

Abstract
Hepatitis C virus (HCV) is a leading cause of liver disease worldwide. With ~170 million individuals infected and current interferon-based treatment having toxic side-effects and marginal efficacy, more effective antivirals are critically needed1. Although HCV protease inhibitors were just FDA approved, analogous to HIV therapy, optimal HCV therapy likely will require a combination of antivirals targeting multiple aspects of the viral lifecycle. Viral entry represents a promising multi-faceted target for antiviral intervention; however, to date FDA-approved inhibitors of HCV cell entry are unavailable. Here we show that the cellular Niemann-Pick C1-Like 1 (NPC1L1) cholesterol uptake receptor is an HCV entry factor amendable to therapeutic intervention. Specifically, NPC1L1 expression is necessary for HCV infection as silencing or antibody-mediated blocking of NPC1L1 impairs cell-cultured-derived HCV (HCVcc) infection initiation. In addition, the clinically-available FDA-approved NPC1L1 antagonist ezetimibe2,3 potently blocks HCV uptake in vitro via a virion cholesterol-dependent step prior to virion-cell membrane fusion. Importantly, ezetimibe inhibits infection of all major HCV genotypes in vitro, and in vivo delays the establishment of HCV genotype 1b infection in mice with human liver grafts. Thus, we have not only identified NPC1L1 as an HCV cell entry factor, but also discovered a new antiviral target and potential therapeutic agent.
HCV is thought to enter cells via receptor-mediated endocytosis beginning with interaction of the viral particle with a series of cell surface receptors, including tetraspanin CD814, scavenger receptor class B member I (SR-BI)5 and tight-junction proteins claudin-1 (CLDN1)6 and occludin (OCLN)7,8, followed by clathrin-mediated endocytosis and fusion between the virion envelope and the endosomal membrane9,10. While the specifics of each interaction are not fully understood, we now recognize that multiple cellular factors as well as many components of the viral particle, not just the viral glycoproteins, participate in the entry process. For example, the HCVcc particle is associated with cellular lipoproteins (e.g. LDL and VLDL)11,12 and enriched in cholesterol13, the latter of which has been shown to be necessary for HCV cell entry13,14. Apart from cholesterol likely functioning in viral membrane stabilization and organization, the dependence of HCV infectivity on cholesterol led us to reason that cholesterol-uptake receptors might play a role in HCV cell entry.
NPC1L1, a 13 transmembrane cell surface cholesterol-sensing receptor (Fig. 1a) expressed on the apical surface of intestinal enterocytes and human hepatocytes, including Huh7 cells (Supplementary Fig. 1), is responsible for cellular cholesterol absorption and whole body cholesterol homeostasis15,16. Similar to what has been observed for other HCV entry factors8, we observed down-regulation of NPC1L1 in HCVcc-infected Huh7 cultures. Specifically, as early as d 4 post-infection (p.i.) NPC1L1 protein levels were markedly reduced and remained down-regulated until the end of the experiment at d 12 p.i. (Fig. 1b). Having observed a correlation between NPC1L1 expression and HCV infection, we next determined if NPC1L1 expression levels affect HCV infection by transfecting Huh7 cells with short interfering RNAs (siRNAs) targeting NPC1L1 or the known HCV entry factors CD81 or SR-BI. Compared to cells transfected with an irrelevant-control siRNA, susceptibility to HCVcc infection was significantly reduced in CD81-, SR-BI- and NPC1L1-silenced cells (Fig. 1c). Inhibition was HCV-specific as silencing of these proteins had no effect on vesicular stomatitis virus G-protein pseudotyped particle (VSVGpp) infection (Supplementary Fig. 2a). Inhibition of HCV also correlated with NPC1L1 mRNA and protein reduction and was confirmed to be NPC1L1-specific and not the result of off-target effects (Fig. 1d,e, Supplementary Figs. 3 and 4a,b). Interestingly, although protein levels were only marginally reduced by siRNA knockdown, the effect on HCV was significant, highlighting the sensitivity of HCV to small changes in NPC1L1 levels. Importantly, since SR-BI mRNA expression has been shown to be reduced by NPC1L1 knockdown in non-hepatic cells17 and SR-BI is an HCV entry factor5, we confirmed that SR-BI expression was not adversely affected by NPC1L1 silencing in Huh7 cells (Supplementary Fig. 4c,d). Finally, NPC1L1 silencing had no effect on HCV subgenomic RNA replication, full length infectious HCVcc RNA replication, or secretion of de novo HCVcc (Supplementary Fig. 5).
Figure 1.

