

Abstract
Structural analysis of the high-mobility group protein B1 (HMGB1)-DNA complex and a docking simulation between glycyrrhetinic acid (GA) and the HMGB1-DNA complex were performed with a software package the Molecular Operating Environment (MOE). An HMGB1-DNA (PDB code: 2GZK) was selected for the 3D structure modeling of the HMGB1-DNA complex. The Site Finder module of the MOE identified 16 possible ligand-binding sites in the modeled HMGB1-DNA complex. The docking simulation revealed that GA possibly inhibits functions of HMGB1 interfering with Lys90, Arg91, Ser101, Tyr149, C230 and C231 in the HMGB1-DNA complex. To the best of our knowledge, this is the first report of an HMGB1-DNA complex with GA, and our data verify that the GA-HMGB1-DNA model can be utilized for application to target HMGB1 for the development of antitumor drugs.
Abbreviations
ASE-Dock - alpha sphere and excluded volume-based ligand-protein docking, CNS - central nervous system, GA - glycyrrhetinic acid, GL - glycyrrhizin, HMGB1 - high-mobility group protein B1, LBS - ligand-biding site, MOE - Molecular Operating Environment, SRY - sex-determining region on the Y chromosome.
Keywords: Antitumor drug, MOE, HMGB1, GA
Background
High-mobility group protein B1 (HMGB1) is an abundant component of chromatin and belongs to a subfamily of the HMG proteins that contain the HMG1 box domains. It is widely distributed in eukaryotic cells from yeast to human, and was originally discovered as a nuclear transcription factor that binds to DNA in the minor groove without sequence specificity. It has been reported that HMGB1 plays important roles in the modulation of transcription, DNA integration and recombination [1]. The structural feature responsible for the DNA-binding properties is the two HMG box domains. Although the binding of an HMG box to DNA is now reasonably well understood, its structural disposition of the two boxes and the intervening linkers remains to be clarified. Further, the role and location of the acidic tail are unclear [2]. HMGB1 has also been identified as an inflammatory cytokine that functions as a late mediator of lethality in sepsis, and consequently its inhibition can be utilized as a promising therapeutic target [3]. Further study revealed that HMGB1 was associated with organ damage in hepatic ischemia-reperfusion injury and an anti-HMGB1 antibody treatment reduced liver damage in the hepatic injury [4]. HMGB1 is phosphorylated by protein kinases and its phosphorylation appears to be important for the proper functioning and turnover rates of HMGB1 [5]. However, the mechanism underlying the ischemiareperfusion- induced liver injury and the functions of HMGB1 remains to be solved. Furthermore, to the best of our knowledge, the physiological significance of the HMGB1 phosphorylation by protein kinases in inflammation has not been elucidated.
Glycyrrhizin (GL) is the principal licorice component and used worldwide in foods and medicines. It is considered to be at least 50 times sweeter than refined sugar and extensively used as a natural sweetener and flavoring additive. Its use in foods has been approved by most national regulatory agencies, and the World Health Organization suggested that consumption of GL at 100 mg/day would be unlikely to cause adverse effects [6]. GL is a saponin compound comprising a triterpenoid aglycone, glycyrrhetinic acid (GA), conjugated to a disaccharide of glucuronic acid. GL is hydrolyzed to GA in the body and in fact GA is the pharmacologically active compound [7]. Owing to the high expectation of the development of more effective and safer medicines in recent years, natural products from plants and their synthetic derivatives have been anticipated to play important roles in creating new and better chemopreventive and therapeutic agents [8]. Several single natural compounds from plants, such as the green tea polyphenol epigallocatechin gallate [9] and apple ursolic acid [10], are being studied as anticancer agents. GA is also used as an antitoxic and immunological regulatory agent for the prevention or treatment of viral infection, inflammation and anaphylaxis [11].
GA has also been investigated for its interactions with HMGB1, and it was demonstrated that GA played an important role in the inhibition of HMGB1 [12]. We have been focusing our attention on GA as a multifunctional agent for the prevention and treatment of cancer, including downregulation of inflammatory responses associated with cancer. Recently, we found that GA demonstrated a significant selective toxicity towards a central nervous system (CNS) tumor cell line [13]. These reports suggest that targeting HMGB1 with GA treatment could be utilized for controlling the development and progression of cancer. However, no structural analysis of the GA-HMGB1-DNA complex has been explored, although a crystal structure of the HMGB1-DNA complex has been reported [14]. Molecular modeling has found widespread utility in the field of drug development [15], and in the present study we will report the structural analysis of the ligand-receptor interaction between GA and the HMGB1-DNA complex by a highly sophisticated software package, the Molecular Operating Environment (MOE) 2010.10 (Chemical Computing Group Inc., Montreal, Canada).
