

Abstract
Screening of the NCI Diversity Set-1 identified PI-083 (NSC-45382) a proteasome inhibitor selective for cancer over normal cells. Focused libraries of novel compounds based on PI-083 chloronaphthoquinone and sulfonamide moieties were synthesized to gain a better understanding of the structure activity relationship responsible for chymotrypsin-like proteasome inhibitory activity. This led to the demonstration that the chloronaphthoquinone and the sulfonamide moieties are critical for inhibitory activity. The pyridyl group in PI-083 can be replaced with other heterocyclic groups without significant loss of activity. Molecular modeling studies were also performed to explore the detailed interactions of PI-083 and its derivatives with the β5 and β6 subunits of the 20S proteasome. The refined model showed an H-bond interaction between the Asp-114 and the sulfonamide moiety of the PI-083 in the β6 subunit.
Keywords: Structure Activity Relationship (SAR), Chymotrypsin-like (CT-L), Proteasome Activity, Naphthoquinone Pharmacophore, β5 and β6 subunits
1. Introduction
Cancer is associated with increased proliferation and/or decreased apoptosis. Both of these processes are regulated by a complex interplay of transcription, protein synthesis, protein-protein interactions, protein phosphorylation, and protein degradation. The 26S proteasome, a large protein complex that consists of the catalytic 20S proteasome1 (molecular weight 700 kDa, also called 20S core particle [CP]) and 19S regulatory particle (molecular weight 900 kDa), has several proteolytic activities defined by different substrate specificities and is responsible for the degradation of more than 80% of intracellular proteins.2
The molecular and functional characteristics of the ubiquitin-proteasome system (UPS) have been studied by several groups3,4,5 and it has been shown that the 26S proteasome is responsible for the degradation of proteins involved in a diverse array of biological processes including cell cycle progression, apoptosis, DNA repair, immune response, signal transduction, transcription, metabolism, and developmental processes. Furthermore, the UPS has been reported to play a crucial role in tumorigenesis3, inflammation4 and autoimmunity.5 Much effort has therefore been dedicated to the discovery of proteasome inhibitors. Indeed, several proteasome inhibitors elicit apoptosis in malignant cells and represent a new class of antineoplastic agents.6 Subsequently, the proteasome has emerged as a promising molecular target for new cancer therapeutics.7 In 2003, Bortezomib (Velcade™, PS-341),8 a covalent but reversible peptidomimetic with a boronic acid moiety (Figure 1), was approved by the FDA for the treatment of multiple myeloma and mantle cell lymphoma. However, side effects and tumor cell resistance against Bortezomib demand the development of improved and selective proteasome inhibitors.9 Several new proteasome inhibitors, such as carfilzomib (PR- 171, a synthetic peptide),10 CEP-1877011 (a boronic acid derivative), and the natural product salinosporamide A (NPI-0052, monochlorinated compound)12 are in phase I and II clinical trials (Figure 1). Recent preclinical trials with the irreversible proteasome inhibitor salinosporamide A suggested a significantly stronger and prolonged effect on the chymotrypsin-like (CT-L) and trypsin-like (T-L) activities of the proteasome compared to Bortezomib.12 Other examples of proteasome inhibitors include natural products13 that possess reactive functional groups such as aldehydes (tyropeptin A), β-lactones (omuralide), epoxyketones (epoxomicin, eponemycin), cyclic peptides (TMC-95A), macrocyclic vinylketones (syringolin A, glidobactin A), and synthetic peptides (MG-132, PS-519 and TMC-95 analogs). Other natural products, such as (-)- epigallocatechin 3-gallate [(-)-EGCG)], the most abundant catechin (in green tea) and genistein (a soy isoflavone) has been suggested to act as a chemoprotective and anti-cancer agent by inhibiting the CT-L activity of the proteasome in vitro.14 Small, drug-like synthetic proteasome inhibitors that are selective for cancer over normal cells are rare, but clearly would have a potential advantage over the existing inhibitors listed above.
Figure 1.

Proteasome inhibitors; Bortezomib (clinically approved covalent and reversible inhibitor). CEP- 18770 (reversible inhibitor), carfilzomib and salinosporamide A (irreversible inhibitors) currently in phase I and II clinical trials. PI-083 (reversible inhibitor identified from the NCI diversity set-1).
As part of our many efforts to identify proteasome inhibitors, we screened the NCI diversity set-1 and other NCI libraries against CT-L activity of the 20S of the proteasome and identified PI-08315 (Figure 1). PI-083 has a unique skeleton, demonstrates good inhibition of CT-L activity of the proteasome and is selective for malignant over normal cells in vitro and in vivo.15 Interestingly, similar naphthoquinone scaffolds have been previously studied as potent anti-diabetic agents16 and inhibitors of protein tyrosine phosphatases such as CD4517 and CDC25B.18 In this study we investigated possible approaches to modulate the PI-083 template through the design of focused compound libraries to gain a better understanding of the structure-activity relationships (SAR) responsible for proteasome inhibitory activity.
2. Chemistry
First, in-house synthesis of PI-083 (NSC-45382) was carried out using a literature protocol19 to provide the material with >95% purity (as determined by HPLC) to confirm the inhibitory activity (IC50 = 1.0 ± 0.6 µM) and the structure (shown in Figure 1). The structure of PI-083 was confirmed using 1H & 13C NMR and high resolution mass spectrometry. The synthesis of the initial PI-083 compound library involved modification of the sulfonamide moiety, where functionality is present for rapid analog synthesis. Commercially available ‘sulfapyridine-like’ building blocks with diverse electronic properties, for example hydrogen-bond donor/acceptor, charge-transfer, dipolar interactions and steric properties were employed to explore the CT-L inhibitory activities (Figure 2). Based on molecular docking, the predicted binding interactions of PI-083 in the β5 and β6 subunits of the proteasome (Figure 2) suggest favorable interactions with Thr-21, Asp-114, Ala-49, Gly-47 and Thr-1. We were able to introduce diverse chemical and electronic properties to target compound libraries 2, 3, 6 and 13 (Schemes 1, 2 and 3) to exploit these interactions. Further diversity was also introduced via naphthoquinone ring substituents.
Figure 2.

A. Modifications around PI-083 for library synthesis and predicted binding interactions of PI-083 in the β5 and β6 subunits of the 20S proteasome. B. Overlay of PI-083 (cyan, docked pose) with the Bortezomib (magenta, X-ray crystal pose) in the β5 and β6 units of the 20S proteasome C. PI-083 overlaid in the β5 and β6 subunits of the 20S proteasome. D. PI-083 (cyan, stick representation) and analogs (line representations) shown in Table 1 overlaid in the β5 and β6 subunits of the proteasome. E. Surface model of the 20S proteasome with PI-083. The protein surface of the proteasome is colored according to electrostatic potential; positively charged areas are colored in blue, and negatively charged areas are colored in red. For PI-083, carbon atoms are colored in cyan, oxygen in red, nitrogen in blue, hydrogen in white, and sulfur in yellow. Images were created by PyMol20.
Scheme 1.

Synthesis of focused libraries of PI-083 using commercially available building blocks. Reagents and conditions: a (i) when R2, R3 = Cl, 95% EtOH, reflux at 115°C, 3 days. (ii) when R2 = Me, R3 = H, anhydrous dioxane, 10% ytterbium triflate, 125°C, 3 days. (iii) when R2, R3 = H, 90% EtOH, reflux, 3 days. b appropriate amine, EtOH, microwave, 140°C, 20 min.
Scheme 2.

Reagents and conditions: a NaBH3CN, AcOH, MeOH, 0 °C-r.t., 1–2 h, b 95% EtOH, sealed tube, reflux, 115°C, 3 days. c (i) RBr (or RI), DIPEA, DMF, microwave 160 °C, 15 min. or (ii) RI, DIPEA, DMF, Ar, , r.t., 4–24 h. d DMF/MeOH (4:1, 0.2 g in 30 mL), H-cube H2, 10% Pd/C, 40 bar, r.t., flow rate = 1.0 mL/min
Scheme 3.

Reagents and conditions: a Pyridine, DCE, microwave 150°C, 10 min. b NiCl2·6H2O, MeOH/THF 1:1, NaBH4, 0 °C, 15~30 min. c, 2,3-Dichloronaphthoquinone, 95% EtOH, sealed tube, reflux, 115°C, 3 days; d 5-aminotetrazole, Na2CO3, H2O, r.t., 24 h e THF, 0°C, 30 min., f MeOH (0.3g in 10 mL), H-cube, H2, 40 bar, 10% Pd/C, 30°C, flow rate = 1 mL/min.
The library 2 was synthesized from commercially available aniline sulfonamide building blocks and 2,3- dichloro-1,4-naphthoquinone, 1,4-naphthoquinone or 2-methyl-1,4-naphthoquinone using the protocol employed for PI-083 (Scheme 1). The synthetic protocol for compounds 2k-2o (Scheme 1) was validated using commercially available building blocks with 2-methyl-1,4-naphthoquinone and ytterbium trifluoromethanesulfonate in anhydrous dioxane under reflux. The crude reaction mixtures were purified by SiO2 chromatography to obtain the desired compounds with low to moderate yields. To study the effects of hydrophobic and hydrophilic substitutions at the 3-position of the naphthoquinone ring in PI-083 (R4, Scheme 1), a set of analogs was generated via a two-step synthesis. These final compounds with amine groups at the 3- position (3, Scheme 1) were prepared in moderate yields with microwave-assisted heating reactions of PI-083 with appropriate secondary amines.
Intermediates 5a and 5b were generated via reductive amination of the commercially available sulfapyridine 4 with requisite aldehydes as shown in Scheme 2. Attempts to react 5a and 5b with commercially available 2,3-dichloronaphthoquinone to obtain alkylated amine analogs of PI-083 were not successful. In an alternative approach, PI-083 was reacted with either an alkyl bromide or an alkyl iodide in DMF (at room temperature or with microwave heating). Under these conditions, we observed the alkylation of the sulfonamide group of PI-083 (R2 in library 6, Scheme 2). The library 6 was purified using flash chromatography to obtain the desired compounds with greater than 95% purity as assessed by 1H NMR and LCMS analysis. Intermediates 7 were synthesized in good yields via coupling commercially available 5- and 6-nitro-2,3-dichloro-1,4-naphthoquinone with the sulfapyridine 4 in refluxing ethanol. Compounds 7a and 7b were obtained as mixtures of regioisomers approximately 1:2 ratios (assessed by 1H NMR). The 5-nitro-2,3-dichloronaphthoquinone is reported to be more reactive towards amines affording regioisomeric mixtures of mono-substituted products.21 Reduced products of 8 (a mixture of regioisomers approximately 1:5 ratio after purification using SiO2 chromatography) were obtained from 7a using the hydrogenation conditions described in the Scheme 2. Attempts to separate the pure isomers of compounds 7 or 8 by chromatography were not successful.
The possible binding interactions of the sulfonamide moiety of PI-083 with the proteasome were further investigated via the synthetic modifications outlined in Scheme 3. A series of nitrosulfonamide building blocks 11 were generated using standard reagents in good yield by coupling (microwave-assisted heating or room temperature) commercially available sulfonyl chlorides and anilines. The corresponding amine intermediates 12 were obtained in good yields via NiCl2/NaBH4 mediated reduction.22 The final library 13 was prepared as described previously by reacting anilines with 2,3-dichloronaphthoquinone with > 95% purity. Starting from 4-nitrobenzenesulfonyl chloride and 5-aminotetrazole, intermediate 11l was obtained as reported in the literature23 (Scheme 3). The 4-nitrobenzenesulfonylguanyl azide 11l intermediate was reduced to N-(4-aminobenzenesulfonyl) guanidine with NaBH4 in the presence of NiCl6 to obtain 12l in good yield. The synthesis and the purity of library 13 were confirmed by 1H NMR and LCMS analysis. The inhibitory activities of potent compounds from libraries 2, 3, 6 and 13 are summarized in Table 1.
Table 1.
PI-083 derivatives with moderate activities.
 | |||||
|---|---|---|---|---|---|
| Compound | R’ | R” |
In vitro IC50(µM)a |
||
| CT-L activity | T-L activity | PGPH activity | |||
|
Bortezomib (Figure 1) |
- | - | 0.009 ± 0.006 | 7.0 ± 0.24 | 0.48 ± 0.021 |
| PI-083 | 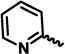 |
H | 1.0 ± 0.63 | 4.5 ± 1.4 | 4.5 ± 1.2 |
| 2s | H | 3.3 ± 0.30 | 8.15 ± 0.84 | 14.3 ± 1.60 | |
| 2h | 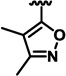 |
H | 3.9 ± 0.50 | 9.25 ± 0.45 | 21.2 ± 0.79 |
| 2f | 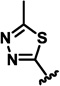 |
H | 6.4 ± 0.5 | 10.7 ± 0.5 | 13.4 ± 1.1 |
| 2d | 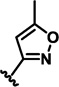 |
H | 11.2 ± 3.4 | 13.2 ± 0.2 | 16.7 ± 0.5 |
| 13h | 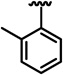 |
OMe | 13.7 ± 0.35 | 24.3 ± 0.04 | 30 ± 1.5 |
| 13j | OMe | 15.3 ± 0.89 | 23.3 ± 0.94 | 35 ± 1.90 | |
| 2b | 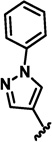 |
H | 16.3 ± 6.5 | 20.65 ± 1.45 | 25.6 ± 1.39 |
| 13e | 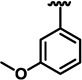 |
H | 17 ± 1 | 24.55 ± 0.55 | 43.5 ± 2.5 |
| 2g | 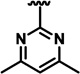 |
H | 19 ± 0.39 | 20.9 ± 0.9 | 50.25 ± 2.25 |
| 13d | 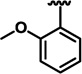 |
H | 20.8 ± 0.25 | 24.65 ± 1.0 | 49.25 ± 1.75 |
| 13c | 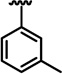 |
H | 20.8 ± 0.3 | 55 ± 2 | > 100 |
Tested in triplicate at least twice.
3. Results and Discussion
PI-083 was identified from the NCI diversity set-1 as a CT-L proteasome inhibitor and confirmed as a hit compound. In this study, we synthesized several focused libraries around the PI-083 scaffold; pyridine, sulfonamide and the chloronaphthoquinone moieties to gain a better understanding of the SAR responsible for CT-L proteasome inhibitory activity. To assess the ability of the synthesized compounds to inhibit the CT-L proteolytic activity of purified rabbit 20S proteasome, a fluorometric assay was utilized.24 Commercially available pyridine sulfonamide (sulfapyridine), 2,3-dichloronaphthoquinone, 2-methyl-1,4-naphthoquinone and 1,4-naphthoquinone building blocks themselves showed no proteosome CT-L activity (IC50 > 100 µM).
Starting from the pyridine end of the PI-083 scaffold, we have demonstrated replacing pyridine with hydrogen (2t, Scheme 2) or amines bearing small hydrophobic units such as methyl, ethyl, and isopropyl (13mr, Scheme 3) resulted in loss of inhibitory activity (IC50 > 100 µM). Replacing the pyridine with hydrophobic electron withdrawing groups such as chloro- or fluoro-phenyl units (2u, 2v and 2w, Scheme 2) also resulted in loss of inhibitory activity (IC50 > 100 µM). Compounds 13m-r and 2u-w suggest non aromatic hydrophobic groups and electron withdrawing aromatic hydrophobic groups are not tolerated in this region. Our docking suggests Asp-114 is able to H-bond with PI-083 in the β6 subunit (See the modeling section). The Asp-114 interactions are also observed crystallographically for Bortezomib (Figure 2B, PDB+ ID: 2F16).25 Previously, we reported15 the pyridine in PI-083 interacting with Asp-114 in the β6 subunit via a water molecule might be responsible for CT-L activity. In the focused library synthesis, the rationale for replacement of the pyridine with basic units was to probe interactions with Asp-114 in the β6 subunit (Figure 2). However, since our original report, the modeling software we employ has become more sophisticated allowing examination of different tautomers and ionization states as well as improved calculation of partial charges using the applications Ligprep26, Epik27 and QM Polarized Ligand Docking28 (QPLD) respectively. Previously these applications were not available to us. This enabled modeling of a form of PI-083 with an anionic sulfonamide nitrogen. These improvements led to refinement of our previous model by taking better account of the pKa for the deprotonation of the sulfonamide nitrogen of PI-083 which was calculated by Epik to be 6.7. As a result, the construct used for modeling (see the modeling section) suggests a hydrogen bond between protonated Asp- 114 in the β6 subunit and the negatively charged sulfonamide moiety of the PI-083 scaffold (Figure 2A). The resonance stabilization of the sulfonamide anion by the adjacent pyridine of the PI-083 in its bioactive conformation could be leading to inhibition of the CT-L proteasome activity (resonance stabilization should increase the acidity of the sulfonamide group). This evolution of the model provided insights into the SAR of several PI-083 derivatives. It is possible that compounds possessing heterocyclic moieties shown in Table 1 such as thiazole (2s), oxazole (2d and 2h), thiadiazole (2f) and pyrazole (2b) are able to stabilize the negative charge on the sulfonamide moiety is contributing to CT-L inhibitory activity (IC50 = 3–16 µM). Compounds with pyrimidine moieties (2e, 2i) that contain H-bond acceptor residues showed weak activity (IC50 = 100 µM). However, compound 2g; with methyl pyrimidine showed improved activity (IC50 = 19 µM). Compounds with phenyl groups with small hydrophobic residues such as 13d, 13e, 13h and 13j showed moderate activity (IC50 = 13–20 µM) suggesting these compounds might be undergoing non-specific hydrophobic interactions in this area. The region marked as “Groove G’ (Figure 2E) is a hydrophobic pocket in the β6 subunit, and it is possible compounds 13d-13j are able to partially occupy this region in their binding conformation. The overall binding affinity was not improved when the pyridyl group in PI-083 was replaced with aromatic hydrophobic groups (compounds 13a-13k, Scheme 3, IC50 > 100 µM).
