

Abstract
Medicinal plants and marine sources are important elements of indigenous medical systems worldwide. The natural drugs from medicinal plants and marine sources have received considerable interest in treatment of diabetes and inflammation. Based on literature, alpha glucosidase, aldose reductase and PTP1B enzymes were chosen as anti-diabetes targets and PLA2 was chosen for the anti-inflammatory target. In our study, plant and bromophenols (BPs) inhibitors were screened using High Throughput Virtual screening (HTVS) followed by Induced Fit Docking (IFD) studies were carried out against diabetes and inflammation targets. The IFD result of natural inhibitors has showed favorable docking score, glide energy and hydrogen bonds interactions with the active site residues. Some of the natural inhibitors successively satisfied all the in silico parameters among the others and seem to be potent inhibitors against diabetes and inflammation.
Keywords: Diabetes, Aldose reductase, Protein tyrosine phosphatase-1B, alpha glucosidase, phospholipase A2, HTVS, IFD
Background
For a long time, the use of natural products with therapeutic properties is considered as ancient as human civilization where minerals, plant and animal products were the main sources of drugs [1, 2]. In the recent years, the natural drugs from medicinal plants and marine sources are considered as an effective treatment against diabetes and inflammation. Many of the currently available exemplary sources of drugs have been derived directly or indirectly from medicinal plants. The ethnobotanical information reports that around 800 plants may possess anti-diabetic properties [3] and several medicinal plants have shown effective activities against the diabetes when assessed by available experimental techniques [4–9]. Some of the medicinal plants show potential inhibition against aldose reductase target [10, 11].
Marine algae are other richest sources of structurally diverse natural products [12, 13]. Recent studies report that the marine bromophenols (BPs) inhibitors exhibit a wide spectrum of beneficial biological activities [14–26]. The bromophenols isolated from marine algae have been reported to exhibit potent inhibitory activities agaisnt Protein Tyrosine phosphatase-1B (PTP1B), Alpha-glucosidase [27–29] and PLA2 [30, 31]. Our current study aims to perform a high throughput virtual screening and induced fit docking studies of natural compounds against diabetes and inflammation. While the inhibitor compounds from medicinal plants were docked with aldose reductase enzyme, the marine bromophenols (BPs) inhibitors were treated against PTP1B, Alpha-glucosidase and PLA2 targets. Docking score glide energy and hydrogen bond interactions were analyzed with the docked complexes. These analyses suggest that inhibitors from plants and marine sources can be used for the treatment of diabetes and inflammation.
Methodology
All computational works were performed on CentOS EL-5 workstation using the molecular modeling software Maestro (Schrodinger LLC 2009, USA). GLIDE-5.5 (Grid-based Ligand Docking with Energetics) performs rigid (HTVS) and flexible (IFD) docking between one or more ligand molecules with a macromolecule, usually a protein. PyMOL software was used for graphical visualization, analyzing hydrogen bond interactions and producing quality images.
Ligand and Protein preparation:
Natural compounds (Figure 1) from medicinal plants and marine algae were found to have anti diabetic and anti inflammatory properties. Based on the biological activity of these compounds, some protein targets were chosen for HTVS and IFD studies. These natural compounds along with those already cocrystallized with the chosen protein targets were built using builder panel in Maestro for further calculations. These compounds were subjected into ligand preparation by Ligprep 2.3 module (Schrödinger). Ligprep performs addition of hydrogens, 2D to 3D conversion, realistic bond lengths and bond angles, low energy structure with correct chiralities, ionization states, tautomers, stereochemistries and ring conformation.
Figure 1.
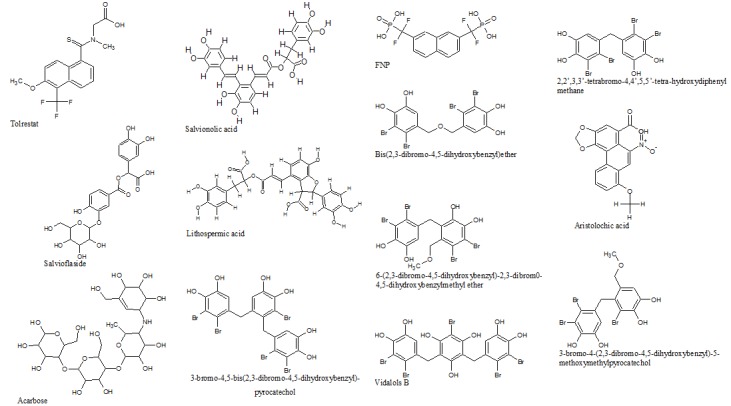
Schematic representation of natural compounds from medicinal plants and marine algae.