Methodology
Modeling of the HMGB1-DNA complex:
Modeling of the 3D structure of the HMGB1-DNA complex was executed as previously reported [13]. In brief, the HMGB1 (NCBI reference sequence: NP_002119.1) and the duplex sequences and their crystal structure coordinates were loaded into the MOE. The primary structure of the HMGB1-DNA complex was aligned, carefully checked to avoid deletions or insertions in conserved regions and corrected wherever necessary. Full energy minimization was applied to the restructured HMGB1-DNA model and the refined model was utilized for further inspection. The qualities of the protein folds of the HMGB1-DNA model were evaluated with the MOE by calculating the effective atomic contact energies, comprising the desolvation free energies required to transfer atoms from water to the interior of the protein. The overall geometric and stereochemical qualities of the final modeled structure of HMGB1 were also examined using Ramachandran plots generated within the MOE.
Binding Site Selection and Exploration:
The binding site selection and exploration for the HMGB1-DNA complex was executed as previously reported [13]. In brief, the Site Finder module of the MOE was used to identify possible ligand-binding pockets within the newly generated 3D structures of HMGB1-DNA. Hydrophobic or hydrophilic alpha spheres served as probes denoting zones of tight atom packing. These alpha spheres were utilized to define potential ligandbinding sites (LBSs) and as centroids for the creation of dummy ligand atoms. The dummy atoms were matched to the corresponding alpha spheres during the characterization of the LBSs in HMGB1-DNA. This method generates bound conformations that approach crystallographic resolutions [16].
Alpha Sphere and Excluded Volume-Based Ligand-Receptor Docking (ASE-Dock):
The docking and analysis of the ligand-receptor interaction between GA and HMGB1-DNA were also performed with ASEDock in the MOE [17]. In the ASE-Dock module, ligand atoms have alpha spheres within 1 Å. Based on this property, concave models are created and ligand atoms from a large number of conformations generated by superimposition with these points can be evaluated and scored by maximum overlap with alpha spheres and minimum overlap with the receptor atoms. The scoring function used by ASE-Dock is based on ligand-receptor interaction energies and the score is expressed as a Utotal value. The Utotal value was utilized for the overall evaluation of the ligand-receptor interaction. The ligand conformations were subjected to energy minimization using the MMF94S force field [18]. From the resulting 500,000 poses, the 200 poses with the lowest Utotal values were selected for further optimization with the MMF94S force field. During the refinement step, the ligand was free to move within the binding pocket. Udock values were also adopted to determine the most reasonably docked GAHMGB1- DNA complex.
Results and Discussion
Modeling of the HMGB1-DNA Structure:
The sequence alignment of HMGB1 (PDB code: 2GZK) and the duplex containing the consensus sequence of the sexdetermining region on the Y chromosome (SRY) [19] is shown in (Figure 1A). The alignment revealed that it contained the residues of the basic linker Lys76 – Lys84 and HMG box B Asp85 – Lys159. The PDB code of 2GZK was selected for the present 3D structure modeling of HMGB1-DNA because the detailed analysis of the HMGB1-DNA complex had been provided [14]. The HMGB1-DNA model with the best packing quality function and full energy minimization was utilized in the present study (Figure 1B).
Figure 1.

(A) The sequence alignment of HMGB1 (PDB code: 2GZK) and the duplex containing the consensus sequence of SRY; (B) The 2D structure of the HMGB1-DNA complex. For HMGB1; Red: α-helix. Blue: turn. For DNA; Green: carbon for G201 – C216. Orange: carbon for G217 – C232. Blue: nitrogen. Purple: phosphorus. Red: oxygen.
Binding Site Selection and Exploration of the HMGB1-DNA Model:
The Site Finder module of the MOE identified 16 possible LBSs in the modeled HMGB1-DNA complex (Figure 2A). The previous report by Stott et al. identified 43 residues that interact with the DNA, including the intercalating residues such as Ile13, Phe97 and Ile116 [14]. Our present results in (Figure 2A) also identified these residues, and the possible ligand-binding pockets within the newly generated 3D structures of the HMGB1-DNA complex are shown in (Figure 2B). The hydrophobic (white) or hydrophilic (red) alpha spheres, which served as probes denoting zones of tight atom packing, revealed that the potential LBS could be found almost anywhere in the HMGB1-DNA complex.
Figure 2.