Next, we investigated the role of the sulfonamide moiety by replacing the hydrogen with hydrophobic groups. Library 6 (Scheme 2), derived from alkylation of the sulfonamide moiety with methyl (6a), ethyl (6b), substituted benzyl (6c, 6d, 6e and 6f) and naphthyl (6g) derivatives lacked inhibitory activities (IC50 > 100 µM). This observation suggests the H-bond interaction of the sulfonamide hydrogen of the PI-083 with the β6 Asp 114 subunit of the proteasome is crucial to retain the CT-L inhibitory activity of PI-083 and further validates the docking results from the refined structural model (Figure 2).
Finally we assessed the contribution of the chloronaphthoquinone moiety of PI-083 to its CT-L inhibitory activity. Our dockings resulted in a pose for PI-083 in which the napthoquinone carbonyl groups hydrogen bond with Gly-47 and Thr-21 creating a hydrogen bond network similar to Bortezomib. Bortezomib forms dual hydrogen bonds with both Gly-47 and Thr-21 where hydrogen bonds are formed with backbone nitrogens and carbonyl groups (Figure 2B). We found the chlorine at the 2-position of the naphthoquinone moiety is essential for activity. Replacement of the chloride in PI-083 with methyl (2k, 2l, 2m, 2n, 2o) or hydrogen (2p, 2q, 2r) is detrimental to in vitro CT-L inhibitory activity (IC50>100). It is conceivable that PI-083 undergoes Michael type nucleophilic attack with Thr-1 in the β5 subunit and inhibits proteasome via a covalent modification. This is consistent with the docking results (Figure 2) that place the chlorinated electrophilic carbon of PI-083 3.22 Å away from the Thr-1 hydroxyl group in the β5 subunit (Figure 2A). Attempts to generate hydronaphthoquinone of PI-083 via reduction of the naphthoquinone moiety were not successful. The hydronaphthoquinone oxidizes back to naphthoquinone instantaneously (experimental details not reported here). The nitro and amino derivatives of PI-083 (7 and 8 respectively, mixtures of isomers shown in Scheme 2) did not lead to any appreciable inhibitory activities in the in vitro enzymatic assay, suggesting that the nitro and amino groups on 5- and 6-position of the chloronaphthoquinone are not tolerable.
Additionally, we determined the ability of PI-083 and its analogs to inhibit T-L and peptidyl glutamyl peptide hydrolase (PGPH) activities in vitro15 (Table 1). Bortezomib was used as a control and none of the compounds shown in Table 1 demonstrated higher potency towards T-L or PGPH than the CT-L activity. Our data also showed (not reported here) that PI-083 does not inhibit (IC50 > 30 µM) purified Calpain, (calcium-dependent non-lysosomal cysteine protease), but inhibited all three proteasomal (CT-L, T-L and PGPH) activities with similar potency15. As reported previously, we have shown that PI-083 inhibits proteasome activity in vitro and in vivo.15 Treatment of MCF-7 cells with PI-083 resulted in inhibition of the CT-L activity of the proteasome with an IC50 value of 6 µM and PI-083 also inhibited cell viability with an IC50 value of 2.31 ± 0.10 µM15. We found PI-083 inhibits proliferation and induces cell death in three different human tumor cell lines (breast, pancreatic and ovarian), but not in their normal/immortalized counterparts.15 Our studies indicated that PI-083 induces apoptosis in cancer cell lines derived from prostate, lung and multiple myeloma, in addition to the human tumor cells lines mentioned above.15 Furthermore, PI-083 suppresses the growth of human breast and lung tumors implanted as xenografts into nude mice, and is efficient in inhibiting proliferation and survival of primary cells derived from patients with multiple myeloma.15
To investigate whether PI-083-mediated proteasome inhibition is reversible, we performed a dialysis29 experiment with PI-083 and Bortezomib, a covalent reversible proteasome inhibitor that was used as an internal control. Figure 3 shows that in the absence of dialysis, PI-083 and Bortezomib were able to inhibit the CT-L activity of the 20S proteasome by 88% and 99% respectively. During dialysis, the CT-L activity started to recover at the 1 hr mark in the PI-083 treated sample. By contrast, in the Bortezomib treated samples, CT-L activity recovery did not begin until 4 hrs. These results suggest that both PI-083 and Bortezomib behave similarly, but that PI-083 appears to be more rapidly released and/or is slower to attach. It is likely that PI-083 behaves as a covalent reversible CT-L inhibitor.
Figure 3.

Recovery of CT-L activity upon dialysis of the 20S proteasome-compound complexes after pre-incubation with PI-083 (■) and Bortezomib (●).
4. Molecular Modeling
GLIDE 5.030 was employed for docking of the ligands described herein into a structure of the β5, β6 subunits of the 20S yeast proteasome with Bortezomib bound obtained from the Protein Data Bank31 (PDB ID: 2F16) and appropriately prepared for docking calculations (hydrogen atoms added, appropriate histidine tautomer and protonation state, restrained energy minimization, etc.). To obtain a reasonable sampling of poses, 100 top ranking poses were kept for each structure in the ligand set, which had been docked using GLIDE in standard precision (SP) mode. Poses with the smallest distance between Thr-1 oxygen and the chloro-carbon of the naphthoquinone moiety were chosen for subsequent docking with GLIDE Extra Precision32 (XP), which allows for more precise calculations of binding energy, poses, hydrophobic interactions, and expulsion of water from pockets. GLIDE XP was employed with QPLD (QM Polarized Ligand Docking) for calculation of partial charges “on the fly” using the B3LYP density functional method in order to adequately account for charge delocalization of the sulfonamide moiety. Poses with lowest energy that resulted in Thr-1 oxygen and chloro-carbon in naphthoquinone distances less than 5Å were considered. All structures were viewed, created, and modified with Schrödinger's Maestro 8.533. PI-083 and its analogs were processed using LigPrep 2.2 and tautomers and structures with ionization states appropriate for a pH range of 5.0 to 9.0 were generated. The pKa values for the sulfonamide nitrogen of the compounds 2b-2s shown in Table 1 were less than 7.91 (data not shown) as calculated by Epik. Thus, at physiological pH substantial fraction of sulfonamides 2b-2s will exist in the anionic deprotonated from. Interestingly, the pKa calculated by Epik for the sulfonamide substituent in 2t is 9.5, which means that the compound would predominately exist in the protonated (conjugate acid) form at pH 7.6 that may explain its lack of activity. When 2t in its protonated form was docked to the B5/B6 subunits of the proteasome, no poses were observed that met our distance criterion of a 5 Å separation between the oxygen atom of the Thr-1 hydroxyl group and the carbon atom to which the chlorine is attached in the naphthoquinone ring
A low energy pose of PI-083 is depicted in Figure 2B with interactions modified from the ones we previously reported.15 Former docking studies were performed with an earlier version of the GLIDE docking software34 and prior to the availability of LigPrep in our lab. LigPrep 2.2 along with Epik was used to generate tautomers and alternative protonation states for PI-083, which includes the anionic form of the sulfonamide nitrogen. Consequently, a low energy pose was generated where the anionic sulfonamide nitrogen formed a hydrogen bond (N-O distance: 2.8 Å) with protonated Asp-114. (Note: the X-ray structure of the proteasome- Bortezomib complex is consistent with protonated Asp-114, and it is reasonable to assume that it may well be protonated in the proteasome-PI-083 complex). In the previous model, the pyridyl nitrogen15 of PI-083 is 3.3 Å from one of the oxygens of Asp-114 suggesting an electrostatic interaction. The interactions of the refined model slightly shift the pose of PI-083 allowing for a better angle of nucleophilic attack by Thr-1 on the chloronaphthoquinone group. Furthermore, PI-083 does not interact with the nearby water in the refined model but does form hydrogen bonds between the carbonyl groups of the naphthoquinone and Thr-21 and Gly-47. In order to determine whether the water molecule near Asp-114 (crystallographically determined for the proteasome-Bortezomib complex) was in an energetically favorable position in our docked proteasome-PI-083 model, MacroModel35 was used to sample possible alternative configurations of this water molecule. With the β5 and β6 coordinates held frozen, the water molecule close to Asp-114 and a crystallographically determined water molecule hydrogen bonded to the first one were allowed to freely rotate and translate during a Monte Carlo simulation performed on our docked model of PI-083 bound to the β5, β6 subunits of the 20S proteasome. Out of 100 low energy configurations generated, 76 retained a water molecule in the location closest to Asp-114 observed crystallographically. The XP pose of PI-083 places the electrophilic carbon, to which chlorine is attached, 3.22 Å away from the oxygen of Thr-1 with reasonable positioning for nucleophilic attack (Figure 2A). Low energy XP poses of the active analogs of PI-083 (Table 1) are observed to have similar binding modes to PI-083 (Figure 2D).
As previously stated, our dialysis experiments suggest that PI-083 behaves as a covalent but reversible proteasome inhibitor. Our docking results suggest that all of the active compounds, which contain a chloro substituent at the 2-position of the naphthoquinone ring, can adopt a low energy docking pose that is poised for covalent bond formation with Thr-1. All of the active molecules may, indeed, form a covalent bond with Thr-1 but we have not yet shown this to be the case experimentally. Our modeling studies suggest that it is possible for all of the active compounds to be involved in formation of a pre-organized complex that subsequently leads to covalent bond formation. We have also performed covalent docking of PI-083 to the β5, β6 subunits of the 20S proteasome using GOLD 4.1 (not shown here). Three poses were obtained that are all qualitatively similar to the pose presented in Figure 2B; however PI-083 has been translated by ~1.5 Å and rotated slightly due to presence of the covalent bond between carbon-2 of the naphthoquinone ring and the hydroxyl-oxygen atom of Thr-1. The hydrogen-bond between Asp-114 and the sulfonamide nitrogen atom is no longer present but the pyridine ring is still located in Groove G (Figure 2E) in the S3 pocket.
5. Conclusions
In summary, novel naphthoquinone derivatives of PI-083 were prepared via several routes. The SAR indicates that the inhibitory activity appears very sensitive to changes around the molecule. The chlorine and sulfonamide groups of PI-083 appear to be essential for activity. The pyridyl group can be replaced with heterocyclic moieties without significant reduction of activity in in-vitro. The replacement of the pyridyl unit with aromatic groups (with hydrophobic and hydrophilic characteristics, 13a-k) or small hydrophobic units (13m-r) were not tolerable. PI-083 has been shown to be more selective in inhibiting proliferation, inducing cell death and apoptosis for breast, ovarian and pancreatic cancer cells over their normal counterparts.15 In nude mice, PI-083 was efficient in inhibiting the growth of human tumor xenografts derived from breast and lung cancer cells.15 Altogether our data suggest PI-083 has potential for further development as an anti-cancer agent.
6. Experimental
6.1. General
All reagents were purchased from commercial suppliers and used without further purification. Melting points were determined using a Barnstead international melting point apparatus and remain uncorrected. 1H NMR spectra were recorded on a Varian Mercury 400 MHz spectrometer with Acetone-d6, CDCl3 or DMSO-d6 as the solvent. 13C NMR spectra are recorded at 100 MHz. All coupling constants are measured in Hertz (Hz) and the chemical shifts (δH and δC) are quoted in parts per million (ppm) relative to TMS (δ 0), which was used as the internal standard. Liquid chromatography mass spectroscopy (LCMS) and High resolution mass spectroscopy (HRMS) were carried out on an Agilent 6210 LC/MS (ESI-TOF). For LCMS and HRMS the compounds were eluted between 2–5 minutes using Rapid Resolution Cartridge (2.1×30 mm, particle size 3.5 µm) from Agilent Technologies. LCMS was used to detect ions of mass 100–1000 Da, and single peak was observed in the chromatogram after purification. Low resolution mass spectroscopy (LRMS) was carried out using Agilent single quad G1956A. HPLC was carried out using Jasco UV-2075 plus uv-vis detector (column: ultra C18, 5µm, 150 mm×4.6 mm). H-Cube® (ThalesNano) continuous-flow hydrogenation reactor was used for hydrogenation reactions. Microwave reactions were performed in CEM Discover 908005 model and Biotage initiator 8 machines. Thin layer chromatography was performed using silica gel 60 F254 plates (Fisher), with observation under UV when necessary. Anhydrous solvents were used as purchased: dichloromethane (anhydrous, 99.8% contains 50–150 ppm hydrocarbon as stabilizer from Aldrich), dimethyl formamide (anhydrous, 99.9% from Aldrich), tetrahydrofuran (anhydrous, 99.9%, inhibitor free, Aldrich), acetonitrile (anhydrous, 99.8%, Aldrich), toluene (anhydrous, 99.8%, Aldrich), methanol (anhydrous, 99.8%, Aldrich), ethanol (absolute, 99.5%, Aldrich).
6.2. General procedures for synthesis of library 2 and library 3 (Scheme 1)
6.2.1. Synthesis of compounds 2a-2j and 2s-2w
The starting material 2,3-dichloronaphthoquinone (700 mg, 3.08 mmol) and appropriate commercially available sulfonamide anilines (0.5 molar equivalents) were suspended in 95% ethanol (15 mL) and heated at 115°C for 3 days to obtain mixtures of red/orange precipitates. The reaction mixtures were cooled to room temperature and the resultant precipitates were filtered and washed with ethanol (5 times, total volume approximately 15–20 mL). The crude products obtained were rinsed with EtOAc (5 mL), DCM (5–10 mL), MeOH (5–10 mL) to remove remaining starting materials and quick acetone in DCM (50:50 mix, 5 mL) rinse was able to remove the impurities when ethanol wash was not sufficient to remove the impurities. The required pure compounds (95% by 1H NMR) in the library 2 were isolated as red or orange solids between 5–98% yields.
6.2.1.1. 4-(3-Chloro-1,4-dioxo-1,4-dihydronaphthalen-2-ylamino)-N-(pyridin-2-yl)benzenesulfonamide PI-083 (2a)
Orange solid (940 mg, 53%); Mp: 265–267 °C (lit. 262 °C decomposed).19 1H NMR (400 MHz, DMSO-d6) δ 9.48 (s, 1H disappeared on D2O shake), 8.03-8.00 (m, 3H), 7.85 (dt, J = 7.6, 1.6 Hz, 1H), 7.80 (dt, J = 7.6, 1.2 Hz, 1H), 7.74 (d, J = 8.4 Hz, 2H), 7.69 (apparent dt J = 7.6, 1.6 Hz, 1H), 7.15 (d, J = 8.0 Hz, partially overlapped, 2H), 7.12 (m, partially overlapped, 1H) 6.86 (br t, 1H); 13C NMR (100 MHz, DMSO-d6) δ 180.5, 177.6, 143.3, 135.4, 134.1, 132.3, 131.1, 127.7, 127.4, 127.2, 126.8, 122.8, 121.9, 118.9; LCMS (ES+) 440 (M+H)+; HRMS (ES+) m/z calculated for C21H15ClN3O4S (M+H)+ 440.0466, found 440.0465; HPLC 99% (Rt = 1.80, 10% water in acetonitrile).
6.2.1.2. 4-(3-Chloro-1,4-dioxo-1,4-dihydronaphthalen-2-ylamino)-N-(1-phenyl-1H-pyrazol-4-yl)benzenesulfonamide (2b)
Orange solid (198 mg, 24%); Mp: 230–231 °C; 1H NMR (400 MHz, DMSO-d6) δ 10.30 (s, 1H disappeared on D2O shake), 9.60 (s, 1H disappeared on D2O shake), 8.05 (d, J = 6.8 Hz, 2H), 7.88 (dt, J = 7.2, 1.2 Hz, 1H), 7.84 (dt, J = 7.2, 1.6 Hz, 1H), 7.58-7.56 (m, 3H), 7.48-7.41 (m, 4H), 7.38-7.36 (m, 1H), 7.19 (d, J = 8.8 Hz, 2H), 5.79 (d, J = 2.0 Hz, 1H), LRMS (ES+) 505 (M+H)+; HRMS (ES+) m/z calculated for C25H18ClN4O4S (M+H)+ 505.0731, found 505.0735.