Alpha glucosidase (2ZQ0) [32], aldose reductase (2FZD) [33] and PTP1B (1KAK) [28] enzymes were chosen as anti-diabetes targets. PLA2 (1FV0) [34] enzyme was subjected for antiinflammatory target. All these structures were retrieved from Protein Data Bank and taken into the protein preparation wizard where changes such as addition of hydrogen atoms, assigning correct bond orders, fixing of the charges and orientation of groups were incorporated into the raw PDB structure. Optimization of hydrogen bonds and amino acid flips were also assigned iteratively. Non-hydrogen atoms were energy minimized until the average root mean square deviation reaches 0.3Å. All the above mentioned steps were performed using Glide, Prime, QSite, Liasion and MacroModel in the Schrödinger modules.
High Throughput Virtual Screening (HTVS):
HTVS is an important method, which helps in screening one or more compounds from a set of compounds. HTVS needs previously calculated receptor grid and one or more ligand structures. When a ligand is docked, the active site of the receptor and its properties will be calculated on a grid by different set of fields which provide accurate scoring function along with their energy. This method facilitates the ligands to bind in more than one possible and meaningful conformation but the receptor is rigid. However, van der Waals radii of nonpolar atoms were estimated to decrease close contact penalties between the active site residues and the ligand. Using HTVS, 42 compounds (data not shown) from medicinal plants were screened against aldose reductase and 7 marine algae compounds (data not shown) were screened against Phospholipase A2, alpha-glucosidase and PTP1B. The best fit compounds have been chosen for each target by docking score and glide energy. The best compounds (Figure 2) were used for IFD.
Figure 2.
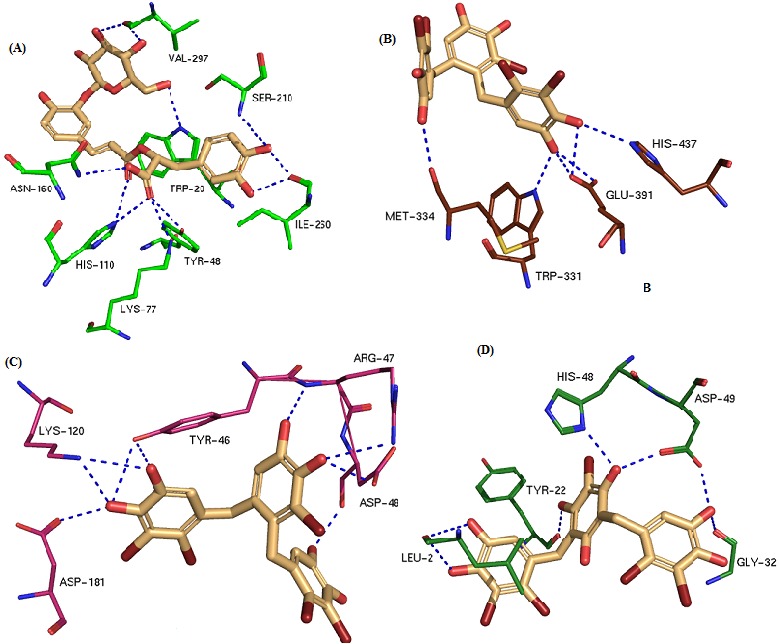
Hydrogen bond interactions (blue dot line) a) Salvioflaside- aldose reductase complex b) 3-bromo-4,5-bis(2,3-dibromo-4,5- dihydroxybenzyl)-pyrocatechol - alpha-glucosidase complex c) 3-bromo-4,5-bis(2,3-dibromo-4,5-dihydroxybenzyl)-pyrocatechol – PTP1B complex d) Vidalol B - Phospholipase A2.