NPC1L1 plays a role in HCVcc infection. (a) NPC1L1 topology. (b) Immunoblot of NPC1L1, HCV NS3, and β-actin in Huh7 cells mock-infected or infected with HCVcc at an MOI of 3.0 FFU cell−1 over the course of 12 d. (c–e) Huh7 cells were mock-transfected or transfected with irrelevant control (siCon), SR-BI-specific, CD81-specfic, or NPC1L1-specific siRNAs and subsequently infected with HCVcc at an MOI of 0.05 FFU cell−1 at indicated times post-transfection. (c) Forty-eight h p.i. HCV RNA was quantified by RTqPCR and data normalized to GAPDH. Results are graphed as a percentage of infection achieved in siCon-transfected cultures. (d) NPC1L1 transcript levels were quantified by RTqPCR, normalized to GAPDH and are graphed as a percentage of the maximum number of copies determined in siCon-transfected cultures at each time point examined. (e) Immunoblot of NPC1L1 and β-actin protein expression in siCon-transfected (–) and siNPC1L1-transfected cultures (+). (f,g) Huh7 cells were treated with 36 µg ml−1 of indicated antibodies for 1 h prior to and during HCVcc infection at an MOI of 0.05 FFU cell−1. HCV RNA levels were determined by RTqPCR analysis 24 (f) or 48 (f and g) h p.i. Data were normalized to GAPDH levels and results are graphed as a percentage of infection achieved in respective IgG control-treated cultures. In all cases, significant differences relative to controls (one-way ANOVA and Tukey’s post hoc t test) are denoted as * P < 0.05 or ** P <0.01. All results are graphed as means ± SD for triplicate samples. The data presented are representative of three independent experiments.
Because siRNA-mediated knockdown of NPC1L1 suggested inhibition of HCV infection at a step before RNA replication or secretion, we next assessed the susceptibility of HCV infection to antibody-mediated blocking of cell surface NPC1L1. Compared to cells treated with irrelevant IgG control antibodies, HCVcc infection was significantly reduced in cells treated with an antibody specific for the known HCV cell entry factor CD81 (Fig. 1f). When we incubated cells with an NPC1L1-specific antibody, HCVcc infection was similarly reduced (Fig. 1f) and inhibition was HCV-specific as antibody-mediated blocking had no effect on VSVGpp entry (Supplementary Fig. 2b,c). To map NPC1L1-specific entry domains, we treated cells with antibodies targeting each of the three large extracellular loops (LELs) of NPC1L1 and observed that HCV infection was reduced only when NPC1L1 LEL1, but not LEL2 or LEL3, was blocked (Fig. 1g). Thus, NPC1L1 silencing and antibody-mediated blocking of NPC1L1 LEL1 reduced HCV infection as effectively as targeting other known HCV cell entry factors.
Ezetimibe is a 2-azetidinone class of drug that has been FDA-approved as a cholesterol-lowering medication18. Since ezetimibe has been shown to be a direct inhibitor of NPC1L1 internalization19,20, we next used this high affinity, specific pharmacological agent as an alternate means of targeting NPC1L1 pre-, co- or post-infection while additionally evaluating its anti-HCV potential. Specifically, we performed HCVcc foci-reduction assays and quantified foci (i.e. clusters of ≥ 5 HCV E2-positive cells) following ezetimibe treatment. Ezetimibe reduced HCVcc foci formation in a dose-dependent manner when present prior to infection and then removed (Fig. 2a) or only during virus inoculation (Fig. 2b). However, when we added ezetimibe to cells post-infection (Fig. 2c), the initiation of HCV-positive foci was unaffected as would be expected for a viral entry inhibitor. Notably, the highest dose of ezetimibe (25 µM) reduced the foci size observed (i.e. 1 – 3 HCV E2-positive cells per foci) which resulted in a reduced number of ≥ 5 HCV E2-positive foci being counted suggesting NPC1L1 may also affect HCV cell-to-cell spread (data not shown). Dose-responsive time-of-addition-dependent inhibition of HCV infection was also evident when HCV RNA levels were measured (Supplementary Fig. 6). Importantly, ezetimibe sensitivity was also observed across a panel of HCVcc inter-genotypic clones containing the structural region of diverse HCV genotypes (1 – 7)21 (Fig. 2d). Finally, because NPC1L1 and SR-BI are both involved in cellular cholesterol uptake and SR-BI has been reported to be a rate-limiting HCV cell entry factor22, we over-expressed SR-BI prior to ezetimibe treatment to confirm that the dependence on NPC1L1 could not be overcome (Supplementary Fig. 7). Likewise, we confirmed that the potent anti-viral effect of ezetimibe was not due to drug-mediated cytotoxicity (Supplementary Figs. 2d,e and 8a), changes in cell proliferation (Supplementary Fig. 8b), reduction of the other known HCV cell surface receptors (Supplementary Fig. 8c–g), inhibition of HCV RNA replication (Supplementary Fig. 9a–c), or inhibition of virus secretion (Supplementary Fig. 9d). Hence, the data support the conclusion that direct pharmacological inhibition of NPC1L1 reduces HCV infection by directly inhibiting viral cell entry.
Figure 2.