6.2.1.3. N-(4-(3-chloro-1,4-dioxo-1,4-dihydronaphthalen-2-ylamino)phenylsulfonyl)benzamide (2c)
Red solid (58 mg, 8%); Mp: 216–217 °C; 1H NMR (400 MHz, DMSO-d6) δ 12.44 (br s, 1H disappeared on D2O shake), 9.60 (s, 1H disappeared on D2O shake), 8.05-8.03 (m, 2H), 7.88-7.80 (m, 6H), 7.61 (t, J = 7.2 Hz, 1H), 7.47 (t, J = 8.0 Hz, 2H), 7.24 (d, J = 8.4 Hz, 2H); LRMS (ES+) 467 (M+H)+; HRMS (ES+) m/z calculated for C23H16ClN2O5S (M+H)+ 467.0463, found 467.0468.
6.2.1.4. 4-(3-Chloro-1,4-dioxo-1,4-dihydronaphthalen-2-ylamino)-N-(5-methylisoxazol-3-yl)benzenesulfonamide (2d)
Dark orange solid (97 mg, 14%); Mp: 215–217 °C; 1H NMR (400 MHz, DMSO-d6) δ 11.32 (s, 1H), 9.55 (s, 1H), 8.05-8.02 (m, 2H), 7.88-7.80 (m, 2H), 7.71 (d, J = 8.8 Hz, 2H), 7.20 (d, J = 8.8 Hz, 2H), 6.13 (s, 1H), 2.28 (s, 3H); LRMS (ES+) 444 (M+H)+; HRMS (ES+) m/z calculated for C20H15ClN3O5S (M+H)+ 444.0415, found 444.0418.
6.2.1.5. 4-(3-Chloro-1,4-dioxo-1,4-dihydronaphthalen-2-ylamino)-N-(5-methoxypyrimidin-2-yl)benzenesulfonamide (2e)
Red solid (332 mg, 46%); Mp: 269–271 °C; 1H NMR (400 MHz, DMSO-d6) δ 11.38 (s, 1H), 9.52 (s, 1H), 8.28 (s, 2H), 8.03 (br t, J = 6.0 Hz, 2H), 7.89-7.81 (m, 4H), 7.18 (d, J = 8.4 Hz, 2H), 3.77 (s, 3H); LRMS (ES+) 471 (M+H)+; HRMS (ES+) m/z calculated for C21H16ClN4O5S (M+H)+ 471.0524, found 471.0526.
6.2.1.6. 4-(3-Chloro-1,4-dioxo-1,4-dihydronaphthalen-2-ylamino)-N-(5-methyl-1,3,4-thiadiazol-2-yl)benzenesulfonamide (2f)
Red solid (696 mg, 98%); Mp: 220 °C decomposed; 1H NMR (400 MHz, DMSO-d6) δ 13.92 (s, 1H disappeared on D2O shake), 9.53 (s, 1H disappeared on D2O shake), 8.03 (br d, J = 8.0 Hz, 2H), 7.84 (dt, J = 7.6, 0.8 Hz, 1H), 7.83 (dt, J = 6.4, 1.2 Hz, 1H), 7.66 (d, J = 8.8 Hz, 2H), 7.18 (d, J = 8.8 Hz, 2H), 4.35 (br s, 1H), 2.45 (s, 3H); LRMS (ES+) 461 (M+H)+; HRMS (ES+) m/z calculated for C19H14ClN4O4S2 (M+H)+ 461.0139, found 461.0131.
6.2.1.7. 4-(3-Chloro-1,4-dioxo-1,4-dihydronaphthalen-2-ylamino)-N-(4,6-dimethylpyrimidin-2-yl)benzenesulfonamide (2g)
Reddish-brown solid (326 mg, 45%); Mp: 210 °C decomposed; 1H NMR (400 MHz, DMSO-d6) δ 9.52 (s, 1H), 8.04 (m, 2H), 7.89-7.80 (m, 4H), 7.71 (d, J = 12.0 Hz, 1H), 7.29 (s, 1H), 7.21 (d, J = 8.0 Hz, 2H), 2.31 (s, 6H) (Note: approximately 10% base line impurities present between 8.10 to 7.80 ppm); LRMS (ES+) 469 (M+H)+; HRMS (ES+) m/z calculated for C22H18ClN4O4S (M+H)+ 469.0731, found 469.0734.
6.2.1.8. 4-(3-Chloro-1,4-dioxo-1,4-dihydronaphthalen-2-ylamino)-N-(3,4-dimethylisoxazol-yl)-benzenesulfonamide (2h)
Orange solid (133 mg, 19%); Mp: 208–210 °C; 1H NMR (400 MHz, DMSO-d6) δ 10.92 (br s, 1H), 9.61 (s, 1H), 8.04 (d, J = 7.6 Hz, 2H), 7.91-7.81 (m, 2H), 7.62 (d, J = 8.8 Hz, 2H), 7.21 (d, J = 8.8 Hz, 2H), 2.07 (s, 3H), 1.60 (s, 3H); LRMS (ES+) 458 (M+H)+; HRMS (ES+) m/z calculated for C21H17ClN3O5S (M+H)+ 458.0572, found 458.0578.
6.2.1.9. 4-(3-Chloro-1,4-dioxo-1,4-dihydronaphthalen-2-ylamino)-N-(2,6-dimethoxypyrimidin-4-yl)-benzenesulfonamide (2i)
Orange solid (299 mg, 65%); Mp: 220–222 °C; 1H NMR (400 MHz, DMSO-d6) δ 11.48 (br s, 1H), 9.56 (s, 1H), 8.03 (dt, J = 8.0, 1.6 Hz, 2H), 7.91-7.80 (m, 4H), 7.21 (d, J = 8.8 Hz, 2H), 5.93 (s, 1H), 3.78 (s, 3H), 3.74 (s, 3H); LRMS (ES+) 501 (M+H)+; HRMS (ES+) m/z calculated for C22H18ClN4O6S (M+H)+ 501.0630, found 501.0634.
6.2.1.10. 4-(3-Chloro-1,4-dioxo-1,4-dihydronaphthalen-2-ylamino)-N-(4-methylpyrimidin-2-yl)-benzenesulfonamide (2j)
Dark red solid (446 mg, 65%); Mp: 246–248 °C; 1H NMR (400 MHz, DMSO-d6) δ 9.53 (s, 1H), 8.31 (d, J = 4.4 Hz, 1H), 8.04-8.02 (m, 2H), 7.87-7.81 (m, 4H), 7.18 (d, J = 8.0 Hz, 2H), 6.90 (s, 1H), 2.30 (s, 3H); LRMS (ES+) 455 (M+H)+; HRMS (ES+) m/z calculated for C21H16ClN4O4S (M+H)+ 455.0575, found 455.0581.
6.2.1.11. 4-(3-Chloro-1,4-dioxo-1,4-dihydronaphthalen-2-ylamino)-N-(thiazol-2-yl)benzene-sulfonamide (2s)
Orange red solid (145 mg, 65%); Mp: 286–288°C; 1H NMR (400 MHz, DMSO-d6) δ 12.72 (br s, 1H), 9.51 (s, 1H), 8.04-8.02 (m, 2H), 7.86 (dt, J = 7.4, 1.5 Hz, 1H), 7.81 (dt, J = 7.4, 1.5 Hz, 1H), 7.67 (d, J = 8.7 Hz, 2H), 7.24 (d, J = 4.6 Hz, 1H), 7.17 (d, J = 8.7 Hz, 2H), 6.76 (d, J = 4.6 Hz, 1H); LCMS (ES+) 446 (M35Cl+H)+, 448 (M37Cl+H)+; HRMS (ES+) m/z calculated for C19H13ClN3O4S2 (M+H)+ 446.0030, found 446.0045.
6.2.1.12. 4-(3-Chloro-1,4-dioxo-1,4-dihydronaphthalen-2-ylamino)benzenesulfonamide (2t)
Red solid (24 mg, 38.9%); Mp: >300°C; 1H NMR (400 MHz, DMSO-d6) δ 9.54 (br s, 1H), 8.06 (d, 7.4 Hz, 1H), 7.87-7.80 (m, 2H), 7.72 (d, J = 8.5 Hz, 2H), 7.30 (br s, 2H), 7.23 (d, J = 8.5 Hz, 2H); 13C NMR (100 MHz, DMSO-d6) δ 180.7, 177.7, 143.5, 142.9, 139.2, 135.5, 134.2, 132.4, 131.2, 127.3, 126.9, 126.4, 123.0, 118.6; LCMS (ES+) 363 (M35Cl+H)+, 365 (M37Cl+H)+; HRMS (ES+) m/z calculated for C16H12ClN2O4S (M+H)+ 363.0201, found 363.0192.
6.2.1.13. 4-(3-Chloro-1,4-dioxo-1,4-dihydronaphthalen-2-ylamino)-N-(4-chlorophenyl)benzenesulfonamide (2u)
Yellow solid (180 mg, 76%); Mp: 272 °C decomposed; 1H NMR (400 MHz, DMSO-d6) δ 10.29 (s, NH, 1H, disappeared in D2O shake), 9.51 (s, NH,1H disappeared in D2O shake), 8.02 (ddd, J = 7.0, 3.2, 1.3 Hz, 2H), 7.83 (dtd, J = 19.8, 7.4,1.3 Hz, 2H), 7.60 (d, J = 8.8 Hz, 2H), 7.27 (d, J = 8.8 Hz, 2H), 7.14 (d, J = 8.8 Hz, 2H), 7.07 (d, J = 8.8 Hz, 2H); LCMS (ES-) 471 (M-H)−; HRMS (ES-) m/z calculated for C22H14Cl2N2O4S (M-H)− 470.9979, found 470.9991.
6.2.1.14. 4-(3-Chloro-1,4-dioxo-1,4-dihydronaphthalen-2-ylamino)-N-(2,4-difluorophenyl)benzenesulfonamide (2v)
Gold yellow solid (146 mg, 62%); Mp: 250–252 °C; 1H NMR (400 MHz, DMSO-d6) δ 9.98 (s, NH,1H, disappeared in D2O shake), 9.55 (s, NH, 1H, disappeared in D2O shake), 8.03 (dd, J = 7.6, 1.4 Hz, 2H), 7.86 (dt, J = 8.0, 1.2 Hz, 1H), 7.81 (dt, J = 7.6,1.2 Hz, 1H), 7.53 (d, J = 8.7 Hz, 2H), 7.24-7.20 (m, 2H), 7.15 (d, J = 8.7 Hz, 2H), 7.04-6.95 (m, 1H); LCMS (ES-) 473 (M35Cl-H)−, 475 (M37Cl-H)−; HRMS (ES-) m/z calculated for C22H13ClF2N2O4S (M-H)− 473.0180, found 473.0197.
6.2.1.15. 4-(3-Chloro-1,4-dioxo-1,4-dihydronaphthalen-2-ylamino)-N-(2,4-dichlorophenyl)benzenesulfonamide (2w)
Orange solid (180 mg, 70%); Mp: 258 °C decomposed; 1H NMR (400 MHz, DMSO-d6) δ 10.00 (s, NH, 1H, disappeared in D2O shake), 9.56 (s, NH, 1H, disappeared in D2O shake), 8.03 (dd, J = 7.3, 1.6 Hz, 2H), 7.87 (dt, J = 7.2, 1.6 Hz, 1H), 7.81 (dt, J = 7.2, 1.6 Hz, 1H), 7.56-7.54 (m, 3H), 7.38 (dd, J = 8.7, 2.4 Hz, 2H), 7.27 (d, J = 8.7 Hz, 2H), 7.16 (d, J = 8.7 Hz, 2H); LCMS (ES-) 505 (M35Cl-H)−, 507 (M37Cl-H)−, HRMS (ES-) m/z calculated for C22H13Cl3N2O4S (M-H)− 504.9589, found 504.9600.
6.2.2. General procedure for synthesis of compounds 2k-2o
2-Methyl-1,4-naphthoquinone (100 mg, 0.58 mmol) and ytterbium trifluoromethanesulfonate (15 mg, 0.024 mmol) were added to anhydrous dioxane (5.0 mL) followed by sulfapyridine (144 mg, 0.58 mmol). The reaction mixture was heated under reflux for 24 h and the TLC analysis indicated depletion of starting material. The crude reaction mixtures were purified by SiO2 chromatography to obtain the desired compounds.
6.2.2.1. 4-(3-Methyl-1,4-dioxo-1,4-dihydronaphthalen-2-ylamino)-N-(pyridin-2-yl)benzenesulfonamide (2k)
Orange solid (74 mg, 20%); Mp: 220–222 °C; 1H NMR (400 MHz, DMSO-d6) δ 8.94 (s, 1H), 7.98 (d, J = 7.6 Hz, 2H), 7.82 (dt, J = 7.6, 1.2 Hz, 1H), 7.77 (dt, J = 7.6, 1.2 Hz, 1H), 7.71 (d, J = 8.4 Hz, 2H), 7.09 (d, J = 8.4 Hz, 1H), 6.99 (d, J = 8.8 Hz, 2H), 6.87 (br s, 1H), 1.70 (s, 3H); LCMS (ES+) 420 (M+H)+; HRMS (ES+) m/z calculated for C22H18N3O4S (M+H)+ 420.1012, found 420.1019.
6.2.2.2. N-(4-(3-methyl-1,4-dioxo-1,4-dihydronaphthalen-2-ylamino)phenylsulfonyl) benzamide (2l)
Dark orange solid (12 mg, 26%); Mp: 235–237 °C; 1H NMR (400 MHz, DMSO-d6) δ 12.35 (br s, 1H), 9.05 (s, 1H), 8.00 (d, J = 6.8 Hz, 2H), 7.83-7.68 (m, 6H), 7.59 (t, J = 8.0 Hz, 1H), 7.46 (t, J = 7.6 Hz, 2H), 7.05 (d, J = 8.8 Hz, 2H), 1.78 (s, 3H); LCMS (ES+) 447 (M+H)+; HRMS (ES+) m/z calculated for C24H19N2O5S (M+H)+ 447.1009, found 447.1014.
6.2.2.3. N-(5-methoxypyrimidin-2-yl)-4-(3-methyl-1,4-dioxo-1,4-dihydronaphthalen-2-ylamino)-benzenesulfonamide (2m)
Orange solid (11.2 mg, 15%); Mp: 215–217 °C; 1H NMR (400 MHz, DMSO-d6) δ 8.95 (s, 1H disappeared on D2O shake), 8.24 (s, 2H disappeared on D2O shake), 8.00-7.97 (m, 2H), 7.83 (dt, J = 7.6, 1.6 Hz, 1H), 7.80-7.77 (m, 3H), 7.00 (d, J = 8.8 Hz, 2H), 3.75 (s, 3H), 1.70 (s, 3H); LCMS (ES+) 451 (M+H)+; HRMS (ES+) m/z calculated for C22H19N4O5S (M+H)+ 451.1071, found 451.1075.
6.2.2.4. N-(2,6-dimethoxypyrimidin-4-yl)-4-(3-methyl-1,4-dioxo-1,4-dihydronaphthalen-2-ylamino)- benzenesulfonamide (2n)
Dark orange solid (14 mg, 16%); Mp: 255–257 °C; 1H NMR (400 MHz, DMSO-d6) δ 11.38 (br s, 1H), 9.02 (s, 1H), 7.99-7.97 (m, 2H), 7.82 (dt, J = 8.8, 1.2 Hz), 7.79-7.74 (m, 4H), 7.028 (d, J = 8.8 Hz, 2H), 5.91 (s, 1H), 3.76 (s, 3H), 3.73 (s, 3H), 1.72 (s, 3H); LCMS (ES+) 481 (M+H)+; HRMS (ES+) m/z calculated for (M+H)+ C23H21N4O6S 481.1176, found 481.1182.
6.2.2.5. N-(3,4-dimethylisoxazol-5-yl)-4-(3-methyl-1,4-dioxo-1,4-dihydronaphthalen-2-ylamino)- benzenesulfonamide (2o)
Orange solid (21 mg, 27%); Mp: 235–238 °C decomposed; 1H NMR (400 MHz, DMSO-d6) δ 10.84 (br s, 1H disappeared on D2O shake), 9.06 (s, 1H disappeared on D2O shake), 8.00 (d, J = 7.6 Hz, 2H), 7.86-7.77 (m, 2H), 7.57 (d, J = 8.8 Hz, 2H), 7.04 (d, J = 8.8 Hz, 2H), 2.06 (s, 3H), 1.73 (s, 3H), 1.59 (s, 3H); LCMS (ES+) 438 (M+H)+; HRMS (ES+) m/z calculated for (M+H)+ C22H19N3O5S 438.1118, found 438.1116.
