics System, Schrödinger, LLC. [Google Scholar]
- 14.A Hinchliffe ISIS/Draw for Windows. Electron. J Theor Chem. (version 1.2W) 1996;1:79. [Google Scholar]
- 15.N Kannan, S Vishveshwara. Protein Eng. 2000;13:753. doi: 10.1093/protein/13.11.753. [DOI] [PubMed] [Google Scholar]
- 16.SM Butterfield, et al. J Am Chem Soc. 2002;124:9751. [Google Scholar]
- 17.CA Hunter, et al. J Mol Biol. 1991;218:837. doi: 10.1016/0022-2836(91)90271-7. [DOI] [PubMed] [Google Scholar]
- 18.L Serrano, et al. J Mol Biol. 1991;218:465. doi: 10.1016/0022-2836(91)90725-l. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.