Induced Fit Docking (IFD):
In many proteins, conformational changes take place in the side-chain or backbone or both, when a ligand binds at the active site. Therefore, receptor is generated by calculating close conforms to the shape and binding mode of the ligand. This is known as IFD. In our study, IFD, one of the main complicating factors predicts accurate ligand binding modes and concomitant structural movements in the receptor using modules of Glide and Prime. In the workspace, each energy minimized complex was loaded and the ligand was selected to specify the active site. All the screened compounds were docked with respective protein target. van der Waals radii of nonpolar atoms of the receptor and ligand were scaled by a factor of 0.50 and 20 conformational poses were calculated. In IFD results, the best conformation was chosen for further calculations based on the docking score, Glide energy and none bonded interactions.
Results and Discussion
Aldose reductase:
The co-crystal ligand, Tolrestat was redocked at the active site of aldose reductase. The docked complex had a docking score of -8.57 and glide energy of -45.60 kcal/mol. The ligand has three hydrogen bond interactions with Tyr 48, His 110 and Ser 302 at the active site. The docked complex has been superimposed with crystal structure of aldose reductase and the rmsd is 0.18Å.
Out of the many screened medicinal compounds, the IFD complex of Salvionolic acid ligand with the target enzyme had eight hydrogen bond interactions with active site residues such as His 110, Trp 111, Gln 183, Ser 210, Ile 260 and Ser 302. In particular, Ile 260 and Ser 260 have two hydrogen bond interactions with the ligand. This complex resulted with a docking score of -12.45 and glide energy of -81.08 kcal/mol.
For Salvioflaside - aldose reductase complex formation a docking score of -11.53 and glide energy of -82.70 kcal/mol were observed. At the active site, His 110, Ile 260 and Val 297 residues have two hydrogen bond interactions with the ligand. Altogether this complex has nine hydrogen bond interactions with Trp 20, Lys 77, His 110, Asn 160, Ile 260 and Val 297.
Another plant ligand Lithospermic acid docked at the active site of aldose reductase has a docking score of -12.07 and glide energy of -75.25 kcal/mol. The ligand has five hydrogen bond interactions with Lys 21, Trp 111, Asn 160, Gly 183 and Ser 302 at the active site.
Alpha-glucosidase:
Alpha-Acarbose inhibitor was redocked at the active site of the alpha-glucosidase and the docking score is -8.33 and Glide energy is -85.66 kcal/mol. This complex has eleven hydrogen bond interactions between the ligand and the active site residues. Pro 215 and Arg 529 have two hydrogen interactions with the ligand and remaining residues like Ser 270, Trp 331, Glu 391, Trp 400, Glu 439, His 507 and Phe 536 have one interaction each. The docked complex was superimposed with the crystal structure of alpha-glucosidase and the rmsd is 0.21Å. Best fit of the 3-bromo-4,5-bis(2,3-dibromo-4,5- dihydroxybenzyl)-pyrocatechol ligand at the active site of alpha-glucosidase's showed the docking score of −9.19 and Glide energy of −75.33 kcal/mol. This inhibitor has totally six hydrogen bond interactions, Trp 331, Met 334, Glu 391 and Tyr 533 residues have one interaction each and Ser 217 has two interactions with the bound ligand.
Protein tyrosine phosphatase 1 B:
FNP inhibitor redocked at the active site of PTP1B has a docking score of -10.46 and Glide energy of -100.31 kcal/mol. Arg 47 and Ser 216 have two interactions and Arg 221 has three interactions with FNP. The remaining active residues have one hydrogen bond interaction each with the ligand. This complex has been stabilized with thirteen hydrogen bond interactions between the ligand and the active site residues. The superimposition of crystal and docked complexes has rmsd of 0.15Å. 2,2',3,3'-tetrabromo-4,4',5,5'-tetra-hydroxydiphenyl methane bound PTP1B complex has a docking score of -8.26 and Glide energy of -52.03 kcal/mol. This ligand has four hydrogen bond interactions with the active site residues Asp 48 (two H-bonds), Phe 182 and Glu 262.