6.2.3. General procedure for synthesis of compounds 2p-2r
The 1-4-naphthoquinone (500 mg, 3.16 mmol) was suspended in EtOH: water (90:10, 10.0 mL) followed by addition of the requisite amine (1.58 mmol, 0.5 equivalents). The reaction mixture was heated under reflux for 2 days. The reaction mixtures turned orange brown from bright orange mixture. The precipitates obtained were filtered and rinsed with EtOH (2–5 mL), EtOAc (5 mL) and acetone (2–5 mL). The required products were obtained as orange/brown solids.
6.2.3.1. N-(4-(1,4-dioxo-1,4-dihydronaphthalen-2-ylamino)phenylsulfonyl)benzamide (2p)
Brown/orange solid (181 mg, 26%); Mp: 294–296 °C; 1H NMR (400 MHz, DMSO-d6) δ 12.57 (br s, 1H), 9.52 (s, 1H), 8.06 (d, J = 7.6 Hz, 1H), 7.99 (d, J = 8.4 Hz, 2H), 7.95 (d, J = 7.2 Hz, 1H), 7.86-7.78 (m, 4H), 7.66-7.59 (m, 3H), 7.47 (t, J = 7.2 Hz, 2H), 6.41 (s, 1H); LCMS (ES+) 433 (M+H)+; HRMS (ES+) m/z calculated for C23H17N2O5S (M+H)+ 433.0853, found 433.0857.
6.2.3.2. 4-(1,4-Dioxo-1,4-dihydronaphthalen-2-ylamino)-N-(5-methylisoxazol-3-yl)benzenesulfonamide (2q)
Brown/Orange solid (319 mg, 49%); Mp: 290–192 °C; 1H NMR (400 MHz, DMSO-d6) δ 11.43 (br s, 1H), 9.47 (s, 1H), 8.06 (d, J = 6.4 Hz, 1H), 7.94 (d, J = 7.6 Hz, 1H), 7.88-7.77 (m, 4H), 7.60 (d, J = 8.8 Hz, 2H), 6.37 (s, 1H), 6.14 (s, 1H), 2.28 (s, 3H);); LCMS (ES+) 410 (M+H)+; HRMS (ES+) m/z calculated for C20H16N3O5S (M+H)+ 410.0805, found 410.0811.
6.2.3.3. 4-(1,4-Dioxo-1,4-dihydronaphthalen-2-ylamino)-N-(pyridin-2-yl)benzenesulfonamide (2r)
Orange solid (148 mg, 20%); Mp: 286 °C decomposed; 1H NMR (400 MHz, DMSO-d6) δ 9.41 (s, 1H), 8.05 (d, J = 7.6 Hz, 1H), 7.99-7.94 (m, 2H), 7.90-7.87 (m, 3H), 7.79 (t, J = 7.6 Hz, 1H), 7.72 (t, J = 7.6 Hz, 1H), 7.53 (d, J = 8.4 Hz, 2H), 7.15 (d, J = 8.4 Hz, 1H), 6.86 (br s, 1H), 6.30 (s, 1H); LRMS (ES+) 406 (M+H)+; HRMS (ES+) m/z calculated for C21H16N3O4S (M+H)+ (M+H)+ 406.0856, found 406.0860.
6.2.4. Synthesis of compounds 3a-3d
The starting material PI-083 (40 mg, 0.040 mmol) was suspended in EtOH (2.0 mL) in a microwave vial and the requisite amine (5 molar equivalents) was added. The reaction mixture was irradiated in a CEM microwave reactor for 20–25 minutes at 140°C (100 W). The resulting crude mixture was purified by SiO2 chromatography using EtOAc and Hexane (gradient elution) to obtain the required compounds.
6.2.4.1. 4-(3-(Dimethylamino)-1,4-dioxo-1,4-dihydronaphthalen-2-ylamino)-N-(pyridin-2-yl)-benzenesulfonamide (3a)
Orange solid (55 mg, 33%); Mp: 118–120 °C; 1H NMR (400 MHz, DMSO-d6) δ 8.42 (s, 1H), 8.03 (br s, 1H), 7.94-7.89 (m, 2H), 7.76-7.71 (m, 2H), 7.66-7.65 (m, 1H), 7.61-7.59 (d, J = 8.0, 2H), 7.06 (d, J = 8.4 Hz, 1H), 6.87-6.85 (m, 1H), 6.85-6.83 (d, J = 8.0 Hz, 2H, partially overlapped), 2.66 (s, 6H); LRMS (ES+) 449 (M+H)+.
6.2.4.2. 4-(3-(Benzyl(methyl)amino)-1,4-dioxo-1,4-dihydronaphthalen-2-ylamino)-N-(pyridin-2-yl)-benzenesulfonamide (3b)
Dark blue solid (15.8 mg, 79%); Mp: 135–140 °C; 1H NMR (400 MHz, DMSO-d6) δ 8.58 (s, NH, 1H, disappeared in D2O shake), 8.03 (d, J = 4.0 Hz, 1H), 7.93-7.90 (m, 2H), 7.78-7.72 (m, 2H), 7.67-7.62 (m, 3H), 7.31 (d, J = 4.4 Hz, 1H), 7.22-7.13 (m, 3H), 7.08 (d, J = 8.8 Hz, 1H), 7.03 (d, J = 6.8 Hz, 2H), 6.96 (d, J = 8,8 Hz, 2H), 6.85 (t, J = 6.0 Hz, 1H), 3.85 (s, 2H), 2.75 (s, 3H); LRMS (ES+) 525 (M+H)+; HRMS (ES+) m/z calculated for C29H25N4O4S (M+H)+ 525.1591, found 525.1588.
6.2.4.3. 4-(3-(4-Methylpiperazin-1-yl)-1,4-dioxo-1,4-dihydronaphthalen-2-ylamino)-N-(pyridin-2-yl)- benzenesulfonamide (3c)
Dark red solid (16.2 mg, 90%); Mp: 222–224 °C; 1H NMR (400 MHz, DMSO-d6) δ 8.48 (s, 1H), 8.02 (d, J = 4.0 Hz, 1H), 7.93-7.90 (m, 2H), 7.78-7.71 (m, 2H), 7.66-7.62 (m, 3H), 7.07 (d, J = 8.4 Hz, 1H), 6.91 (d, J = 8.8 Hz, 2H), 6.85 (t, J = 6.0 Hz, 1H), 3.09 (br t, 4H), 1.98-1.97 (m, 4H), 1.87 (s, 3H); LRMS (ES+) 504 (M+H)+; HRMS (ES+) m/z calculated for C26H26N5O4S (M+H)+ 504.1700, found 504.1713.
6.2.4.4. 4-(3-Morpholino-1,4-dioxo-1,4-dihydronaphthalen-2-ylamino)-N-(pyridin-2-yl)benzenesulfonamide (3d)
Dark blue solid (15.0 mg, 77%); Mp: 220–222 °C; 1H NMR (400 MHz, DMSO-d6) δ 8.57 (s, 1H), 8.00 (br s, 1H), 7.96-7.91 (m, 2H), 7.67-7.61 (m, 3H), 7.06 (d, J = 8.4 Hz, 1H), 6.95 (d, J = 8.8 Hz, 2H), 6.84 (br s, 1H), 3.14 (d, J = 4.0 Hz, 4 H), 3.06 (d, J = 4.0 Hz, 4H); LRMS (ES+) 491 (M+H)+; HRMS (ES+) m/z calculated for C25H23N4O5S (M+H)+ 491.1384, found 491.1386.
6.3. General procedure for synthesis of sulfanilamide derivatives 5a and 5b. 36
Sodium cyanoborohydride (0.36 g, 5.65 mmol) was added to a mixture of requisite aldehyde (1.1 equivalents), sulfapyridine (1.00 g, 4.01 mmol), and acetic acid (0.67 g, 11.23 mmol) in methanol (13 mL) at 0 °C and the reaction mixture was warmed to r.t., stirred for 1 h. The reaction was quenched with KHSO4 (5% aqueous solution, 10 mL), and extracted with ethyl acetate (3×20 mL). The organic phase was washed with sat. NaHCO3, brine, dried (Na2SO4), and concentrated. The crude product was purified by flash chromatography (SiO2 MeOH in DCM gradient elution).
6.3.1. 4-(Benzylamino)-N-(pyridin-2-yl)benzenesulfonamide (5a)
White solid (246 mg, 27%); Mp: 163–165 °C; 1H NMR (400 MHz, Acetone-d6) δ 9.99 (br s, 1H disappeared on D2O shake), 8.24 (ddd, J = 4.8, 2.0, 0.8 Hz, 1H), 7.67-7.60 (m, 3H), 7.32-7.24 (m, 5H), 7.19-7.17 (m, 1H), 6.93 (ddd, J = 7.2, 4.8, 0.8 Hz, 1H), 6.64 (d, J = 9.2 Hz, 2H), 6.33 (apparent t, J = 5.6 Hz, 1H disappeared on D2O shake), 4.35 (d, J = 5.6 Hz, 2H, doublet changed to a singlet on D2O shake); LRMS (ES+) 340 (M+H)+; HRMS (ES+) m/z calculated for C18H18N3O2S (M+H)+ 340.1114, found 340.1131.
6.3.2. 4-(Butylamino)-N-(pyridin-2-yl)benzenesulfonamide (5b)
White solid (658 mg, 54%); Mp: 128–130 °C; 1H NMR (400 MHz, CDCl3), 13.15 (br s, 1H), 8.40 (apparent dd, J = 5.6, 0.8 Hz, 1H), 7.62 (d, J = 8.8 Hz, 2H), 7.56 (dt, J = 8.0, 2.0 Hz, 1H), 7.35 (d, J = 8.8 Hz, 1H), 6.75 (t, J = 6.4 Hz, 1H), 6.47 (d, J = 8.8 Hz, 2H), 4.30 (br s, 1H, disappeared on D2O shake), 3.04 (br t, 2H), 1.52 (p, J = 7.3 Hz, 2H), 1.32 (h, J = 7.3 Hz, 2H), 0.88 (t, J = 7.4 Hz, 3H); LRMS (ES+) 306 (M+H)+; HRMS (ES+) m/z calculated for C15H20N3O2S (M+H)+ 306.1271, found: 306.1311.
6.4. Synthesis of sulfapyridine naphthoquinone derivatives 6a and 6b
N,N-Di-isopropylethylamine [DIPEA] (1.2 molar equivalents) was added to a solution of PI-083 in anhydrous DMF (10 mL/mmol) under inert conditions. Appropriate alkyl iodide (1.2 molar equivalents) was added to the reaction mixture after 5 min., and the reaction was stirred at r.t. for two days. The reaction mixture was concentrated under reduced pressure and purified by flash chromatography (SiO2, EtOAc in hexane, gradient elution) to obtain the required pure product.
6.4.1. 4-((3-Chloro-1,4-dioxo-1,4-dihydronaphthalen-2-yl)(methyl)amino)-N-(pyridin-2-yl) benzenesulfonamide (6a)
Orange solid (13 mg, 25%); Mp: 152–154 °C; 1H NMR (400 MHz, CDCl3) δ 8.27 (ddd, J = 4.8, 2.0, 0.8 Hz, 1H), 8.19 (dd, J = 7.6, 1.2 Hz, 1H), 8.13 (dd, J =7.6, 1.2 Hz, 1H), 7.79 (dt, J = 7.6, 1.6 Hz, 1H), 7.72 (dt, J = 7.6, 1.6 Hz, 1H), 7.73-7.66 (m, 3H), 7.53 (d, J = 8.4 Hz, 2H), 7.13 (ddd, J = 6.8, 4.8, 1.2 Hz, 1H), 7.01 (d, J = 8.4 Hz, 2H), 3.28 (s, 3H); LRMS (ES+) 454 (M+H)+; HRMS (ES+) m/z calculated for C22H17ClN3O4S (M+H)+ 454.0623, found 454.0638.
6.4.2. 4-((3-Chloro-1,4-dioxo-1,4-dihydronaphthalen-2-yl)(ethyl)amino)-N-(pyridin-2-yl) benzenesulfonamide (6b)
Orange solid, (60 mg, 38%); Mp: 180–182 °C; 1H NMR (400 MHz, CDCl3) δ 8.32 (ddd, J = 4.8, 2.0, 0.8 Hz, 1H), 8.20 (dd, J = 7.6, 1.2 Hz, 1H), 8.13 (dd, J = 7.6, 1.2 Hz, 1H), 7.80 (dt, J = 7.6, 1.2 Hz, 1H), 7.73 (dt, J = 7.6, 1.2 Hz, 2H), 7.67 (br s, 1H, disappeared on D2O shake), 7.59-7.55 (m, 3H), 7.16 (ddd, J = 7.2, 4.8, 1.6 Hz, 1H), 7.01 (d, J = 8.4 Hz, 2H), 3.84 (q, J = 7.2 Hz, 2H), 1.12 (t, J = 7.2 Hz, 3H). LRMS (ES+) 468 (M+H)+; HRMS (ES+) m/z calculated for C23H19ClN3O4S (M+H)+ 468.0779, found 468.0800.
6.5. General procedure for synthesis of sulfapyridine naphthoquinone derivatives (6c, 6d, 6e, 6f, 6g)
The PI-083 (100 mg, 0.23 mmol), appropriate alkyl halide (0.27 mmol, 1.2 molar equivalents) and DIPEA (35 mg, 0.27 mmol, 1.2 molar equivalents) were mixed in anhydrous DMF (2 mL). The reaction mixture was heated at 160°C for 15 min. in a microwave reactor (for 6f, 6g, the reactions were heated for 30 min. at 160°C). The reaction mixtures were then concentrated under reduced pressure and the products were purified using flash chromatography (SiO2, EtOAc in hexane, gradient elution).
6.5.1. 4-(Benzyl(3-chloro-1,4-dioxo-1,4-dihydronaphthalen-2-yl)amino)-N-(pyridin-2-yl) benzenesulfonamide (6c)
Orange solid (56 mg, 47%); Mp: 78–80 °C; 1H NMR (400 MHz, CDCl3) δ 8.27 (ddd, J = 4.0, 2.0, 1.2 Hz, 1H), 8.20 (dd, J = 7.6, 1.2 Hz, 1H), 8.14 (td, J = 7.6, 1.6 Hz, 1H), 7.8-7.59 (m, 7H), 7.48 (dd, J = 8.0, 0.8 Hz, 1H), 7.30 (d, J = 7.2 Hz, 2H), 7.24-7.15 (m, 3H), 7.09-7.03 (m, 3H), 5.01 (s, 2H); LRMS (ES+) 530 (M+H)+; HRMS (ES+) m/z calculated for C28H21ClN3O4S (M+H)+ 530.0936, found 530.0936
6.5.2. 4-((3-Chloro-1,4-dioxo-1,4-dihydronaphthalen-2-yl)(4-nitrobenzyl)amino)-N-(pyridin-2-yl)benzenesulfonamide (6d)
Orange solid (75 mg, 58%); Mp: 80–82 °C; 1H NMR (400 MHz, CDCl3) δ 8.24 (ddd, J = 4.8, 1.6, 0.8 Hz, 1H), 8.20 (dd, J = 7.6, 0.8 Hz, 1H), 8.14 (dd, J = 7.6, 1.2 Hz, 1H), 8.09 (d, J = 8.8 Hz, 2H), 7.81 (dt, J = 7.6, 1.6 Hz, 1H), 7.76-7.60 (m, 4H), 7.57 (d, J = 8.8 Hz, 2H), 7.51 (d, J = 8.8 Hz, 2H), 7.10 (ddd, J = 7.2, 4.8, 1.2 Hz, 1H), 7.03 (d, J = 8.4 Hz, 2H), 5.11 (s, 2H); LRMS (ES+) 575 (M+H)+; HRMS (ES+) m/z calculated for C28H20ClN4O6S (M+H)+ 575.0787, found 575.0795.