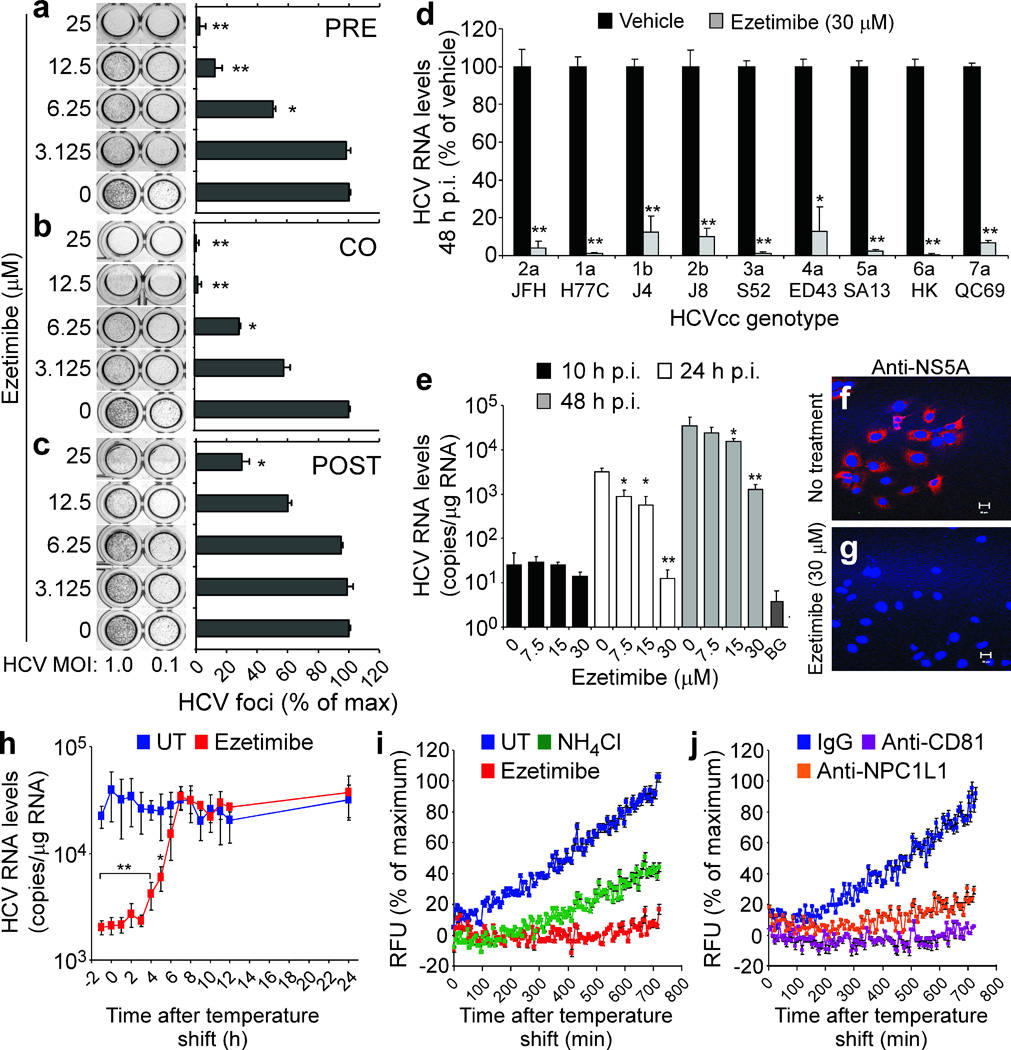
Ezetimibe-mediated inhibition of NPC1L1 reduces HCV entry at a post-binding pre-fusion step. (a–c) Huh7 cells were vehicle-treated or treated with increasing concentrations of ezetimibe (a) for 6 h prior to infection and then removed (PRE), (b) for 12 h coincident with viral inoculation and then removed (CO), or (c) following viral inoculation (POST) with HCVcc at an MOI of 1.0 or 0.1 FFU cell−1. HCV foci were quantified 72 h p.i. and are expressed as a percentage of the foci obtained in vehicle-treated (0 µM) cultures ± SD (n = 3). (d–g) Huh7 cells were treated with vehicle or indicated concentrations of ezetimibe beginning 1 h prior to and during infection with (d) HCVcc containing the structural region of the indicated genotypes or (e–g) HCVcc JFH-1 at an MOI of 0.1 FFU cell−1. HCV RNA was quantified by RTqPCR at the indicated times p.i. and data normalized to GAPDH. Results are graphed as a percentage of infection in vehicle-treated cultures or as mean HCV RNA copies/μg total cellular RNA ± SD (n = 3). Assay background (BG) = HCV RNA level detected in uninfected samples. (f,g) Indirect immunofluorescence analysis of HCV NS5A in (f) vehicle-treated and (g) ezetimibe-treated cultures 24 h p.i. Scale bar = 20 µm. (h) Synchronized infections in Huh7 cells were treated with vehicle or ezetimibe (30 µM) at the indicated times. Thirty hours p.i. RNA was harvested. HCV RNA was quantified by RTqPCR, normalized to GAPDH and displayed as mean HCV RNA copies/μg total cellular RNA ± SD (n = 3). Significant reduction in HCV relative to vehicle-treated cultures (one-way ANOVA and Tukey’s post hoc t test) is denoted as * P < 0.05 or ** P <0.01. (i,j) Huh7 cells were treated with vehicle (UT), NH4Cl (10 mM), ezetimibe (30 µM), IgG control antibody (36 µg ml−1), anti-CD81 antibody (36 µg ml−1), or anti-NPC1L1 LEL1 antibody (36 µg ml−1) beginning 1 h prior to inoculation with HCVccDiD (MOI of 5.0 FFU cell−1). HCV fusion was measured by DiD dequenching every 6 min. Results are graphed as a percentage of maximum background-corrected relative fluorescence units (RFU) achieved in vehicle-treated or IgG control-treated cultures. All data presented are representative of three independent experiments.