The best fit of 3-bromo-4,5-bis(2,3-dibromo-4,5- dihydroxybenzyl) pyrocatechol at the active site of PTP1B has shown five hydrogen bond interactions with Arg 47, Asp 48( two H-bonds), Lys 120 and Asp 181 residues. Docking score of - 9.84 and glide energy of -67.94 kcal/mol were observed for the complex formation. Bis(2,3-dibromo-4,5-dihydroxybenzyl) ether ligand has three hydrogen bond interactions with the active site residues (Tyr 20, Lys 120 and Gln 262) of PTP1B. Docking score of -7.45 and glide energy of -50.02 kcal/mol were observed in this complex.
Phospholipase A2:
Aristolochic acid ligand was redocked at the active site of the PLA2 which has the docking score of -8.18 and glide energy of - 48.52 kcal/mol. The ligand has three hydrogen bond interactions with active site residues Gly 30, His 48 and Asp 49. The docked complex superimposed with crystal structure of PLA2 and the rmsd is 0.17Å. 6-(2,3-dibromo-4,5- dihydroxybenzyl)-2,3-dibrom0-4,5-dihydroxybenzylmethyl ether ligand has also three hydrogen bond interactions with His 48, Asp 49 and Lys 69 residues. This complex has the docking score of -8.51 and glide energy of -60.71 kcal/mol.
The docked Vidalol B-PLA2 complex showed the docking score of -9.65 and glide energy of -73.71 kcal/mol. The vidalol B ligand has maintained the hydrogen bond interaction with His 48 and Asp 49 and additionally has two more interactions with active site residues (Tyr 22 and Leu 2). 3-bromo-4-(2,3-dibromo- 4,5-dihydroxybenzyl)-5-methoxymethylpyrocatechol bound at the active site of PLA2 has showed the docking score of -8.27 and glide energy of -59.35 kcal/mol. This ligand has the hydrogen bond interactions with His 48 and Asp 49 residues.
Conclusion
In this study, natural inhibitors from medicinal plants and marine algae were screened for anti-diabetic and antiinflammatory properties. Based on the screening, few of the ligands were subjected for induced fit docking studies and were compared using docking score, glide energy and hydrogen bond interactions. Along with Tolrestat, the natural compounds Salvionolic acid, Salvioflaside and lithospermic were docked at the active site of aldo-reductase. Based on the IFD studies, salvioflaside was selected as the potent anti-diabetic compound for the target aldose reductase compared to tolrestat which was reported to have some side effects.
Although, docking studies with Acarbose show better results than 3-bromo-4,5-bis(2,3-dibromo-4,5-dihydroxybenzyl)- pyrocatechol, due to the side effects of the Acarbose, the new compound, an analog of the compound from the marine algae appears to be the best in the IFD results of alpha-glucosidase. The cocrystal ligand (FNP) of PTP1B inhibitor has the best score and energy but with the IC50 value of 25 µM. 3-bromo-4, 5-bis(2, 3-dibromo-4,5-dihydroxybenzyl) pyrocatechol seems to be the next alternative according to glide energy but with better IC50 value than FNP (IC50=1.7 µM).
Out of the four ligands docked with PLA2 against inflammation, it is found that the new compound Vidalol B is better than the anti-inflammatory drug aristolochic acid from another natural product. The induced fit docking studies thus support the use of natural products for curing inflammation and diabetes.
Supplementary material
Acknowledgments
SS thanks the University Grants Commission (UGC), Government of India, New Delhi, for Fellowship for Meritorious students. The authors thank Bioinformatics infrastructure facility of University of Madras supported by DBT. UGC is gratefully acknowledged for the SAP program.