6.5.3. 4-((3-Chloro-1,4-dioxo-1,4-dihydronaphthalen-2-yl)(naphthalen-2-ylmethyl)amino)-N-(pyridin-2-yl)benzenesulfonamide (6e)
Orange solid (60 mg, 45%); Mp: 165–167 °C; 1H NMR (400 MHz, CDCl3) δ 8.28 (ddd, J = 4.8, 2.0, 0.8 Hz, 1H), 8.20 (apparent dd, J = 7.6, 1.2 Hz, 1H), 8.13 (apparent dd, J = 7.6, 1.2 Hz, 1H), 7.81 (dt, J = 7.6, 1.2 Hz, 1H), 7.76-7.71 (m, 7H), 7.65-7.57 (m, 3H), 7.51-7.49 (m, 2H), 7.42-7.39 (m, 2H), 7.08-7.03 (m, 2H), 5.15 (s, 2H); LRMS (ES+) 580 (M+H)+; HRMS (ES+) m/z calculated for C32H23ClN3O4S (M+H)+ 580.1092, found 580.1089
6.5.4. 4-((3-Chloro-1,4-dioxo-1,4-dihydronaphthalen-2-yl)(4-methylbenzyl)amino)-N-(pyridin-2-yl)benzenesulfonamide (6f)
Orange solid (50 mg, 41%); Mp: 99–101 °C; 1H NMR (400 MHz, CDCl3) δ 8.27 (ddd, J = 4.8, 2.0, 0.8 Hz, 1H), 8.21 (dd, J = 7.6, 1.2 Hz, 1H), 8.15 (dd, J = 7.6, 1.2 Hz, 1H), 7.80 (dt, J =7.6, 1.2 Hz, 1H), 7.74 (dt, J = 7.6, 1.2 Hz, 1H), 7.64-7.60 (m, 3H), 7.46 (td, J = 8.4, 0.8 Hz, 1H), 7.19 (d, J = 8.0 Hz, 2H), 7.09-7.01 (m, 5H), 4.96 (s, 2H), 2.25 (s, 3H); LRMS (ES+) 544 (M+H)+; HRMS (ES+) m/z calculated for C29H23ClN3O4S (M+H)+ 544.1092, found 544.1101.
6.5.5. 4-((3-Chloro-1,4-dioxo-1,4-dihydronaphthalen-2-yl)(4-(trifluoromethyl)benzyl) amino)-N-(pyridin-2-yl)benzenesulfonamide (6g)
Orange solid (62 mg, 46%); Mp: 157–159 °C; 1H NMR (400 MHz, CDCl3) δ 8.25 (apparent d, J = 4.8 Hz, 1H), 8.21 (d, J = 7.6 Hz, 1H), 8.14 (d, J = 8.0 Hz, 1H), 7.83-7.64 (m, 4H), 7.59-7.55 (m, 3H), 7.48 (d, J = 8.0 Hz, 2H), 7.44 (d, J = 8.0 Hz, 2H), 7.11-7.03 (m, 2H), 5.01 (s, 2H); 19F NMR: δ −62.93 (s); LRMS (ES+) 598 (M+H)+; HRMS (ES+) m/z calculated for C29H20ClF3N3O4S (M+H)+ 598.0810, found 598.0816.
6.6. Synthesis of compounds 7a and 7b
6.6.1. Mixture of regio-isomers of 4-(3-chloro-6-nitro-1,4-dioxo-1,4-dihydronaphthalen-2-ylamino)-N-(pyridin-2-yl)benzenesulfonamide and 4-(3-chloro-7-nitro-1,4-dioxo-1,4-dihydronaphthalen-2-ylamino)-N-(pyridin-2-yl)benzenesulfonamide (7a)
A well-stirred suspension of sulfapyridine (0.229 g, 0.919 mmol) and 2,3-dichloro-6-nitronaphthalene-1,4-dione (0.5 g, 1.84 mmol) in a mixture of 95% EtOH in water (10.0 mL) was heated in a sealed tube at 115°C for three days. The resultant orange precipitate was filtered and washed with hot ethanol (5×5 mL), acetone (3×5 mL), and dried under reduced pressure to afford the title compound as an orange solid (0.393 g, 88%). 1H NMR indicated formation of 2:1 regio-isomers. 1H NMR (400 MHz, DMSO-d6) δ 9.76 (s, 1H, [δ 9.72 minor isomer shown]) 8.64-8.56 (m, 2H), 8.28-8.25 (m, 1H), 8.02 (br s, 1H), 7.78 (d, J = 8.4 Hz, 2H), 7.71 (t, J = 7.2 Hz, 1H), 7.22 (d, J = 8.4 Hz, 2H), 7.15 (d, J = 8.0 Hz, 1H), 6.88 (br t, J = 6.4 Hz, 1H); LRMS (ES+) 485 (M+H)+; HRMS (ES+) m/z calculated for C21H14ClN4O6S (M+H)+ 485.0317, found 485.0330.
6.6.2. Mixture of regio-isomers of 4-(3-chloro-5-nitro-1,4-dioxo-1,4-dihydronaphthalen-2-ylamino)-N-(pyridin-2-yl)benzenesulfonamide and 4-(3-chloro-8-nitro-1,4-dioxo-1,4-dihydronaphthalen-2-ylamino)-N-(pyridin-2-yl)benzenesulfonamide (7b)
This compound was prepared according to the procedure described for compound 7a, except using 2,3-dichloro- 5-nitronaphthalene-1,4-dione to obtain 7b as an orange solid (0.178 g, 92%). 1H NMR indicated formation of 2:1 regio-isomers. 1H NMR (400 MHz, DMSO-d6) δ 9.67 (s, 1H, [δ 9.73 minor isomer shown]), 8.25 (dd, J = 7.6, 1.2 Hz, 1H, [δ 8.23 minor isomer shown]), 8.13 (d, J = 8.4 Hz, 1H), 8.07-7.97 (m, 2H), 7.77 (dd, J = 8.4, 3.2 Hz, 2H), 7.72 (t, J = 8.4 Hz, 1H), 7.23-7.15 (m, 3H), 6.88 (br t, J = 5.6 Hz, 1H); LCMS (ES+) 485 (M+H)+; HRMS (ES+) m/z calculated for C21H14ClN4O6S (M+H)+ 485.0317, found 485.0327.
6.6.3. Mixture of regio-isomers of 4-(6-amino-3-chloro-1,4-dioxo-1,4-dihydronaphthalen-2-ylamino)-N- (pyridin-2-yl)benzenesulfonamide and 4-(7-amino-3-chloro-1,4-dioxo-1,4-dihydronaphthalen-2-ylamino)- N-(pyridin-2-yl)benzenesulfonamide (8)
Compound 7a (0.213 g, 0.44 mmol) was dissolved in a mixture of DMF:MeOH (4:1, 30 mL) and passed through the H-cube apparatus at a rate of 1mL/min (40 bar pressure, 10% Pd/C as catalyst, room temperature). The resultant solution was evaporated and dried under reduced pressure to obtain a red solid. This product was purified using SiO2 flash chromatography (MeOH/DCM, 5–10% gradient elution) to obtain the required reduced product as a red solid (50 mg, 25%). 1H NMR indicated 1:5 ratio of regio-isomers after SiO2 purification. 1H NMR (400 MHz, DMSO-d6) δ 9.18 (s, 1H [δ 9.40 minor isomer]), 8.03 (br s, 1H), 7.75 (d, J = 7.6 Hz, 2H), 7.73 (d, J = 6.4 Hz, 2H), 7.17-7.15 (m, 2H), 7.06 (d, J = 8.8 Hz, 2H), 6.86-6.77 (m, 2H), 6.54 (s, 2H disappear on D2O shake); LCMS (ES+) 455 (M+H)+; HRMS (ES+) m/z calculated for C21H16ClN4O4S (M+H)+ 455.0575, found 455.0588.
6.7. Synthesis of library 11a-11l
6.7.1. 4-Nitro-N-o-tolyl-benzenesulfonamide (11a)
A solution of 4-nitrobenzenesulfonyl chloride (200 mg, 0.90 mmol), o-toluidine (106 mg, 0.99 mmol), and pyridine (79 mg, 0.08 mL, 0.99 mmol) in 1,2-dichloroethane (5.0 mL) was heated to 150°C for 10 minutes in the microwave reactor. A 1M HCl solution was added until the pH was 2, and the acidified aqueous layer was separated and extracted with DCM (3×10 mL). The combined organic fractions were washed successively with brine and water then dried over Na2SO4 and evaporated to dryness to afford the title compound as a peach colored solid (127 mg, 47%). Mp: 157–158°C (lit. 157–159°C)37; 1H NMR (400 MHz, CDCl3) δ 8.28 (d, J = 9.0 Hz, 2H), 7.89 (d, J = 9.0 Hz, 2H), 7.28-7.27 (m, 1H), 7.20-7.12 (m, 3H), 6.37 (br s, 1H), 2.01 (s, 3H).
6.7.2. 4-Nitro-N-phenyl-benzenesulfonamide (11b)
This compound was prepared according to the procedure described for compound 11a except using aniline to obtain required product as an off-white solid, (222 mg, 87%). Mp: 170–171°C (lit. 174–176°C)37; 1H NMR (400 MHz, CDCl3) δ 8.28 (d, J = 8.8 Hz, 2H), 7.91 (d, J = 8.4 Hz, 2H), 7.29 (t, J = 6.8 Hz, 2H), 7.21 (t, J = 7.2Hz, 1H), 7.22-7.18 (m, 1H), 7.08 (d, J = 8.4 Hz, 2H), 6.57 (br s, 1H).
6.7.3. 4-Nitro-N-m-tolyl-benzenesulfonamide (11c)
This compound was prepared according to the procedure described for compound 11a except using m-toluidine to obtain required product as a light tan solid, (264 mg, 100%); Mp: 136–138°C (lit. 138–139°C)37; 1H NMR (400 MHz, CDCl3) δ 8.28 (d, J = 8.8 Hz, 2H), 7.92 (d, J = 8.8 Hz, 2H), 7.15 (t, J = 7.8 Hz, 1H), 7.00 (d, J = 7.6 Hz, 1H), 6.90 (br s, 1H), 6.85 (d, J = 8.4 Hz, 1H), 6.56 (br s, 1H).
6.7.4. 4-Nitro-N-p-tolyl-benzenesulfonamide (11d)
This compound was prepared according to the procedure described for compound 11a except using p-toluidine to obtain required product as a yellow solid, (256 mg, 97%). Mp: 170–172°C (lit. 184–184.5°C)38; 1H NMR (400 MHz, CDCl3) δ 8.27 (d, J = 9.0 Hz, 2H), 7.88 (d, J = 9.0 Hz, 2H), 7.08 (d, J = 8.4 Hz, 2H), 6.94 (d, J = 8.4 Hz, 2H), 6.41 (br s, 1H), 2.30 (s, 3H).
6.7.5. N-(2-methoxyphenyl)-4-nitro-benzenesulfonamide (11e)
This compound was prepared according to the procedure described for compound 11a except using o-anisidine. Recrystallization from DCM/hexanes obtained the required product as white crystals, (164 mg, 59%). Mp: 141–143°C; 1H NMR (400 MHz, CDCl3) δ 8.24 (d, J = 8.8 Hz, 2H), 7.91 (d, J = 8.8 Hz, 2H), 7.56 (dd, J = 8.0, 1.6 Hz, 1H), 7.11 (dt, J =7.9, 1.5 Hz, 1H), 7.02 (br s, 1H), 6.94 (dt, J = 7.6, 1.2 Hz, 1H), 6.74 (dd, J = 8.0, 1.2 Hz, 1H), 3.62 (s, 3H).
6.7.6. N-(3-methoxyphenyl)-4-nitrobenzenesulfonamide (11f)
This compound was prepared according to the procedure described for compound 11a except using m-anisidine. Recrystallization from DCM/hexanes obtained the required product as brown-yellow needles (202 mg, 73%). Mp: 96–98°C; 1H NMR (400 MHz, CDCl3) δ 8.29 (d, J = 9.0 Hz, 2H), 7.94 (d, J = 9.0 Hz, 2H), 7.16 (t, J = 8.0 Hz, 1H), 6.73-6.70 (m, 2H), 6.60-6.58 (m, 2H), 3.76 (s, 3H).
6.7.7. N-(4-methoxyphenyl)-4-nitrobenzenesulfonamide (11g)
This compound was prepared according to the procedure described for compound 11a except using p-anisidine to obtain the required product as a light brown solid (278 mg, 100 %). Mp: 173–175°C (lit. 187–189°C)37; 1H NMR (400 MHz, CDCl3) δ 8.28 (dd, J = 9.2, 2.4 Hz, 2H), 7.85 (dd, J = 9.2, 2.0 Hz, 2H), 6.97 (dd, J = 8.6, 2.2 Hz, 2H), 6.80 (dd, J = 8.6, 2.2 Hz, 2H), 6.36 (br s, 1H), 3.78 (s, 3H).
6.7.8. 2-Methoxy-4-nitro-N-o-tolyl-benzenesulfonamide (11h)
This compound was prepared according to the procedure for compound 11a except using 2-methoxy-4 nitrobenzenesulfonyl chloride. Recrystallization from DCM/hexanes obtained the required product as brown crystals (202 mg, 79%). Mp: 143–145°C; 1H NMR (400 MHz, CDCl3) δ 8.03 (d, J = 8.8 Hz, 1H), 7.85–7.88 (m, 2H), 7.14-7.11 (m, 2H), 7.06-7.04 (m, 2H), 6.76 (br s, 1H), 4.13 (s, 3H), 2.26 (s, 3H).
6.7.9. 2-Methoxy-4-nitro-N-m-tolyl-benzenesulfonamide (11i)
This compound was prepared according to the procedure described for compound 11a except using 2-methoxy- 4-nitrobenzenesulfonyl chloride and m-toluidine as starting materials. Recrystallization from DCM/hexanes obtained the required product as gold-brown needles (236 mg, 92%). Mp: 142–145°C; 1H NMR (400 MHz, CDCl3) δ 8.03 (d, J = 9.2 Hz, 1H), 7.84-7.81 (m, 2H), 7.28 (br s, 1H), 7.07 (t, J = 7.6, 1H), 6.90-6.83 (m, 3H), 4.16 (s, 3H), 2.25 (s, 3H).
6.7.10. 2-Methoxy-4-nitro-N-p-tolyl-benzenesulfonamide (11j)
This compound was prepared according to the procedure described for compound 11a except using 2-methoxy- 4-nitrobenzenesulfonyl and p-toluidine as starting materials. Re-crystallization from DCM/hexanes gave the required product as yellow-brown needles (199 mg, 78%). Mp: 126–129°C; 1H NMR (400 MHz, CDCl3) δ 7.98 (d, J = 8.4 Hz, 1H), 7.84 (d, J = 2.0 Hz, 1 H), 7.81 (dd, J = 8.8, 2.0 Hz, 1H), 7.00 (d, J = 8.2 Hz, 2H), 6.94 (d, J = 8.4 Hz, 2H), 7.10 (br s, 1H), 4.17 (s, 3H), 2.23 (s, 3H).
6.7.11. 2-Methoxy-N-(4-methoxyphenyl)-4-nitrobenzenesulfonamide (11k)
This compound was prepared according to the procedure described for compound 11a except using 2-methoxy- 4-nitrobenzenesulfonyl and p-anisidine as starting materials. Re-crystallization from DCM/hexanes gave the required product as gold-brown needles (213 mg, 79%). Mp: 117–119°C; 1H NMR (400 MHz, CDCl3) δ 7.93 (d, J = 8.4 Hz, 1H), 7.88 (d, J = 2.0 Hz, 1H), 7.82 (dd, J = 8.4, 2.0 Hz, 1H), 6.97 (d, J = 8.8 Hz, 2H), 7.00 (br s, 1H), 6.72 (d, J = 9.2 Hz, 2H), 4.19 (s, 3H), 3.72 (s, 3H).
6.7.12. 4-Nitro-N-(1-azidoformimidoyl)benzenesulfonamide (11l)
This compound was prepared according to the literature procedure23 to afford the title compound as a yellow solid (682 mg, 51%). 1H-NMR (400 MHz, DMSO-d6) δ 9.08 (br, 1H), 8.39 (d, J = 8.8 Hz, 2H), 8.15 (d, J = 8.8 Hz, 2H), 8.00 (br s, 1H); LCMS (ES-) 269 (M-H)−; HRMS (ES-) m/z calculated for C7H5N6O4S (M-H)− 269.0099, found 269.0094.
6.8. General procedure for synthesis of 11m to 11r
The sulfonyl chloride (1.0 g, 4.51 mmol) was dissolved in THF (10 ml) and appropriate amine (3 molar equivalents) was added to the reaction mixture. The reaction was stirred 30 min. at room temperature until TLC (30% ethyl acetate in hexane) indicated completion of the reaction. The reaction mixture was acidified with 1 M HCl (pH=2) at 0 °C and the solvent was evaporated under reduced pressure and the white solid obtained at this point was triturated in ethyl acetate/hexane and filtered. The product obtained was washed with water and dried under vacuum to afford a white solid. Upon acidification to pH=2, some reactions produced oily mixtures. These oily mixtures were diluted with water (10 mL) and extracted twice with ethyl acetate (20 mL). The organic layer was separated dried (Na2SO4), filtered and the filtrate was evaporated and dried under vacuum to afford the desired compound as a solid.