We next assessed if ezetimibe inhibits HCVcc binding or a post-binding step by examining cell-associated HCV RNA and protein expression from internalized RNA in vehicle- and ezetimibe-treated HCVcc-infected cultures. At 10 h p.i., a time before detectable HCV RNA replication occurs (Supplementary Fig. 10), ezetimibe did not affect bound/cell-associated HCV RNA levels (Fig 2e). In contrast, at later time points, HCV RNA expansion (Fig. 2e) and de novo NS5A protein expression (Fig. 2f,g) were reduced in ezetimibe-treated cultures, suggesting HCV can efficiently bind to ezetimibe-treated cells, but that a post-binding step is prevented. To further test this hypothesis and determine when during the entry process NPC1L1 functions, we assessed the ability of ezetimibe to block HCVcc infection when added at various times post 4 °C virus binding. Ezetimibe retained inhibitory activity after temperature shift to 37 °C for up to 5 h (half-maximal inhibition at 4 h), confirming that NPC1L1 functions post-binding likely late in viral entry (Fig. 2h).
To determine if ezetimibe acts prior to fusion, we developed a fluorescence-based HCVcc fusion assay. Specifically, we labeled HCVcc with the hydrophobic fluorophore DiD23, which incorporates into biological membranes and at high concentrations is self-quenching. Upon fusion of viral and target membranes, the DiD fluorophores diffuse away from each other causing dequenching, and the progression or inhibition of fusion can be measured in real time (Supplementary Fig. 11). Compared to NH4Cl, an inhibitor of endosomal acidification9, ezetimibe more potently inhibited HCVccDiD fusion, such that by 12 h post-binding only ~10% HCVccDiD dequenching was measured in ezetimibe-treated cultures as compared to vehicle-treated controls (Fig. 2i). Analogously, antibody-mediated inhibition of both CD81 and NPC1L1 also reduced HCVccDiD fusion (Fig. 2j), indicating that the inhibition observed in ezetimibe-treated wells (Fig. 2i) was not drug-specific. Similar results were also observed using HCVccDiD alternatively purified by iodixanol density gradient centrifugation (Supplementary Fig. 12a,b).
Since not all viral membrane-incorporated DiD is self-quenched, DiD can also serve as a fluorescent tag to monitor virions during cell entry24. Taking advantage of this, we performed fluorescence microscopy analysis of HCVccDiD−infected cultures and noted that while little DiD was observed on the surface of vehicle-treated cells, indicative of successful viral entry and fusion, markedly more DiD was observed on the surface of ezetimibe-treated cells (Supplementary Fig. 12c,d). Together with the DiD-fusion data, this indicates that inhibition of NPC1L1 prevents HCVcc cell entry at or prior to virion:host cell fusion.
Since antibody-mediated blocking of only NPC1L1 LEL1 (Fig. 1g) reduced HCVcc infection, LEL1 has been shown to bind cholesterol20,25 and infectious HCV particles are enriched in cholesterol13,26, we next investigated whether the dependence of HCV cell entry on NPC1L1 might be related to the cholesterol contained within the HCV virions13. To address this hypothesis, we utilized JFH-1-based viruses that differ in their virion-associated cholesterol content and assessed their relative dependence on NPC1L1. Specifically, we show that HCV JFH-1 pseudotype particles (JFHpp) contain 94% less cholesterol than HCVcc JFH-1 while the cell culture-adapted virus JFH-1G451R, which has a distinct density profile27, contains ~50% more cholesterol than HCVcc JFH-1 (Fig. 3a). These cholesterol profiles are consistent with the fact that HCVpp are produced from 293T embryonal kidney cells, which do not produce cholesterol-associated lipoproteins28, and are therefore compositionally distinct from HCVcc JFH-1, while HCVcc JFH-1G451R contains a glycine-to-arginine mutation (G451R) in the viral E2 glycoprotein resulting in production of HCVcc having a narrower density range with a higher average mean density27. As expected, when CD81 was silenced, both HCVpp (Fig. 3b) and HCVcc JFH-1G451R (Fig. 3c) cell entry was reduced; however, when NPC1L1 was silenced or inhibited by ezetimibe, the cholesterol-scarce JFHpp exhibited NPC1L1-independent cell entry and insensitivity to ezetimibe inhibition (Fig. 3b and d, respectively). In contrast, the cholesterol-abundant HCVcc JFH-1G451R exhibited enhanced NPC1L1-dependent cell entry and hypersensitivity to ezetimibe-mediated inhibition (Fig. 3c and e, respectively). Together, these data reveal a correlation between the amount of virion-associated cholesterol and dependence on NPC1L1 for HCV cell entry.
Figure 3.