(A) The 16 possible LBSs identified by the Site Finder module of the MOE; (B) The identified possible LBSs depicted with the hydrophobic (white) or hydrophilic (red) alpha spheres, which serves as probes denoting zones of tight atom packing. For HMGB1; Red: α-helix. Blue: turn. For DNA; Green: carbon for G201 – C216. Orange: carbon for G217 – C232. Blue: nitrogen. Purple: phosphorus. Red: oxygen.
Docking Simulation of GA to the HMGB1-DNA Complex:
It has been reported that GA interacts with HMGB1 and that it has inhibitory effects on HMGB1 [12, 20]. We recently found that GA demonstrated significant growth inhibitory activity toward a CNS tumor cell line [13], suggesting that GA can be utilized for the antitumor drug development. However, whether or not GA can bind to the HMGB1-DNA complex is not known. Further, if GA binds to the HMGB1-DNA complex, the precise mode of the binding of GA needs to be elucidated. To this end, the ASE-Dock [17] was performed and showed that GA bound at the No. 9 site (Figure 2A) in the HMGB1-DNA complex (Figure 3A). The ligand docking score (Udock value) for GA was -59.5 kcal/mol. The enlarged figure of the LBS in the GA-HMGB1-DNA complex revealed that the hydroxyl group of GA at C3 and the oxygen at C11 formed hydrogen bonds with Lys90, and the distance between Lys90 and the OH was 2.58 Å while that for the C=O oxygen was 2.85 Å (Figure 3B). There has been a report exhibiting that Lys90 was identified as an important residue for high-binding of the B domain and that it contacted the T229 phosphate group [14]. This report and our present results suggest that GA can inhibit binding of HMGB1 to DNA interfering with the contact between Lys90 and T229.
Contacts of GA with the HMGB1-DNA Complex in Detail:
To create ligand-receptor interaction plots for the GA-HMGB1- DNA complex, the Ligand Interactions module of the MOE was used, which provided a clearer arrangement of the putative key intermolecular interactions that aid in the interpretation of the 3D juxtaposition of the ligand GA and the LBS in the HMGB1- DNA complex (Figure 4). Our present results ascertained the presence of the hydrogen bond between the hydroxyl group of GA at C3 and Lys90. The C=O oxygen at C11 was also identified as the element that formed the hydrogen bond with Lys90. Further, the hydroxyl group at C29 was also identified as forming a hydrogen bond with C231. It has been reported that C231 had contacts with Ser101 and Lys104 and that Ile116 was the intercalating residue that interacted with C231 [14]. This report and our results suggest that GA can interfere with the formation of the HMGB1-DNA complex at C231. Arg91 was also identified as the critical residue for proper binding of HMGB1 to DNA, and it made contacts with C206, C207, G227 and A228 [14]. The present results revealed that GA had interaction with Arg91, which suggests that GA possibly inhibit HMGB1 interfering with Arg91 to make contact with DNA. Tyr149 in HMGB1 contacted with T207 [14] and our current results showed that GA interacted with Tyr149, indicating that GA interferes with Tyr149. C230 has also been reported to make contacts with Pro93 and Phe97 in HMGB1 [14]. The present study also identified GA as having interaction with C230, which suggests that GA interferes with C230.
Figure 4.

The ligand-receptor interaction plots for the GA-HMGB1-DNA complex created by the Ligand Interactions module of the MOE.
Conclusion
Examination of the HMGB1-DNA complex provides considerable insight into the binding mechanism of HMGB1 to DNA and suggests approaches by which HMGB1 inhibitors with greater selectivity may be attainable. Further, detailed analysis of the ligand-receptor interaction is of great significance in designing in silico HMGB1-inhibiotor models for successful development of antitumor drugs. The main objective in the present study was to determine whether or not GA can act as an inhibitor of HMGB1 and to elucidate the precise mode of GA binding to the HMGB1-DNA complex. Analyses of the structural properties of the HMGB1-DNA complex and the docking simulations of the GA-HMGB1-DNA pose suggest that GA possibly inhibits HMGB1 interfering with Lys90, Arg91, Ser101, Tyr149, C230 and C231 in the HMGB1-DNA complex. To the best of our knowledge, this is the first report of an HMGB1- DNA model with GA as a potential inhibitor.
Figure 3.