6.8.1. N-methyl-4-nitrobenzenesulfonamide 11m
White solid (0.67 g, 68%); Mp: 105–106 °C;. 1H NMR (400 MHz, CDCl3) δ 8.38 (apparent d, J = 8.8 Hz, 2H), 8.06 (apparent d, J = 8.8 Hz, 2H), 4.52 (broad s, 1H), 2.74 (d, J = 5.2 Hz, 3H), 1.58 (s, 3H); LCMS (ES-) m/z 215 (M-H)−; HRMS (ES-) calculated for C7H7N2O4S (M-H)− 215.0132, found 215.0148
6.8.2. N-ethyl-4-nitrobenzenesulfonamide 11n
Yellow solid (0.406 g, 78%); Mp: 99–100 °C; 1H NMR (400 MHz, CDCl3) δ 8.37 (d, J = 8.8 Hz, 2H), 8.06 (d, J = 8.8 Hz, 2H), 4.53 (br s, 1H), 3.10-3.06 (m, 2H), 1.15 (t, J = 7.2 Hz, 3H); LCMS (ES-) m/z 229 (M-H)−; HRMS (ES-) m/z calculated for C8H9N2O4S (M-H)− 229.0289, found 229.0304.
6.8.3. N-isopropyl-4-nitrobenzenesulfonamide 11o
Yellow solid (1.031 g, 94%); Mp: 112–113 °C; 1H NMR (400 MHz, CDCl3) δ 8.36 (d, J = 8.8 Hz, 2H), 8.07 (d, J = 8.4 Hz, 2H), 4.44 (d, J = 8.0 Hz, 1H), 3.61-3.52 (m, 1H), 1.12 (d, J = 6.8 Hz, 6H); LCMS (ES-) m/z 243 (MH)−; HRMS (ES-) calculated for C9H11N2O4S (M-H)− 243.0445, found 243.0465.
6.8.4. N-methyl-3-nitrobenzenesulfonamide 11p
White solid (0.67 g, 69%); Mp: 120–121 °C; 1H NMR (400 MHz, CDCl3) δ 8.71 (t, J = 2.0 Hz, 1H), 8.45 (ddd, J = 8.4, 2.0, 0.8 Hz, 1H), 8.20 (ddd, J = 8.0, 1.6, 0.8 Hz, 1H), 7.76 (t, J = 8.0 Hz, 1H), 2.74 (d, J = 9.2 Hz, 3H); LCMS (ES-) m/z 215 (M-H); HRMS (ES-) calculated for C7H7N2O4S (M-H)− 215.0132, found 215.0147.
6.8.5. N-ethyl-3-nitrobenzenesulfonamide 11q
Yellow solid (0.48g, 92%); Mp: 86–87 °C; 1H NMR (400 MHz, CDCl3) δ 8.71 (apparent t, J = 2.0 Hz, 1H), 8.44 (ddd, J = 8.8, 2.0, 1.2 Hz, 1H), 8.21 (ddd, J = 7.6, 1.6, 1.2 Hz, 1H), 7.75 (t, J = 8.4 Hz, 1H), 4.58 (br s, 1H), 3.11-3.06 (m, 2H), 1.63 (t, J = 7.2 Hz, 3H ; LCMS (ES-) m/z 229 (M-H)−; HRMS (ES-) m/z calculated for C8H9N2O4S (M-H)− 229.0289, found 229.0298.
6.8.6. N-isopropyl-3-nitrobenzenesulfonamide 11r
White solid (0.962 g, 87%); Mp: 67–68 °C; 1H NMR (400 MHz, CDCl3) δ 8.73 (t, J = 2.0 Hz, 1H), 8.43 (ddd, J = 8.4, 2.0, 0.8 Hz, 1H), 8.21 (ddd, J = 8.0, 2.0, 1.2 Hz, 1H), 7.74 (t, J = 8.0 Hz, 1H), 4.43 (br s, 1H), 3.62-3.53 (m,1H), 1.13 (d, J = 6.8 Hz, 6H); LCMS (ES-) m/z 243 (M-H)−; HRMS (ES-) calculated for C9H11N2O4S (M-H)− 243.0445, found 243.0462.
6.9. Synthtsis of Library 12a-12l
6.9.1. 4-Amino-N-o-tolyl-benzenesulfonamide (12a)
To a solution of 4-nitro-N-o-tolyl-benzenesulfonamide 11a in a mixture of MeOH/THF (1:1, 4.0 mL), was added nickel chloride hexahydrate (163 mg, 0.68 mmol) at 0°C under constant stirring. Sodium borohydrate (52 mg, 1.37 mmol) was added portion wise and the reaction was monitored by TLC (60% hexanes/40% ethyl acetate). The solvent was removed in vacuo and the remaining black solid was re-suspended in EtOAc and filtered using a pad of celite and washed with EtOAc until the filtrate, when visualized under UV light, showed no product. The solvent was removed under vacuum affording the title compound as an off white solid (0.045 g, 100%). 1H NMR (400 MHz, CDCl3) δ 7.42 (d, J = 8.8 Hz, 2H), 7.24 (d, J = 8.0 Hz, 1H), 7.07 (dt, J = 7.2, 2.4, 1H), 7.01-6.95 (m, 2H), 6.52 (d, J = 8.8 Hz, 2H), 6.35 (br s, 1H), 4.05 (br s, 2H), 1.95 (s, 3H).
6.9.2. 4-Amino-N-phenyl-benzenesulfonamide (12b)
This compound was prepared according to the procedure described for compound 12a except using 11b to obtain the required product as a pale yellow solid (109 mg, 70%). Mp: 180–182°C (lit. 260.5-261.5°C)38; 1H NMR (400 MHz, CDCl3) δ 7.52 (d, J = 8.8 Hz, 2H), 7.24-7.21 (m, 2H), 7.10 (t, J = 7.6 Hz, 1H), 7.04 (dd, J = 8.8, 1.2 Hz, 2H), 6.58 (d, J = 8.8 Hz, 2H), 6.32 (br s, 1H), 4.08 (br s, 2H).
6.9.3. 4-Amino-N-m-tolyl-benzenesulfonamide (12c)
This compound was prepared according to the procedure described for compound 12a except using 11c to obtain the required product as a light yellow solid (160 mg, 74%). Mp: 117–120°C (lit. 132.5-133°C)38; 1H NMR (400 MHz, CDCl3) δ 7.47 (d, J = 8.6 Hz, 2H), 7.03 (t, J = 7.6 Hz, 1H), 6.84-6.81 (m, 2H), 6.76 (br d, J = 8.0 Hz, 1H), 6.52 (d, J = 8.6 Hz, 2H), 6.26 (br s, 1H), 4.01 (br s, 2H), 2.20 (s, 3H).
6.9.4. 4-Amino-N-p-tolyl-benzenesulfonamide (12d)
This compound was prepared according to the procedure for compound 12a except using 11d to obtain the required product as an off-white solid (225 mg, 99%). Mp: 174–176°C (lit. 190–190.5°C)38; 1H NMR (400 MHz, CDCl3) δ 7.43 (d, J = 8.8 Hz, 2H), 6.96 (d, J = 8.4 Hz, 2H), 6.86 (d, J = 8.4 Hz, 2H), 6.52 (d, J = 8.8 Hz, 2H), 6.14 (br s, 1H), 4.00 (br s, 2H), 2.20 (s, 3H).
6.9.5. 4-Amino-N-(2-methoxyphenyl)benzenesulfonamide (12e)
This compound was prepared according to the procedure described for compound 12a except using 11e to obtain the required product as a white solid (85 mg, 57%). 1H NMR (400 MHz, CDCl3) δ 7.47 (d, J = 8.6 Hz, 2H), 7.42 (dd, J = 7.8, 1.1 Hz, 1H), 6.94 (dt, J = 8.0, 1.6 Hz, 1H), 6.92 (br s, 1H), 6.81 (dt, J = 7.8, 1.1 Hz, 1H), 6.67 (dd, J = 8.2, 1.1 Hz, 1H), 6.49 (d, J = 8.6 Hz, 2H), 3.60 (s, 3H).
6.9.6. 4-Amino-N-(3-methoxyphenyl)benzenesulfonamide (12f)
This compound was prepared according to the procedure described for compound 12a except using 11f to obtain the required product as a pale yellow solid (151 mg, 83%). Mp: 142–145°C; 1H NMR (400 MHz, CDCl3) δ 7.49 (d, J = 8.8 Hz, 2H), 7.05 (t, J = 8.0 Hz, 1H), 6.61-6.50 (m, 5H), 6.32 (br s, 1H), 3.679 (s, 3H).
6.9.7. 4-Amino-N-(4-methoxyphenyl)benzenesulfonamide (12g)
This compound was prepared according to the procedure described for compound 12a except using 11g to obtain the required product as a light yellow solid, (197 mg, 79%). Mp: 150°C, decomposed; 1H NMR (400 MHz, CDCl3) δ 7.45 (d, J = 8.8 Hz, 2H), 6.96 (d, J = 8.8 Hz, 2H), 6.76 (d, J = 9.2 Hz, 2H), 6.58 (d, J = 8.4 Hz, 2H), 6.09 (br s, 1H), 4.07 (br s, 2H), 3.76 (s, 3H).
6.9.8. 4-Amino-2-methoxy-N-m-tolyl-benzenesulfonamide (12h)
This compound was prepared according to the procedure described for compound 12a except using 11h to obtain the required product as an orange-brown solid in (169 mg, 66 %). 1H NMR (400 MHz, CDCl3) δ 7.50 (d, J = 8.4 Hz, 1H), 6.99 (t, J = 7.8 Hz, 1H), 6.82 (br s, 1H), 6.79-6.73 (m, 2H), 6.65 (br s, 1H), 6.11 (d, J = 2.0 Hz, 1H), 6.09 (s, 1H), 3.99 (br s, 2H), 3.86 (s, 3H), 2.18 (s, 3H).
6.9.9. 4-Amino-2-methoxy-N-p-tolyl-benzenesulfonamide (12i)
This compound was prepared according to the procedure described for compound 12a except using 11i to obtain the required product as a light yellow solid (130 mg, 93%). Mp: 158–160°C; 1H NMR (400 MHz, CDCl3) δ 7.52 (d, J = 8.8 Hz, 1H), 6.98 (d, J = 8.2 Hz, 2H), 6.93 (d, J = 8.2 Hz, 2H) 6.70 (br s, 1H), 6.16 (s, 1H), 6.14 (appd, J = 2.0 Hz, 1H), 4.05 (br s, 2H), 3.95 (s, 3H), 2.23 (s, 3H).
6.9.10. 4-Amino-2-methoxy-N-o-tolyl-benzenesulfonamide (12j)
This compound was prepared according to the procedure described for compound 12a except using 11j to obtain the required product as a yellow solid (63 mg, 93%). Mp: 189–191 °C; 1H NMR (400 MHz, CD3OD) δ 7.30 (d, J = 8.8 Hz, 1H), 7.10-7.06 (m, 2H), 7.01-6.97 (m, 2H), 6.31 (d, J = 1.6 Hz, 1H), 6.12 (dd, J = 8.8, 2.0 Hz, 1H), 3.30 (s, 3H), 2.22 (s, 3H).
6.9.11. 4-Amino-2-methoxy-N-(4-methoxyphenyl)benzenesulfonamide (12k)
This compound was prepared according to the procedure described for compound 12a except using 11k to obtain the required product as a yellow solid (141 mg, 77%). Mp: 51–54°C; 1H NMR (400 MHz, CDCl3) δ 7.46 (d, J = 8.4 Hz, 1H), 6.97 (d, J = 8.8 Hz, 2H), 6.73 (d, J = 8.8 Hz, 2H), 6.61 (br s, 1H), 6.19 (d, J = 1.6 Hz, 1H), 6.14 (dd, J = 8.2, 2.2 Hz, 1H), 4.06 (br s, 2H), 3.98 (s, 3H), 3.72 (s, 3H).
6.9.12. 4-Amino-N-carbamimidoylbenzenesulfonamide (12l)
This compound was prepared according to the procedure for compound 12a except using 11l to afford the title compound as a grey solid (80 mg, 13.2%). Mp: 169–171°C; 1H-NMR (400 MHz, DMSO-d6) δ 7.37 (d, J = 8.3 Hz, 2H), 6.55 (br, 4H), 6.53 (d, J = 8.3 Hz, 2H), 5.69 (br, 2H); LCMS (ES+) 215 (M+H)+, 237 (M+Na)+, 451 (2M+Na)+; HRMS (ES+) m/z calculated for C7H11N4O2S (M+H)+ 215.0597, found 215.0595.
6.10. General procedure for synthesis of 12m to12r
6.10.1. 4-Amino-N-methylbenzenesulfonamide 12m
N-methyl-4-nitrobenzenesulfonamide 11m (0.143 g, 0.66 mmol) was dissolved in MeOH/THF (1:1 solution, 12 ml) and nickel chloride(11) hexahydrate (0.627 g, 2.64 mmol) was added to the solution at 0 °C under constant stirring. Sodium borohydride (0.199 g, 5.28 mmol) was added portion wise over 15 minutes. The reaction mixture was stirred for additional 5 minutes until TLC (50% ethyl acetate in hexane) indicated completion of the reaction and the solvent was evaporated to obtain a black solid. The solid was suspended in ethyl acetate, filtered through a pad of celite, and washed with ethyl acetate until the filtrate showed no product on t.l.c (under uv visualization). The filtrate was evaporated and dried to obtain the desired compound as a yellow solid (0.103 g, 84%); Mp: 108–109 °C [lit. 98–99 °C]39; 1H NMR (400 MHz, DMSO-d6) δ 7.36 (d, J = 8.8 Hz, 2H), 6.90 (q, J = 4.8 Hz, 1H), 6.58 (d, J = 8.8 Hz, 2H), 5.91(s, 2H), 2.29 (d, J = 4.8 Hz, 3H); LCMS (ES+) m/z 187 (M+H)+, 156 (M-CH3NH)+; HRMS (ES+) calculated for C7H11N2O2S (M+H)+ 187.0536, found 187.0527.
6.10.2. 4-Amino-N-ethylbenzenesulfonamide 12n
N-ethyl-4-nitrobenzenesulfonamide (0.3 g, 1.3 mmol) was dissolved in methanol and passed through H-cube apparatus (Full H2 mode, 30 bar pressure and 10% Pd/C catalyst cartridge at 25 °C, flow rate 1.0 mL/min). The reaction was monitored by TLC (40% ethyl acetate in hexane). After completion, the solvent was evaporated and dried under vacuum to afford a white solid (0.229 g, 88%); Mp: 98–99 °C [lit. 107 °C]40; 1H NMR (400 MHz, DMSO-d6) δ 7.37 (d, J = 6.8 Hz, 2H), 7.00 (t, J = 5.6 Hz, 1H), 6.58 (d, J = 8.8 Hz, 2H), 5.88 (s, 2H), 2.69-2.62 (m, 2H), 0.92 (t, J = 7.2 Hz, 3H); LCMS (ES+) m/z 201 (M+H)+, 156 (M-C2H5NH)+, HRMS (ES+) calculated for C8H13N2O2S (M+H)+ 201.0692, found 201.0694.
6.10.3. 4-Amino-N-isopropylbenzenesulfonamide 12o
This compound was prepared according to the procedure described for 12n except using N-isopropyl-4-nitrobenzenesulfonamide 11o (0.3 g, 1.23 mmol) and H-cube settings at 30 °C and pressure at 40 bars to obtain a yellowish-orange solid (0.29 g, 97%); Mp: 117–118 °C, [lit. 116.5–117 °C]41; 1H NMR (400 MHz, DMSO-d6) δ 7.55 (d, J = 8.8 Hz, 2H), 7.19 (d, J = 7.2 Hz, 1H), 6.85 (d, J = 8.8 Hz, 2H), 3.17-3.09 (m,1H), 0.89 (d, J = 6.8 Hz, 6H); LCMS (ES+) m/z 215 (M+H)+, 156 (M-C3H7NH)+; HRMS (ES+) calculated for C9H15N2O2S (M+H)+ 215.0849, found 215.0849.
6.10.4. 3-Amino-N-methylbenzenesulfonamide 12p
This compound was prepared according to the procedure described for 12m except using 11p N-methyl-3-nitrobenzenesulfonamide (0.04 g, 0.185 mmol). Yellow oil (0.034 g, 99%); 1H NMR (400 MHz, CDCl3) δ 7.69 (ddd, J = 7.8, 1.6, 1.1 Hz, 1H), 7.60 (t, J = 1.8 Hz, 1H), 7.50 (t, J = 7.9 Hz, 1H), 7.31 (ddd, J = 7.9, 2.2, 1.0 Hz, 1H), 4.34 (br s, 2H), 2.70 (d, J = 5.2 Hz, 3H); LCMS (ES+) m/z 187 (M+H)+; HRMS (ES+) calculated for C7H11N2O2S (M+H)+ 187.0536, found 187.0548.