NPC1L1-mediated HCV cell entry is cholesterol-dependent. (a) Cholesterol content of HCVcc JFH-1, HCVcc JFH-1G451R and JFHpp determined by a fluorometric cholesterol quantification assay, after HiTrap™ Heparin HP affinity column purification. Cholesterol content is graphed as cholesterol (nM) per 1×106 genome copies determined by RTqPCR. (b,c) Huh7 cells were mock-transfected or transfected with indicated siRNA, knockdown was confirmed by RTqPCR (data not shown) and cultures were infected with (b) equal titers of JFHpp or VSVGpp or (c) HCVcc JFH-1 or HCVcc JFH-1G451R at an MOI of 0.05 FFU cell−1. JFHpp and VSVGpp infection was determined 72 h p.i and is expressed as relative light units (RLU) ± SD (n = 3). HCV RNA levels were determined by RTqPCR 48 h p.i., normalized to GAPDH and are graphed as a percentage of maximum determined in siCon-transfected cultures. (d,e) Huh7 cells were treated with vehicle or increasing concentrations of ezetimibe beginning 1 h prior to inoculation with (d) equal titers of JFHpp or VSVGpp or (e) HCVcc JFH-1 or HCVcc JFH-1G451R at an MOI of 0.05 FFU cell−1. JFHpp and VSVGpp infection was determined 72 h p.i and is expressed as RLU ± SD (n = 3). HCV RNA was quantified by RTqPCR, normalized to GAPDH and is displayed as mean HCV RNA copies/μg total cellular RNA ± SD. Significant reductions in RNA or RLU values relative to siCon-transfected or vehicle-treated cultures (one-way ANOVA and Tukey’s post hoc t test) are denoted as * P < 0.05 or ** P <0.01.
Finally, to assess the involvement of NPC1L1 in HCV cell entry in vivo we evaluated the ability of ezetimibe to inhibit infection of a genotype 1 clinical isolate in a hepatic xenorepopulation model of acute HCV infection29. Specifically, uPA-SCID mice repopulated with human hepatocytes were pre-treated via oral gavage with ezetimibe (10 mg kg−1 day−1) or diluent alone for a total of 3 weeks, with treatment beginning 2 weeks, 1 week or 2 d prior to challenge with HCV genotype 1b-positive serum (Fig. 4a). Ezetimibe treatment delayed the establishment of HCV infection in mice pre-treated for 2 weeks prior to infection (Fig. 4b, P = 0.0192), confirming the ability of this FDA-approved drug to inhibit HCV infection in vivo. However, when mice were pre-treated for only 1 week prior to infection, ezetimibe was less effective at delaying infection (P = 0.062) and completely ineffective when treatment was initiated 2 d prior to challenge or after infection had been established (data not shown). Specifically, 100% of the nine control diluent-treated mice were HCV serum-positive 1 week following challenge, while 71% (five out of seven) and 43% (three out of seven) of mice treated with ezetimibe for 2 weeks and 1 week prior to infection were HCV-negative, respectively (Fig. 4b,c). Although the majority of ezetimibe-treated mice eventually became HCV-positive, of the five mice in the 2-week ezetimibe pre-treatment group that were HCV-negative at week 1, two were completely protected remaining HCV-negative at weeks 2 and 3 p.i. (and one mouse died during gavage) (Supplementary Fig. 13). Thus, similar to what was recently reported for another potential HCV entry inhibitor, erlotinib30, ezetimibe was able to delay initial infection in vivo. Notably, since NPC1L1 is highly expressed on the apical surface of intestinal enterocytes15,16, a significant amount of orally administered ezetimibe initially binds to these cells following oral administration31. Thus it is plausible that development of alternate non-oral delivery or drug-targeting methods might improve transport of ezetimibe to hepatocytes and increase its anti-HCV efficacy. Nevertheless, demonstration that ezetimibe can delay the establishment of HCV genotype 1 infection in mice confirms the involvement of NPC1L1 in HCV infection in vivo and highlights the therapeutic potential of further pursuing the refinement or development of anti-NPC1L1-based therapies32 for the treatment of HCV.
Figure 4.

Ezetimibe delays the establishment of HCV infection in hepatic xenorepopulated mice. (a) Schematic diagram of experiment in which uPA-SCID mice transplanted with human hepatocytes35 were pre-treated with diluent alone (n = 4 – 5) or ezetimibe (n = 7, 10 mg kg−1 day−1), via oral gavage, starting 2 weeks, 1 week or 2 d prior to infection (indicated by grey bars). The mice were intravenously inoculated on d 0 with HCV human serum containing 1.0×105 genome copies of HCV genotype 1b (indicated by arrow) and treatments were continued as indicated (black bars). (b,c) Serum samples were obtained weekly for three weeks post-infection for HCV RNA determination. Graphed are HCV RNA levels (genome copies ml−1 of serum) one week post-infection from mice pre-treated for (b) two weeks or (c) one week. The lower limit of HCV RNA detection is equal to 102 genomic copies ml−1 of serum. A 2-tailed Fisher’s exact test was performed to compare categorical variables. In all cases P < 0.05 was used to reject the null hypothesis that the distribution of HCV-positive/HCV-negative mice between ezetimibe-treated and nine diluent-treated mice at specific weeks post-infection were the same.
Meanwhile, herein we demonstrate that NPC1L1 is an HCV cell entry factor which functions post-binding, at or prior to fusion. These findings, together with the facts that NPC1L1 is a cellular cholesterol receptor, the HCV particle is enriched in cholesterol, and relative dependence on NPC1L1 is correlated with HCV particle cholesterol levels support and expand upon previous reports suggesting that virion cholesterol plays a role during HCV cell entry13,14,26. Whether NPC1L1 directly interacts with HCV or indirectly participates in HCV entry by removing virion-associated cholesterol to perhaps reveal protected viral glycoprotein binding sites or confer a required conformational change remains to be determined. Lastly, since NPC1L1 is only expressed on the surface of human and primate hepatocytes33,34, this discovery additionally highlights NPC1L1 as a potential HCV tropism determinant, which may facilitate the future development of a small animal model of HCV infection.