Footnotes
Citation:Suhitha et al, Bioinformation 8(23): 1125-1131 (2012)
References
- 1.A de Pasquale. J Ethnopharmacol. 1984;11:1. doi: 10.1016/0378-8741(84)90092-8. [DOI] [PubMed] [Google Scholar]
- 2.SM Rates. Toxicon. 2001;39:603. [Google Scholar]
- 3.FJ Alarcon-Aguilara, et al. J Ethnopharmacol. 1998;61:101. doi: 10.1016/s0378-8741(98)00020-8. [DOI] [PubMed] [Google Scholar]
- 4.AQ Saifi, et al. J Res Indian Med. 1971;6:205. [Google Scholar]
- 5.K Mukherjee, et al. Indian J Exp Biol. 1972;10:347. [PubMed] [Google Scholar]
- 6.TC Coimbra, et al. Fitoterapia. 1992;63:320. [Google Scholar]
- 7.A Kar, et al. J Ethnopharmacol. 1999;64:179. doi: 10.1016/s0378-8741(98)00118-4. [DOI] [PubMed] [Google Scholar]
- 8.MA Jafri, et al. J Ethnopharmacol. 2000;70:309. [Google Scholar]
- 9.JK Grover, et al. J Ethnopharmacol. 2002;81:81. [Google Scholar]
- 10.GH Du, et al. Yao Xue Xue Bao. 1995;30:561. [PubMed] [Google Scholar]
- 11.R Kasimu, et al. Chem Pharm Bull (Tokyo) 1998;46:500. doi: 10.1248/cpb.46.500. [DOI] [PubMed] [Google Scholar]
- 12.SK Kim, YX Li. Adv Food Nutr Res. 2011;64:391. doi: 10.1016/B978-0-12-387669-0.00030-2. [DOI] [PubMed] [Google Scholar]
- 13.KC Guven, et al. Mar Drugs. 2010;8:269. [Google Scholar]
- 14.AA El Gamal. Saudi Pharm J. 2010;8:1. [Google Scholar]
- 15.KY Kim, et al. Phytochemistry. 2008;69:2820. doi: 10.1016/j.phytochem.2008.09.007. [DOI] [PubMed] [Google Scholar]
- 16.KY Kim, et al. J Food Sci. 2010;75:145. [Google Scholar]
- 17.M Ma, et al. J Nat Prod. 2006;69:206. doi: 10.1021/np050343g. [DOI] [PubMed] [Google Scholar]
- 18.XJ Duan, et al. J Nat Prod. 2007;70:1210. doi: 10.1021/np070061b. [DOI] [PubMed] [Google Scholar]
- 19.K Li, et al. Bioorg Med Chem. 2007;15:6627. doi: 10.1016/j.bmc.2007.08.023. [DOI] [PubMed] [Google Scholar]
- 20.K Li, et al. J Nat Prod. 2008;71:28. doi: 10.1021/np070281p. [DOI] [PubMed] [Google Scholar]
- 21.N Xu, et al. Phytochemistry. 2003;62:1221. doi: 10.1016/s0031-9422(03)00004-9. [DOI] [PubMed] [Google Scholar]
- 22.WL Popplewell, et al. Tetrahedron Lett. 2009;50:6814. [Google Scholar]
- 23.HS Lee, et al. J Agric Food Chem. 2007;55:6923. doi: 10.1021/jf071125r. [DOI] [PubMed] [Google Scholar]
- 24.NA Shoeib, et al. J Nat Prod. 2004;67:1445. doi: 10.1021/np0305268. [DOI] [PubMed] [Google Scholar]
- 25.Shi, et al. D Chin Sci Bull. 2008;53:2476. [Google Scholar]
- 26.HT Balaydın, et al. Eur J Med Chem. 2012;54:423. doi: 10.1016/j.ejmech.2012.05.025. [DOI] [PubMed] [Google Scholar]
- 27.S Koren, IG Fantus. Best Pract Res Clin Endocrinol Metab. 2007;21:621. doi: 10.1016/j.beem.2007.08.004. [DOI] [PubMed] [Google Scholar]
- 28.Z Jia, et al. J Med Chem. 2001;44:4584. doi: 10.1021/jm010266w. [DOI] [PubMed] [Google Scholar]
- 29.S Suzen, E Buyukbingol. Curr Med Chem. 2003;10:1329. doi: 10.2174/0929867033457377. [DOI] [PubMed] [Google Scholar]
- 30.DF Wiemer, et al. Experientia. 1991;47:851. doi: 10.1007/BF01922471. [DOI] [PubMed] [Google Scholar]
- 31.M Liu, et al. Mar Drugs. 2011;9:1273. doi: 10.3390/md9071273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.M Kitamura, et al. J Biol Chem. 2008;283:36328. doi: 10.1074/jbc.M806115200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.H Steuber, et al. J Mol Biol. 2006;363:174. [Google Scholar]
- 34.V Chandra, et al. Biochemistry. 2002;41:10914. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.