6.10.5. 3-Amino-N-ethylbenzenesulfonamide 12q
This compound was prepared according to the procedure described for 12n except using N-ethyl-3-nitrobenzenesulfonamide 11q (0.3 g, 1.3 mmol). White solid (0.229 g, 88%); Mp: 98–99 °C; 1H NMR (400 MHz, DMSO-d6) δ 7.31 (t, J = 6.0 Hz, 1H), 7.16 (t, J = 8.0 Hz, 1H), 6.95 (t, J = 2.0 Hz, 1H), 6.84 (ddd, J = 7.6, 1.6, 0.8 Hz, 1H), 6.71 (ddd, J = 8.0, 2.2, 0.8 Hz, 1H), 5.54 (s, 2H), 2.69-2.62 (m, 2H), 0.92 (t, J = 7.2 Hz, 3H); LCMS (ES+) m/z 201 (M+H)+; HRMS (ES+) calculated for C8H13N2O2S (M+H)+ 201.0692, found 201.0695.
6.10.6. 3-Amino-N-isopropylbenzenesulfonamide 12r
This compound was prepared according to the procedure described for 12m, except using 11r N-isopropyl-3-nitrobenzenesulfonamide (0.3g, 1.2 mmol). Yellow solid (0.25 g, 96%); Mp: 96–97 °C; 1H NMR (400 MHz, DMSO-d6) δ 7.33 (d, J = 7.2 Hz, 1H), 7.15 (t, J = 8.0 Hz, 1H), 6.96 (s, 1H), 6.86 (d, J = 7.6 Hz, 1H), 6.70 (d, J = 9.2 Hz, 1H), 5.52 (s, 2H), 3.22-3.13 (m,1H), 0.92 (d, J = 6.4 Hz, 6H); LCMS (ES+) m/z 215 (M+H)+; HRMS (ES+) calculated for C9H15N2O2S (M+H)+ 215.0849, found 215.0860.
6.11. Synthesis of Library 13a-13l
6.11.1. 4-(3-Chloro-1,4-dioxo-1,4-dihydro-naphthalen-2-ylamino)-N-o-tolyl-benzenesulfonamide (13a)
A well-stirred suspension of 4-amino-N-o-tolyl-benzenesulfonamide 12a (47 mg, 179 mmol) and 2,3-dichloro-1,4-naphthoquinone (41 mg, 179 mmol) in 95% EtOH in water (10.0 mL) was refluxed at 115 °C for three days. The orange precipitate obtained was filtered and washed with hot ethanol (5×5 ml), concentrated and dried (under vacuum) to afford the title compound (23 mg, 28%). Mp: 265–266 °C; 1H NMR (400 MHz, DMSO-d6) δ 9.55 (br s, 1H), 9.45 (br s, 1H), 8.04 (d, J = 7.6 Hz, 2H), 7.89-7.80 (m, 2H), 7.49 (d, J = 8.4 Hz, 2H), 7.17-7.07 (m, 5H), 6.98-6.96 (m, 1H), 1.97 (s, 3H); LRMS (ES+) 453 (M35Cl+H)+, 455 (M37Cl+H)+; HRMS (ES+) m/z calculated for C23H18ClN2O4S (M+H)+ 453.0670, found 453.0665.
6.11.2. 4-(3-Chloro-1,4-dioxo-1,4-dihydro-naphthalen-2-ylamino)-N-phenylbenzenesulfonamide (13b)
This compound was prepared according to the procedure described for compound 13a except using 12b to obtain the required product as an orange-red solid (79 mg, 41%). Mp: 220–223 °C; 1H NMR (400 MHz, DMSO-d6) δ 10.14 (br s, 1H), 9.50 (br s, 1H), 8.03-8.01 (m, 2H), 7.86-7.79 (m, 2H), 7.60 (d, J = 8.8 Hz, 2H), 7.20 (t, J = 7.6 Hz, 2H), 7.14 (d, J = 8.4 Hz, 2H), 7.06 (d, J = 8.4 Hz, 2H), 7.00 (t, J = 7.6 Hz, 1H); LRMS (ES+) 439 (M35Cl+H)+, 441 (M37Cl+H)+; HRMS (ES+) m/z calculated for C22H16ClN2O4S (M+H)+ 439.0514, found 439.0508.
6.11.3. 4-(3-Chloro-1,4-dioxo-1,4-dihydro-naphthalen-2-ylamino)-N-m-tolylbenzenesulfonamide (13c)
This compound was prepared according to the procedure described for compound 13a except using 12c to obtain the required product as an orange-red solid (139 mg, 56%). Mp: 234–237 °C; 1H NMR (400 MHz, DMSO-d6) δ 10.07 (br s, 1H, disappeared on D2O shake), 9.50 (br s, 1H, disappeared on D2O shake), 8.04-8.01 (m, 2H), 7.88-7.79 (m, 2H), 7.61 (d, J = 8.4 Hz, 2H), 7.14 (d, J = 8.8 Hz, 2H), 7.08 (t, J = 7.6 Hz, 1H), 6.87-6.81 (m, 3H), 2.17 (s, 3H); LRMS (ES+) for 453 (M35Cl+H)+, 455 (M37Cl+H)+; HRMS (ES+) m/z calculated for C23H18ClN2O4S (M+H)+ 453.0670, found 453.0662.
6.11.4. 4-(3-Chloro-1,4-dioxo-1,4-dihydro-naphthalen-2-ylamino)-N-(2-methoxyphenyl)-benzenesulfonamide (13d)
This compound was prepared according to the procedure described for compound 13a except using 12d to obtain the required product as an orange solid (41 mg, 28%). Mp: 198–201 °C; 1H NMR (400 MHz, DMSO-d6) δ 9.51 (br s, 1H), 9.34 (br s, 1H), 8.04-8.02 (m, 2H), 7.88-7.79 (m, 2H), 7.55 (d, J = 8.4 Hz, 2H), 7.20 (d, J = 8.0 Hz, 1H), 7.14-7.08 (m, 3H), 6.90-6.83 (m, 2H), 3.52 (s, 3H); LRMS (ES+) 469 (M35Cl+H)+, 471 (M37Cl+H)+; HRMS (ES+) m/z calculated for C23H18ClN2O5S (M+H)+ 469.0620, found 469.0609.
6.11.5. 4-(3-Chloro-1,4-dioxo-1,4-dihydro-naphthalen-2-ylamino)-N-(3-methoxyphenyl)-benzenesulfonamide (13e)
This compound was prepared according to the procedure described for compound 13a except using 12e to obtain the required product as an orange solid, (135 mg, 53%). Mp: 213–216 °C; 1H NMR (400 MHz, DMSOd6) δ 10.16 (br s, 1H), 9.51 (br s, 1H), 8.04-8.01 (m, 2H), 7.88-7.79 (m, 2H), 7.63 (d, J = 8.4 Hz, 2H), 7.15 (d, J = 8.4 Hz, 2H), 7.11 (t, J = 8.0 Hz, 1H), 6.65-6.63 (m, 2H), 6.58 (apparent dd, J = 8.4, 2.4 Hz, 1H), 3.64 (s, 3H): LRMS (ES+) 469 (M35Cl+H)+, 471 (M37Cl+H)+; HRMS (ES+) m/z calculated for C23H18ClN2O5S (M+H)+ 469.0620, found 469.0611.
6.11.6. 4-(3-Chloro-1,4-dioxo-1,4-dihydro-naphthalen-2-ylamino)-N-(4-methoxyphenyl)- benzenesulfonamide (13f)
This compound was prepared according to the procedure described for compound 13a except using 12f to obtain the required product as a yellow-orange solid (295 mg, 78%). Mp: 233–234 °C; 1H NMR (400 MHz, DMSO-d6) δ 9.76 (br s, 1H), 9.50 (br s, 1H), 8.03 (d, J = 7.2 Hz, 2H), 7.86-7.79 (m, 2H), 7.53 (d, J = 8.8 Hz, 2H), 7.13 (d, J = 8.8 Hz, 2H), 6.94 (d, J = 8.8 Hz, 2H), 6.78 (d, J = 8.8 Hz, 2H), 3.65 (s, 3H); LRMS (ES+) 469 (M35Cl+H)+, 471 (M37Cl+H)+; HRMS (ES+) m/z calculated for C23H18ClN2O5S (M+H)+ 469.0620, found 469.0612.
6.11.7. 4-(3-Chloro-1, 4-dioxo-1,4-dihydro-naphthalen-2-ylamino)-N-p-tolylbenzenesulfonamide (13g)
This compound was prepared according to the procedure described for compound 13a except using 12g to obtain the required product as a yellow-orange solid (245 mg, 63%). Mp: 257–260 °C; 1H NMR (400 MHz, DMSO-d6) δ 9.96 (br s, 1H), 9.49 (br s, 1H), 8.04-8.01 (m, 2H), 7.86-7.79 (m, 2H), 7.58 (d, J = 8.4 Hz, 2H), 7.13 (d, J = 8.8 Hz, 2H), 7.00 (d, J = 8.4 Hz, 2H), 6.94 (d, J = 8.4 Hz, 2H), 2.17 (s, 3H); LRMS (ES+) 453 (M35Cl+H)+, 455 (M37Cl+H)+; HRMS (ES+) m/z calculated for C23H18ClN2O4S (M+H)+ 453.0670, found 453.0661.
6.11.8. 4-(3-Chloro-1,4-dioxo-1,4-dihydro-naphthalen-2-ylamino)-2-methoxy-N-o-tolylbenzenesulfonamide (13h)
This compound was prepared according to the procedure described for compound 13a except using 12h to obtain the required product as an orange-red solid (55 mg, 35%). Mp: 197–200 °C; 1H NMR (400 MHz, DMSOd6) δ 9.46 (br s, 1H), 9.15 (br s, 1H), 8.04 (dd, J = 7.6, 1.2 Hz, 2H), 7.87-7.80 (m, 2H), 7.42 (d, J = 8.4 Hz, 1H), 7.12-7.09 (m, 1H), 7.04-6.97 (m, 3H), 6.89 (d, J = 1.6 Hz, 1H), 6.65 (dd, J = 8.4, 1.6, Hz, 1H), 3.78 (s, 3H), 2.14 (s, 3H); LRMS (ES+) 483 (M35Cl+H)+, 485 (M37Cl+H)+; HRMS (ES+) m/z calculated for (M+H)+ C24H20ClN2O5S 483.0776, found 483.0771.
6.11.9. 4-(3-Chloro-1,4-dioxo-1,4-dihydro-naphthalen-2-ylamino)-2-methoxy-N-p-tolylbenzenesulfonamide (13i)
This compound was prepared according to the procedure described for compound 13a except using 12i to obtain the required product as a red solid (80 mg, 38%). Mp: 186–189 °C; 1H NMR (400 MHz, DMSO-d6) δ 9.69 (br s, 1H), 9.40 (br s, 1H), 8.04-8.01 (m, 2H), 7.86-7.79 (m, 2H), 7.55 (d, J = 8.4 Hz, 1H), 6.97-6.92 (m, 4H), 6.93 (d overlapped, J = 8.8 Hz, 2H), 6.80 (d, J = 1.6 Hz, 1H), 6.65 (dd, J = 8.6, 1.8 Hz, 1H), 3.79 (s, 3H), 2.14 (s, 3H); LRMS (ES+) 483 (M35Cl+H)+, 485 (M37Cl+H)+; HRMS (ES+) m/z calculated for C24H20ClN2O5S (M+H)+ 483.0776, found 483.0769.
6.11.10. 4-(3-Chloro-1,4-dioxo-1,4-dihydro-naphthalen-2-ylamino)-2-methoxy-N-(4-methoxyphenyl)- benzenesulfonamide (13j)
This compound was prepared according to the procedure described for compound 13a except using 12j to obtain the required product as a red solid (118 mg, 54%). Mp: 165–167 °C; 1H NMR (400 MHz, DMSO-d6) δ 9.50 (br s, 1H), 9.40 (br s, 1H), 8.04-8.01 (m, 2H), 7.88-7.79 (m, 2H), 7.48 (d, J = 8.8 Hz, 1H), 6.96 (d, J = 8.8 Hz, 2H), 6.82 (br s, 1H), 6.74 (d, J = 8.8 Hz, 2H), 6.63 (dd, J = 8.6, 1.4 Hz, 1H), 3.82 (s, 3H), 3.63 (s, 3H); LRMS (ES+) 499 (M35Cl+H)+, 501 (M37Cl+H)+; HRMS (ES+) m/z calculated for C24H20ClN2O6S (M+H)+ 499.0725, found 499.0717.
6.11.11. 4-(3-Chloro-1,4-dioxo-1,4-dihydro-naphthalen-2-ylamino)-2-methoxy-N-m-tolylbenzenesulfonamide (13k)
This compound was prepared according to the procedure described for compound 13a except using 12k to obtain the required product as an orange solid, (91 mg 64%). Mp: 264–267 °C; 1H NMR (400 MHz, DMSO-d6) δ 9.81 (br s, 1H), 9.41 (br s, 1H), 8.04-8.01 (m, 2H), 7.83 (dt, J = 7.2, 1.2 Hz, 1H), 7.79 (dt, J = 7.2, 1.2 Hz, 1H), 7.60 (d, J = 8.8 Hz, 1H), 7.03 (t, J = 8.2 Hz, 1H), 6.86-6.84 (m, 2H), 6.80 (d, J = 2.0 Hz, 1H), 6.76 (d, J = 7.6 Hz, 1H), 6.67 (dd, J = 8.6, 1.8 Hz, 1H), 3.78 (s, 3H), 2.15 (s, 3H); LRMS (ES+) 483 (M35Cl+H)+, 485 (M37Cl+H)+; HRMS (ES+) m/z calculated for C24H20ClN2O5S (M+H)+ 483.0803, found 483.0809.
6.11.12. N-Carbamimidoyl-4-(3-chloro-1,4-dioxo-1,4-dihydronaphthalen-2-ylamino)benzenesulfonamide (13l)
This compound was prepared according to the procedure for compound 13a except using compound 12l to afford the title compound as an orange-red solid (35 mg, 54.2%). Mp: 294–295 °C; 1H-NMR (400 MHz, DMSO-d6) δ 9.48 (br s, 1H), 8.05 (d, J = 7.2 Hz, 2H), 7.86-7.80 (m, 2H), 7.65 (d, J = 8.5 Hz, 2H), 7.17 (d, J = 8.5 Hz, 2H), 6.70 (br s, 4H); LCMS (ES+) 405 (M35Cl+H)+, 407 (M37Cl+H)+; HRMS (ES+) m/z calculated for C17H14ClN4O4S (M+H)+ 405.0419, found 405.0408.
6.12. General procedure for synthesis of 13m to13r
Amino-N-alkylbenzenesulfonamides 12m-12r (0.1 g, 0.499 mmol) and 2,3-dichloronaphthoquinone (0.11g, 0.499 mmol) were dissolved in 95% ethanol in water (5 mL). The reaction mixture was heated at 120°C and stirred for 3 days in a sealed tube until TLC (50% ethyl acetate in hexane) indicated completion of the reaction. The solvent was partially evaporated and the solid was filtered and washed with ethanol (20–30 mL). The pure products obtained were dried under vacuum to afford orange or brown solid.
6.12.1. 4-(3-Chloro-1,4-dioxo-1,4-dihydronaphthalen-2-ylamino)-N-methylbenzenesulfonamide (13m)
Orange solid (0.122 g, 61%); Mp: 277–278°C; 1H NMR (400 MHz, DMSO-d6) δ 9.53 (s, 1H disappeared on D2O shake), 8.02 (d, J = 7.6 Hz, 2H), 7.87 (dt, J = 7.6, 1.6 Hz, 1H), 7.80 (dt, J = 7.2, 1.2 Hz, 1H), 7.63 (d, J = 8.8 Hz, 2H), 7.32 (apparent q, J = 4.8 Hz, 1H disappeared on D2O shake), 7.22 (d, J = 8.8 Hz, 2H), 2.37 (d, J = 5.2 Hz, 3H changed to singlet on D2O shake); LCMS (ES+) m/z 377 (M+H)+; HRMS (ES+) calculated for C17H14ClN2O4S (M+H)+ 377.0357, found 377.0355.