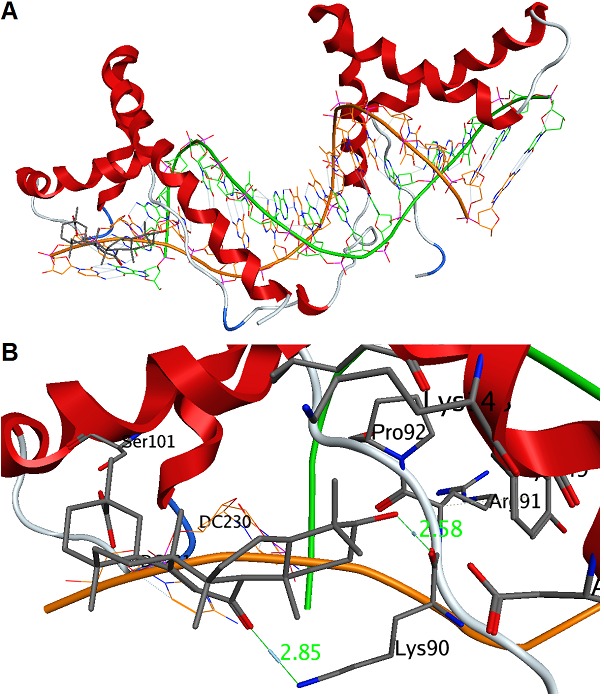
Docking simulation of GA to the HMGB1-DNA complex. (A) ASE-Dock showing that GA binds at the No. 9 site (Figure 2A) in the HMGB1-DNA complex; (B) The enlarged figure of the LBS in the GA-HMGB1-DNA complex. For GA; Gray: carbon. For HMGB1; Red: α-helix. Blue: turn. For DNA; Green: carbon for G201 – C216. Orange: carbon for G217 – C232. Blue: nitrogen. Purple: phosphorus. Red: oxygen.
Acknowledgments
This study was partially supported by a grant-in-aid from the Promotion and Mutual Aid Corporation for Private Schools of Japan.
Footnotes
Citation:Yamaguchi et al, Bioinformation 8(23): 1147-1153 (2012)
References
- 1.JR Klune, et al. Mol Med. 2008;14:476. [Google Scholar]
- 2.JO Thomas, AA Travers. Trends Biochem Sci. 2001;26:167. doi: 10.1016/s0968-0004(01)01801-1. [DOI] [PubMed] [Google Scholar]
- 3.H Yang, et al. Proc Natl Acad Sci USA. 2004;101:296. [Google Scholar]
- 4.A Tsung, et al. J Exp Med. 2005;201:1135. doi: 10.1084/jem.20042614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.JR Wiseniewski, et al. J Biol Chem. 1999;274:20116. [PubMed] [Google Scholar]
- 6.World Health Organization. World Health Organ Tech Rep Ser. 2005;928:1. [PubMed] [Google Scholar]
- 7.RA Isbrucker, GA Burdock. Regul Toxicol Pharmacol. 2006;46:167. doi: 10.1016/j.yrtph.2006.06.002. [DOI] [PubMed] [Google Scholar]
- 8.R Martin, et al. Cancer Res. 2007;67:3741. doi: 10.1158/0008-5472.CAN-06-4759. [DOI] [PubMed] [Google Scholar]
- 9.M Nihal, et al. Int J Cancer. 2005;114:513. doi: 10.1002/ijc.20785. [DOI] [PubMed] [Google Scholar]
- 10.H Yamaguchi, et al. J Health Sci. 2008;54:654. [Google Scholar]
- 11.D Armanini, et al. Exp Clin Endocrinol Diabetes. 2002;110:257. [Google Scholar]
- 12.Ogiku, et al. J Pharmacol Exp Ther. 2011;339:93. doi: 10.1124/jpet.111.182592. [DOI] [PubMed] [Google Scholar]
- 13.H Yamaguchi, et al. J Biol Chem. 2011;286:36888. doi: 10.1074/jbc.M111.265900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stott, et al. J Mol Biol. 2006;360:90. doi: 10.1016/j.jmb.2006.04.059. [DOI] [PubMed] [Google Scholar]
- 15.WL Jorgensen. Science. 2004;303:1813. doi: 10.1126/science.1096361. [DOI] [PubMed] [Google Scholar]
- 16.J Goto, et al. J Med Chem. 2004;47:6804. doi: 10.1021/jm0493818. [DOI] [PubMed] [Google Scholar]
- 17.J Goto, et al. J Chem Inf Model. 2008;48:583. doi: 10.1021/ci700352q. [DOI] [PubMed] [Google Scholar]
- 18.TA Halgren. J Comput Chem. 1996;17:490. [Google Scholar]
- 19.VR Harley, et al. Nucl Acids Res. 1994;22:1500. doi: 10.1093/nar/22.8.1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sakamoto, et al. Biol Pharm Bull. 2001;24:906. doi: 10.1248/bpb.24.906. [DOI] [PubMed] [Google Scholar]