Methods
Cell-culture-propagated HCV (HCVcc)
Plasmids containing the full-length JFH-1 genome (pJFH1)36, full-length JFH-1 genome with a glycine-to-arginine mutation at amino acid residue 451 in the E2 glycoprotein (pJFH-1G451R)37 and the eight intergenotypic clones (described in21) were XbaI linearized, transcribed using MEGAscript T7 (Ambion) and 10 µg in vitro transcribed RNA was electroporated (BioRad) into Huh7 cells38. We generated HCVcc viral stocks by infecting naïve Huh7 cells at a multiplicity of infection (MOI) of 0.01 focus forming units (FFU) cell−1 with medium from Huh7 cells electroporated with in vitro transcribed RNA from pJFH-1-based vectors , as described38.
Treatments and analysis
Huh7 cultures were established as previously described38. We performed RNA silencing experiments by reverse transfection (Lipofectamine™ RNAiMAX, Invitrogen) of Huh7 cells with indicated siRNAs. Transfected cells were infected with HCVcc at indicated times post-transfection. For antibody experiments, we treated cells with 36 µg ml−1 of indicated antibodies prior to and during infection. For ezetimibe inhibition experiments, cells were vehicle-treated or treated with increasing concentrations of ezetimibe prior to infection (PRE), during the time of virus inoculation (CO), and/or following virus inoculation (POST), as indicated. The ezetimibe concentrations of 3.125 – 30 µM (i.e. 1.5 – 12.28 µg ml−1 culture medium) used in this study are consistent with previous published reports19,20,39 and are additionally in line with patient daily intake concentrations of 10 mg day−1 (i.e. 2.0 – 3.3 µg ml−1 of serum). For RTqPCR analysis, we isolated total cellular RNA from triplicate wells at indicated times post-infection or transfection. For HCV E2-positive foci analysis, we fixed infected cells with 4% paraformaldehyde (w/v) 72 h p.i., and immunocytochemical staining for HCV E2 was performed. See Supplementary Methods for further details.
HCV infection in chimeric mice
All mouse studies were conducted with protocols approved by the Ethics Review Committee for Animal Experimentation of the Graduate School of Biomedical Sciences, Hiroshima University, Hiroshima, Japan. Human hepatocyte-transplanted mice generated in severe combined immunodeficient (SCID)/urokinase plasminogen activator (uPA) mice were purchased from PhenixBio40. For acute HCV infection experiments, we stably transplanted male uPA/SCID mice with human hepatocytes (purchased from BD Biosciences)35 and treated them daily with 10 mg kg−1 ezetimibe via oral gavage of a 0.02 mg ml−1 solution of ezetimibe resuspended in corn oil (100 µl 20g−1) for a total of 3 weeks, with treatment initiation beginning at indicated times prior to infection16. Control mice were treated via oral gavage with corn oil alone (100 µl 20g−1). A total of 4 – 7 mice were included in each group. On day 0 we intravenously inoculated mice with HCV human serum containing 1.0×105 copies of HCV genotype 1b. We obtained mouse serum samples on indicated days for HCV RNA or human albumin determination by RTqPCR and Alb-II Kit (Eiken Chemical), respectively.
Statistics
Data are presented as the means ± standard deviation (SD). We determined significant differences by one-way analysis of variance (ANOVA) followed by Tukey's post hoc t test (GraphPad Prism© Software). To compare categorical variables we used a 2-tailed Fisher’s exact test (SPSS v18, Chicago, IL). In all cases a P value < 0.05 was considered statistically significant.
Additional methods
Detailed methodology is described in the Supplementary Methods.
Supplementary Material
Acknowledgements
The authors graciously thank T. Wakita (National Institute of Infectious Diseases, Tokyo, Japan) for JFH1-based plasmids, F Chisari (The Scripps Research Institute, La Jolla, CA) for Huh7 cells, J. Buhk (National Institutes of Health, Bethesda, MD) for JFH-1-based inter-genotypic HCV clones, Y. Ioannou (Mount Sinai School of Medicine, New York, NY) for the antibody (Bsn4052) against NPC1L1, D. Burton and M. Law (The Scripps Research Institute, La Jolla, CA) for the antibody (C1) against the HCV glycoprotein E2 and C. Rice (The Rockefeller Institute, New York, NY) for the antibody (E910) against the HCV nonstructural protein NS5A. The authors would also like to thank P. Corcoran for outstanding technical assistance, H. Dahari for assistance with statistical analyses and T. Layden and S. Cotler for editing the manuscript. We also thank J. Graves of the University of Illinois at Chicago (UIC) Research Resources Center Flow Cytometry laboratory and K. Ma of the UIC Research Resources Center Confocal Microscopy laboratory for technical assistance. This work was supported by the US National Institutes of Health Public Health Service Grants R01-AI070827 (SLU) and R03-AI085226 (SLU), the American Cancer Society Research Scholar Grant RSG-09-076-01 (SLU), the UIC Center for Clinical and Translational Science NIH Grant UL1RR029879, the UIC Council to support Gastrointestinal and Liver Disease (UIC GILD) and a grant from the Ministry of Education, Culture, Sports, Science and Technology-Japan, Ministry of Health, Labour and Welfare-Japan (KC). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Author contributions
B.S.Jr. made the initial discovery. B.S.Jr. and S.L.U. designed the project, analyzed the results and wrote the manuscript. B.S.Jr., N.B., D.N.M, S.H., K.A.M. and X.Y. performed experimental work. B.S.Jr., S.L.U., M.I. and K.C. designed the hepatic xenorepopulation mouse experiments and N.H. performed the in vivo studies. W.A.A. was involved in the initial conception of the project and provided valuable expertise.