6.12.2. 4-(3-Chloro-1,4-dioxo-1,4-dihydronaphthalen-2-ylamino)-N-ethylbenzenesulfonamide (13n)
Orange solid (0.125 g, 62%); Mp: 240–241 °C; 1H NMR (400 MHz, DMSO-d6) δ 9.52 (s, 1H disappeared on D2O shake), 8.03 (d, J = 11.6 Hz, 2H), 7.88-7.79 (m, 2H), 7.65 (d, J = 7.6 Hz, 2H), 7.43 (t, J = 8.0 Hz, 1H disappeared on D2O shake), 7.21(d, J = 6.4 Hz, 2H), 2.79-2.72 (m, 2H, changed to a quartet on D2O shake), 0.94 (t, J = 8.0 Hz, 3H); LCMS (ES+) m/z 391 (M+H)+; HRMS (ES+) calculated for C18H16ClN2O4S (M+H)+ 391.0514, found 391.0502.
6.12.3. 4-(3-Chloro-1,4-dioxo-1,4-dihydronaphthalen-2-ylamino)-N-isopropylbenzenesulfonamide (13o)
Light brown solid (0.32 g, 14%); Mp: 244–245°C; 1H NMR (400 MHz, DMSO-d6) δ 9.53 (s, 1H), 8.03 (dd, J = 7.6, 1.6 Hz, 2H), 7.87 (dt, J = 7.6, 1.6 Hz, 1H), 7.81 (dt, J = 7.2, 2.0 Hz, 1H), 7.68 (d, J = 8.4 Hz, 2H), 7.46 (d, J = 7.2 Hz, 1H), 7.21 (d, J = 8.4 Hz, 2H), 3.24-3.19 (m, 1H), 0.92 (d, J = 6.8 Hz, 6H); LCMS (ES+) m/z 405 (M+H)+; HRMS (ES+) calculated for C19H18ClN2O4S (M+H)+ 405.0670, found 405.0664.
6.12.4. 2-Chloro-3-(3-(methylaminoperoxythio)phenylamino)naphthalene-1,4-dione (13p)
Brown solid (0.28 g, 97%); Mp: 229–230°C; 1H NMR (400 MHz, DMSO-d6) δ 9.52 (s, 1H disappeared on D2O shake), 8.04-8.02 (m, 2H), 7.82 (dt, J = 7.6, 1.2 Hz, 1H), 7.76 (dt, J = 7.6, 1.2 Hz, 1H), 7.53-7.45 (m, 4H changed to 3H on D2O shake), 7.35 (d, J = 4.0 Hz, 1H), 2.79-2.76 (m, 2H), 0.96 (t, J = 7.2, 3H); LCMS (ES+) m/z 377.03 (M+H)+; HRMS (ES+) calculated for C17H14ClN2O4S (M+H)+ 377.0357, found 377.0370.
6.12.5. 2-Chloro-3-(3-(ethylaminoperoxythio)phenylamino)naphthalene-1,4-dione (13q)
Orange solid (0.193 g, 99%); Mp: 239–240°C decomposed; 1H NMR (400 MHz, DMSO-d6) δ 9.51 (s, 1H), 8.04-8.01 (m, 2H), 7.86 (dt, J = 7.6, 1.2 Hz, 1H), 7.81 (dt, J = 7.6, 1.2 Hz, 1H), 7.55 (t, J = 5.6 Hz, 1H), 7.50-7.48 (m, 2H), 7.35-33 (m, 1H), 2.79-2.76 (m, 2H), 0.96 (t, J = 7.2 Hz, 3H); LCMS (ES-) m/z 389 (M-H)−; HRMS (ES-) calculated for C18H16ClN2O4S (M-H)− 389.0369, found 389.0371.
6.12.6. 2-Chloro-3-(3-(isopropylaminoperoxythio)phenylamino)naphthalene-1,4-dione (13r)
Orange solid (0.64 g, 34%); Mp: 211–212°C; 1H NMR (400 MHz, MeOH-d4) δ 8.13 (dd, J = 3.2, 1.2 Hz, 1H), 8.12 (dd, J = 4.8, 1.6 Hz, 1H), 7.83 (dt, J = 9.2, 1.6 Hz, 1H), 7.77 (dt, J = 7.6, 1.2 Hz, 1H), 7.65 (ddd, J = 7.6, 2.0, 1.2 Hz, 1H), 7.56 (t, J = 1.6, 1H), 7.51 (t, J = 8.0 Hz, 1H), 7.34 (ddd, J = 8.0, 2.4, 1.2 Hz, 1H), 3.32 (m, partially overlapped with residual MeOH signal), 1.04 (d, J = 6.4 Hz, 6H); LCMS (ES+) m/z 405.07 (M+H)+; HRMS (ES+) calculated for C19H18ClN2O4S (M+H)+ 405.0670, found 405.0664.
Acknowledgment
This work was supported by NIH grant P01CA118210.
Footnotes
This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Groll M, Ditzel L, Loewe J, Stock D, Bochtler M, Bartunik HD, Huber R. Nature. 1997;386:463. doi: 10.1038/386463a0. [DOI] [PubMed] [Google Scholar]; Groll M, Berkers CR, Ploegh HL, Ovaa H. Structure (Cambridge, MA, US) 2006;14:451–456. doi: 10.1016/j.str.2005.11.019. [DOI] [PubMed] [Google Scholar]
- 2.Burger AM, Seth AK. Eur. J. Cancer. 2004;40:2217. doi: 10.1016/j.ejca.2004.07.006. [DOI] [PubMed] [Google Scholar]; Hjerpe R, Rodriguez MS. Int. J. Biochem. Cell Biol. 2008;40:1126. doi: 10.1016/j.biocel.2007.11.021. [DOI] [PubMed] [Google Scholar]
- 3.Yamasaki L, Pagano M. Curr. Opin. Cell Biol. 2004;16:623. doi: 10.1016/j.ceb.2004.08.005. [DOI] [PubMed] [Google Scholar]
- 4.Ostrowska H. Cell. Mol. Biol. Lett. 2008;13:353. doi: 10.2478/s11658-008-0008-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bennett MK, Kirk CJ. Curr. Opin. Drug Discovery Dev. 2008;11:616. [PubMed] [Google Scholar]
- 6.Adams J. Nat. Rev. Cancer. 2004;4:349. doi: 10.1038/nrc1361. [DOI] [PubMed] [Google Scholar]; Orlowski RZ, Kuhn DJ. Clin. Cancer Res. 2008;14:1649. doi: 10.1158/1078-0432.CCR-07-2218. [DOI] [PubMed] [Google Scholar]
- 7.Voorhees PM, Dees EC, O'Neil B, Orlowski RZ. Clin. Cancer Res. 2003;9:6316. [PubMed] [Google Scholar]; Nalepa G, Rolfe M, Harper JW. Nat. Rev. Drug Discovery. 2006;5:596. doi: 10.1038/nrd2056. [DOI] [PubMed] [Google Scholar]; Zavrski I, Jakob C, Schmid P, Krebbel H, Kaiser M, Fleissner C, Rosche M, Possinger K, Sezer O. Anti-Cancer Drugs. 2005;16:475. doi: 10.1097/00001813-200506000-00002. [DOI] [PubMed] [Google Scholar]
- 8.Jung L, Holle L, Dalton William S. Oncology. 2004;18:4. [PubMed] [Google Scholar]; Lara PN, Jr, Davies AM, Mack PC, Mortenson MM, Bold RJ, Gumerlock PH, Gandara DR. Semin. Oncol. 2004;31:40. doi: 10.1053/j.seminoncol.2003.12.013. [DOI] [PubMed] [Google Scholar]; Adams J. Semin. Oncol. 2001;28:613. doi: 10.1016/s0093-7754(01)90034-x. [DOI] [PubMed] [Google Scholar]
- 9.Bang S-M, Lee JH, Yoon S-S, Park S, Min C-K, Kim C-C, Suh C, Sohn SK, Min Y-H, Lee J-J, Kim K, Seong C-M, Yoon H-J, Cho KS, Jo D-Y, Lee KH, Lee N-R, Kim CS. Int. J. Hematol. 2006;83:309. doi: 10.1532/IJH97.A30512. [DOI] [PubMed] [Google Scholar]
- 10.Sterz J, von Metzler I, Hahne J-C, Lamottke B, Rademacher J, Heider U, Terpos E, Sezer O. Expert Opin. Invest. Drugs. 2008;17:879. doi: 10.1517/13543784.17.6.879. [DOI] [PubMed] [Google Scholar]; Kuhn DJ, Chen Q, Voorhees PM, Strader JS, Shenk KD, Sun CM, Demo SD, Bennett MK, van Leeuwen FWB, Chanan-Khan AA, Orlowski RZ. Blood. 2007;110:3281. doi: 10.1182/blood-2007-01-065888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Piva R, Ruggeri B, Williams M, Costa G, Tamagno I, Ferrero D, Giai V, Coscia M, Peola S, Massaia M, Pezzoni G, Allievi C, Pescalli N, Cassin M, di Giovine S, Nicoli P, de Feudis P, Strepponi I, Roato I, Ferracini R, Bussolati B, Camussi G, Jones-Bolin S, Hunter K, Zhao H, Neri A, Palumbo A, Berkers C, Ovaa H, Bernareggi A, Inghirami G. Blood. 2008;111:2765. doi: 10.1182/blood-2007-07-100651. [DOI] [PubMed] [Google Scholar]
- 12.Fenical W, Jensen Paul R, Palladino Michael A, Lam Kin S, Lloyd GK, Potts Barbara C. Bioorg Med Chem. 2009;17:2175. doi: 10.1016/j.bmc.2008.10.075. [DOI] [PMC free article] [PubMed] [Google Scholar]; Groll M, McArthur KA, Macherla VR, Manam RR, Potts BC. J. Med. Chem. 2009;52:5420. doi: 10.1021/jm900559x. [DOI] [PubMed] [Google Scholar]; Chauhan D, Hideshima T, Anderson KC. Br. J. Cancer. 2006;95:961. doi: 10.1038/sj.bjc.6603406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Piccinini M, Mostert M, Rinaudo MT. Curr. Drug Targets. 2003;4:657–671. doi: 10.2174/1389450033490759. [DOI] [PubMed] [Google Scholar]; Crawford LJ-A, Walker B, Irvine AE. Front. Biosci. 2008;13:4285. doi: 10.2741/3005. [DOI] [PubMed] [Google Scholar]
- 14.Guedat P, Colland F. BMC Biochem. 2007;8(Suppl 1):S14. doi: 10.1186/1471-2091-8-S1-S14. [DOI] [PMC free article] [PubMed] [Google Scholar]; Kazi ADK, Smith DM, Kumar NB, Dou QP. Biochem Pharmacol. 2003;66:965. doi: 10.1016/s0006-2952(03)00414-3. [DOI] [PubMed] [Google Scholar]
- 15.Kazi A, Lawrence H, Guida WC, McLaughlin ML, Springett GM, Berndt N, Yip RML, Sebti SM. Cell Cycle. 2009;8:1940. doi: 10.4161/cc.8.12.8798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ahn JH, Cho SY, Ha JD, Chu SY, Jung SH, Jung YS, Baek JY, Choi IK, Shin EY, Kang SK, Kim SS, Cheon HG, Yang S-D, Choi J-K. Bioorg. Med. Chem. Lett. 2002;12:1941. doi: 10.1016/s0960-894x(02)00331-1. [DOI] [PubMed] [Google Scholar]
- 17.Urbanek RA, Suchard SJ, Steelman GB, Knappenberger KS, Sygowski LA, Veale CA, Chapdelaine MJ. J. Med. Chem. 2001;44:1777. doi: 10.1021/jm000447i. [DOI] [PubMed] [Google Scholar]
- 18.Brun M-P, Braud E, Angotti D, Mondesert O, Quaranta M, Montes M, Miteva M, Gresh N, Ducommun B, Garbay C. Bioorg. Med. Chem. 2005;13:4871. doi: 10.1016/j.bmc.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 19.Calandra JC, Adams EC., Jr J. Am. Chem. Soc. 1950;72:4804. [Google Scholar]; Calandra JC, Adams EC., Jr 2647123. US Patent. 1953
- 20.DeLano WL. The PyMol Molecular Graphics System, version 0.99. Palo Alto, CA: DeLano Scientific; 2004. [Google Scholar]
- 21.Blackburn C. Tetrahedron Lett. 2005;46:1405. [Google Scholar]
- 22.Walz AJ, Sundberg RJ. J Org Chem. 2000;65:8001. doi: 10.1021/jo001080s. [DOI] [PubMed] [Google Scholar]
- 23.Nagy HK, Tomson AJ, Horwitz JP. J. Am. Chem. Soc. 1960;82:1609. [Google Scholar]
- 24.70 ng of purified 20S rabbit proteasome was incubated with 20 µM Suc-Leu-Leu-Val-Tyr-AMC for the CT-L activity, Bz-Val-Gly-Arg-AMC for the T-L activity, and benzyloxycarbonyl Z-Leu-Leu-Glu-AMC for the PGPH activity for 1 hour at 37 °C in 100 µL of assay buffer (50 mM Tris-HCl, pH = 7.6) with or without inhibitors. After incubation, production of hydrolyzed 7-amido-4-methyl-coumarin (AMC) was measured using a WALLAC Victor2 1420 Multilabel Counter with an excitation filter of 355 nm and an emission filter of 460 nm (Perkin Elmer Life Sciences, Turku, Finland). The inhibitory activity of the compounds was calculated based on vehicle control
- 25.Groll M, Berkers CR, Ploegh HL, Ovaa H. Structure (Cambridge, MA, U.S.) 2006;14:451–456. doi: 10.1016/j.str.2005.11.019. [DOI] [PubMed] [Google Scholar]
- 26.LigPrep, version 2.2. New York: Schrödinger LLC; 2005. [Google Scholar]
- 27.Shelley JC, Cholleti A, Frye LL, Greenwood JR, Timlin MR, Uchimaya M. J. Comp. Aided. Mol. Des. 2007;21:681. doi: 10.1007/s10822-007-9133-z. [DOI] [PubMed] [Google Scholar]
- 28.Cho AE, Guallar V, Berne B, Friesner RA. J. Comput. Chem. 2005;26:915. doi: 10.1002/jcc.20222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dialysis using purified rabbit 20S proteasome. To measure the effect of dialysis on CT-L activity, compounds (10 µM) or vehicle (0.1 % DMSO) were added to rabbit 20S proteasome at a final concentration of 3 nM in proteasome assay buffer (50 mM Tris-HCl, pH = 7.6) and incubated at room temperature for 30 min. After 30 min of incubation, proteasome-compound mixtures were added to 10,000 MWCO Thermo Scientific Slide-A-Lyzer Dialysis Cassette (Rockford, IL) and dialyzed against proteasome assay buffer. Immediately (t = 0) and 0.5, 1, 2, 4, and 18 hours of dialysis at 4 °C, samples were removed from the dialysis cassette and the CT-L 20S proteasome activity was determined as described in reference 24. Proteasome activity was normalized against proteasome activity of DMSO control
- 30.Glide, version 5.0. New York: Schrödinger, LLC; 2008. [Google Scholar]
- 31.Bernstein FC, Koetzle TF, Williams GJB, Meyer EF, Jr, Brice MD, Rodgers JR, Kennard O, Shimanouchi T, Tasumi M. J. Mol. Biol. 1977;112:535. doi: 10.1016/s0022-2836(77)80200-3. [DOI] [PubMed] [Google Scholar]
- 32.Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood JR, Halgren TA, Sanschagrin PC, Mainz DT. J. Med. Chem. 2006;49:6177. doi: 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- 33.Maestro, version 8.5. New York: Schrödinger, LLC; 2008. [Google Scholar]
- 34.Glide, version 3.0. New York: Schrödinger, LLC; 2004. [Google Scholar]
- 35.Mohamadi F, Richard NG, Guida WC, Liskamp R, Lipton M, Caufield C, Chang G, Hendrickson T, Still WC. J. Comput. Chem. 1990;11:440. [Google Scholar]
- 36.Miki T, Kori M, Mabuchi H, Banno H, Tozawa R-i, Nakamura M, Itokawa S, Sugiyama Y, Yukimasa H. Bioorg Med. Chem. 2001;10:401. doi: 10.1016/s0968-0896(01)00290-5. [DOI] [PubMed] [Google Scholar]
- 37.Le Pera A, Leggio A, Liguori A. Tetrahedron. 2006;62:6100. [Google Scholar]
- 38.Zheng X, Oda H, Takamatsu K, Sugimoto Y, Tai A, Akaho E, Ali HI, Oshiki T, Kakuta H, Sasaki K. Bioorg. Med. Chem. 2007;15:3299. doi: 10.1016/j.bmc.2006.10.029. [DOI] [PubMed] [Google Scholar]
- 39.Felt P. W. Acta Chemica Scandinavica. 1962;16:297. [Google Scholar]
- 40.Bezzi S, Concilio C, Zambon A. Farmaco (1946–1952) 1950;5:260. [PubMed] [Google Scholar]
- 41.Fishwick BR. 908656. Application: GB,GB Patent. 1962