

Abstract
Understanding how geographical and environmental features affect genetic variation at both the population and individual levels is crucial in biology, especially in the case of pathogens. However, distinguishing between these factors and the effects of historical range expansion on spatial genetic structure remains challenging. In the present study, we investigated the case of Mycosphaerella fijiensis—a plant pathogenic fungus that has recently colonized an agricultural landscape characterized by the presence of potential barriers to gene flow, including several commercial plantations in which disease control practises such as the use of fungicides are applied frequently, and low host density areas. We first genotyped 300 isolates sampled at a global scale on untreated plants in two dimensions over a 50 × 80-km area. Using two different clustering algorithms, no genetic structure was detected in the studied area, suggesting expansion of large populations and/or no influence of potential barriers. Second, we investigated the potential effect of disease control practises on M. fijiensis diversity by comparing populations sampled in commercial vs food-crop plantations. At this local scale, we detected significantly higher allelic richness inside commercial plantations compared with the surrounding food-crop plantation populations. Analysis of molecular variance indicated that 99% of the total genetic variance occurred within populations. We discuss the suggestion that high population size and/or high migration rate between populations might be responsible for the absence of any effect of disease control practises on genetic diversity and differentiation.
Keywords: landscape genetics, gene flow, genetic drift, cultural practises, emergent disease, Mycosphaerella fijiensis
Introduction
Understanding the factors influencing the spatial genetic structure of populations has important implications for evolutionary biology (Holderegger and Wagner, 2008). The recently emerged discipline of ‘landscape genetics'—combining approaches from landscape ecology, population genetics and spatial statistics—investigates the contemporary effects of landscape composition and environmental factors on spatial genetic variation (Manel et al., 2003). Within this context, the overwhelming majority of studies to date have addressed the question of how landscape features affect connectivity or create barriers to gene movement (see Sork and Waits, 2010 for a review). Linear elements, such as rivers, mountain ridges or roads, have been the most frequent barriers to gene flow considered thus far. However, other less obvious ecological factors, for instance those linked to the presence of unsuitable natural habitat, might also influence neutral genetic structure if such factors limit gene flow (McRae and Beier, 2007) or induce genetic drift through a reduction in population size (Hoffmann and Willi, 2008). The effects of ecological factors (for example, variation in temperature, habitat fragmentation and local environment) or contaminants (for example, heavy metals) on neutral genetic structure have been described previously for both plants (for example, Schmidt et al., 2009) and animals (for example, Dionne et al., 2008).
A major challenge in landscape genetic studies is the potential mismatch between different temporal scales in factors reflected by genetic data (Balkenhol et al., 2009). Indeed, genetic data bear signatures of both past and current processes. Several factors implicated in the historical expansion of a species may influence its present spatial genetic patterns (Excoffier et al., 2009). In addition to barriers to gene flow, the natural dispersal ability of a species during range expansion can generate spatial breaks in allele frequencies. For instance, the combination of rare long-distance dispersal (LDD) events accompanied by founder effects followed by natural gradual diffusion is expected to produce a patchy distribution of allele frequencies. This original pattern first gained theoretical support from simulations (Nichols and Hewitt, 1994), but more recently has been observed empirically and named ‘stratified dispersal combination' (Parisod and Bonvin, 2008). Nevertheless, the spatial genetic structures left by species range expansion are not always expected to match with landscape and environmental factors. Thus, fine scale genetic structure analyses in two-dimensional environments could help to disentangle the effects on spatial genetic structures of these two different types of factor.