Conflict of interest
The authors declare competing financial interests: details accompany the full-text HTML version of the paper at www.nature.com/nature.
References
- 1.Uprichard SL. Hepatitis C virus experimental model systems and antiviral drug research. Virol Sin. 2010;25:227–245. doi: 10.1007/s12250-010-3134-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gupta EK, Ito MK. Ezetimibe: the first in a novel class of selective cholesterol-absorption inhibitors. Heart Dis. 2002;4:399–409. doi: 10.1097/00132580-200211000-00011. [DOI] [PubMed] [Google Scholar]
- 3.Garcia-Calvo M, et al. The target of ezetimibe is Niemann-Pick C1-Like 1 (NPC1L1) Proc Natl Acad Sci U S A. 2005;102:8132–8137. doi: 10.1073/pnas.0500269102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pileri P, et al. Binding of hepatitis C virus to CD81. Science. 1998;282:938–941. doi: 10.1126/science.282.5390.938. [DOI] [PubMed] [Google Scholar]
- 5.Scarselli E, et al. The human scavenger receptor class B type I is a novel candidate receptor for the hepatitis C virus. Embo J. 2002;21:5017–5025. doi: 10.1093/emboj/cdf529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Evans MJ, et al. Claudin-1 is a hepatitis C virus co-receptor required for a late step in entry. Nature. 2007;446:801–805. doi: 10.1038/nature05654. [DOI] [PubMed] [Google Scholar]
- 7.Ploss A, et al. Human occludin is a hepatitis C virus entry factor required for infection of mouse cells. Nature. 2009;457:882–886. doi: 10.1038/nature07684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu S, et al. Tight junction proteins claudin-1 and occludin control hepatitis C virus entry and are downregulated during infection to prevent superinfection. J Virol. 2009;83:2011–2014. doi: 10.1128/JVI.01888-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tscherne DM, et al. Time- and temperature-dependent activation of hepatitis C virus for low-pH-triggered entry. J Virol. 2006;80:1734–1741. doi: 10.1128/JVI.80.4.1734-1741.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Meertens L, Bertaux C, Dragic T. Hepatitis C virus entry requires a critical postinternalization step and delivery to early endosomes via clathrin-coated vesicles. J Virol. 2006;80:11571–11578. doi: 10.1128/JVI.01717-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huang H, et al. Hepatitis C virus production by human hepatocytes dependent on assembly and secretion of very low-density lipoproteins. Proc Natl Acad Sci U S A. 2007;104:5848–5853. doi: 10.1073/pnas.0700760104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gastaminza P, et al. Cellular determinants of hepatitis C virus assembly, maturation, degradation, and secretion. J Virol. 2008;82:2120–2129. doi: 10.1128/JVI.02053-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Aizaki H, et al. Critical role of virion-associated cholesterol and sphingolipid in hepatitis C virus infection. J Virol. 2008;82:5715–5724. doi: 10.1128/JVI.02530-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kapadia SB, Barth H, Baumert T, McKeating JA, Chisari FV. Initiation of hepatitis C virus infection is dependent on cholesterol and cooperativity between CD81 and scavenger receptor B type I. J Virol. 2007;81:374–383. doi: 10.1128/JVI.01134-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yu L. The structure and function of Niemann-Pick C1-like 1 protein. Curr Opin Lipidol. 2008;19:263–269. doi: 10.1097/MOL.0b013e3282f9b563. [DOI] [PubMed] [Google Scholar]
- 16.Altmann SW, et al. Niemann-Pick C1 Like 1 protein is critical for intestinal cholesterol absorption. Science. 2004;303:1201–1204. doi: 10.1126/science.1093131. [DOI] [PubMed] [Google Scholar]
- 17.Sane AT, et al. Localization and role of NPC1L1 in cholesterol absorption in human intestine. J Lipid Res. 2006;47:2112–2120. doi: 10.1194/jlr.M600174-JLR200. [DOI] [PubMed] [Google Scholar]
- 18.Bays HE, Neff D, Tomassini JE, Tershakovec AM. Ezetimibe: cholesterol lowering and beyond. Expert Rev Cardiovasc Ther. 2008;6:447–470. doi: 10.1586/14779072.6.4.447. [DOI] [PubMed] [Google Scholar]
- 19.Chang TY, Chang C. Ezetimibe blocks internalization of the NPC1L1/cholesterol complex. Cell Metab. 2008;7:469–471. doi: 10.1016/j.cmet.2008.05.001. [DOI] [PubMed] [Google Scholar]
- 20.Weinglass AB, et al. Extracellular loop C of NPC1L1 is important for binding to ezetimibe. Proc Natl Acad Sci U S A. 2008;105:11140–11145. doi: 10.1073/pnas.0800936105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gottwein JM, et al. Development and characterization of hepatitis C virus genotype 1–7 cell culture systems: role of CD81 and scavenger receptor class B type I and effect of antiviral drugs. Hepatology. 2009;49:364–377. doi: 10.1002/hep.22673. [DOI] [PubMed] [Google Scholar]
- 22.Grove J, et al. Scavenger receptor BI and BII expression levels modulate hepatitis C virus infectivity. J Virol. 2007;81:3162–3169. doi: 10.1128/JVI.02356-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zaitseva E, Yang ST, Melikov K, Pourmal S, Chernomordik LV. Dengue virus ensures its fusion in late endosomes using compartment-specific lipids. PLoS Pathog. 2010;6 doi: 10.1371/journal.ppat.1001131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Coller KE, et al. RNA interference and single particle tracking analysis of hepatitis C virus endocytosis. PLoS Pathog. 2009;5:e1000702. doi: 10.1371/journal.ppat.1000702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang JH, et al. The N-terminal Domain of NPC1L1 Protein Binds Cholesterol and Plays Essential Roles in Cholesterol Uptake. J Biol Chem. 2011;286:25088–25097. doi: 10.1074/jbc.M111.244475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yamamoto M. Structural requirements of virion-associated cholesterol for infectivity, buoyant density and apolipoprotein association of hepatitis C virus. J Gen Virol. 2011;92:2082–2087. doi: 10.1099/vir.0.032391-0. [DOI] [PubMed] [Google Scholar]
- 27.Zhong J, et al. Persistent hepatitis C virus infection in vitro: coevolution of virus and host. J Virol. 2006;80:11082–11093. doi: 10.1128/JVI.01307-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Farquhar MJ, McKeating JA. Primary hepatocytes as targets for hepatitis C virus replication. J Viral Hepat. 2008;15:849–854. doi: 10.1111/j.1365-2893.2008.01051.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kneteman NM, Toso C. In vivo study of HCV in mice with chimeric human livers. Methods Mol Biol. 2009;510:383–399. doi: 10.1007/978-1-59745-394-3_29. [DOI] [PubMed] [Google Scholar]
- 30.Lupberger J, et al. EGFR and EphA2 are host factors for hepatitis C virus entry and possible targets for antiviral therapy. Nat Med. 2011;17:589–595. doi: 10.1038/nm.2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sweeney ME, Johnson RR. Ezetimibe: an update on the mechanism of action, pharmacokinetics and recent clinical trials. Expert Opin Drug Metab Toxicol. 2007;3:441–450. doi: 10.1517/17425255.3.3.441. [DOI] [PubMed] [Google Scholar]
- 32.Betters JL, Yu L. Transporters as drug targets: discovery and development of NPC1L1 inhibitors. Clin Pharmacol Ther. 2010;87:117–121. doi: 10.1038/clpt.2009.209. [DOI] [PubMed] [Google Scholar]
- 33.Davies JP, Scott C, Oishi K, Liapis A, Ioannou YA. Inactivation of NPC1L1 causes multiple lipid transport defects and protects against diet-induced hypercholesterolemia. J Biol Chem. 2005;280:12710–12720. doi: 10.1074/jbc.M409110200. [DOI] [PubMed] [Google Scholar]
- 34.Davis HR, Jr, et al. Niemann-Pick C1 Like 1 (NPC1L1) is the intestinal phytosterol and cholesterol transporter and a key modulator of whole-body cholesterol homeostasis. J Biol Chem. 2004;279:33586–33592. doi: 10.1074/jbc.M405817200. [DOI] [PubMed] [Google Scholar]
- 35.Matsumura T, et al. Amphipathic DNA polymers inhibit hepatitis C virus infection by blocking viral entry. Gastroenterology. 2009;137:673–681. doi: 10.1053/j.gastro.2009.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wakita T, et al. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat Med. 2005;11:791–796. doi: 10.1038/nm1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sabahi A, et al. The rate of hepatitis C virus infection initiation in vitro is directly related to particle density. Virology. 2010;407:110–119. doi: 10.1016/j.virol.2010.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yu X, Uprichard SL. Cell-based hepatitis C virus infection fluorescence resonance energy transfer (FRET) assay for antiviral compound screening. Curr Protoc Microbiol. 2010:5. doi: 10.1002/9780471729259.mc1705s18. Chapter 17, Unit 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ge L, et al. The cholesterol absorption inhibitor ezetimibe acts by blocking the sterol-induced internalization of NPC1L1. Cell Metab. 2008;7:508–519. doi: 10.1016/j.cmet.2008.04.001. [DOI] [PubMed] [Google Scholar]
- 40.Tateno C, et al. Near completely humanized liver in mice shows human-type metabolic responses to drugs. Am J Pathol. 2004;165:901–912. doi: 10.1016/S0002-9440(10)63352-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.