Plant pathogenic fungi make interesting models for such studies, because various factors have a potential influence on their spatial genetic structure. First, the spread of such organisms is inherently a spatial process, often embedded in heterogeneous landscapes (Biek and Real, 2010). Second, in addition to being fragmented by landscape features, pathogen populations in modern agroecosystems are challenged regularly by a variety of cultural practices that aim to reduce plant infection (Stukenbrock and McDonald, 2008). Among them, the intensive use of fungicides is the most common. Such practices represent a major ecological constraint that could influence the neutral genetic structure of pathogen populations for two main reasons: (i) treated areas can act as barriers to gene flow within an agricultural landscape composed of mixed (treated vs untreated) populations; and (ii) genetic drift is expected to occur in treated areas where the application of chemicals generally induces drastic reductions in population size, which could result in decreased genetic diversity. Lastly, historical range expansions combining LDD and gradual diffusion events are frequent in pathogenic fungi (Brown and Hovmøller, 2002), which could also impact current spatial genetic structures. Furthermore, as pathogenic fungi cause serious biological and economic damage to many agroecosystems (Anderson et al., 2004), a detailed understanding of the various factors influencing their spatial genetic structures at the agricultural landscape scale will increase our ability to control plant disease (Archie et al., 2009). To address such questions, landscape genetics appears to be a useful approach that has so far seldom been applied to plant pathogen populations (Biek and Real, 2010).
Black leaf streak disease (BLSD) in Southwest Cameroon constitutes an interesting case study with which to disentangle the effect of various factors on the spatial genetic structure of a plant pathogen. BLSD is a recent worldwide pandemic on banana and plantains that spread originally from Southeast Asia (Jones, 2000). This very devastating foliar disease is caused by the ascomycete fungus Mycosphaerella fijiensis that spreads by three modes: (i) the movement of infected plant material and the dispersal of spores produced through either (ii) sexual (ascospores) or (iii) asexual (conidia) reproduction (Robert et al., 2012). The expansion of this disease in Cameroon is recent as it was reported for the first time in 1980 from neighboring Gabon (Fouré and Lescot, 1988). On the other hand, the agricultural landscape in this part of Cameroon is characterized by the co-existence and geographical proximity of large commercial banana plantations and many food-crop plots of dessert bananas and plantains distributed continuously as small fields or backyard gardens. In commercial banana plantations, BLSD is controlled mainly through two cultural practices (De Lapeyre de Bellaire et al., 2010). First, multisite protectant fungicides preventing pathogen penetration into the leaves are applied weekly. These fungicides have a multisite mode of action and resistance has not been detected to date. Second, the necrotic stages where ascospores are produced before being released into the air are eliminated physically. Conversely, in food-crop plots, plantain and dessert bananas are grown extensively with no control of BLSD and, particularly, without fungicide application. Furthermore, the agricultural landscape in this area is also characterized by the presence of large commercial plantations of rubber and oil palm, where the density of banana and plantain (the hosts of M. fijiensis) is very low. Such an heterogeneous agricultural landscape is rare and provides an interesting opportunity to study the effect of cultural practices on the connectivity of a pathogenic fungus.
In this Cameroonian area, only two studies have examined the spatial genetic structure of M. fijiensis populations sampled from food-crop bananas. The first detected a pattern of isolation by distance (IBD) along a 300-km-long transect, suggesting a continuous range expansion of the disease at the regional scale through the natural and gradual dispersal of spores (Halkett et al., 2010). However, a break in allelic frequencies was detected in the middle of this transect. A second spatial analysis of population structure was therefore conducted recently within a 30 × 40-km area including the region of this genetic discontinuity (Rieux et al., 2011). This latter study allowed the spatial mapping of this sharp genetic discontinuity but did not reveal any influence of landscape features such as a high mountain, natural forest or a broad river, on this spatial genetic structure. The genetic discontinuity was better explained as a relict of the recent history of M. fijiensis expansion in the country (for example, a stratified dispersal combination scenario) rather than to have resulted from contemporary gene flow. These studies raised questions regarding the frequency of occurrence of such phenomena at the spatial scale considered. Furthermore, because the studied area did not contain any commercial banana plantations, the potential effect of cultural practises like fungicide treatments on the spatial genetic structure could not be evaluated.
In the present study, we focused on the heterogeneity existing within an agricultural landscape. We asked whether a major ecological constraint induced by specific cultural practices (intensive fungicide use and regular elimination of ascospores sources) used for efficient pathogen population control could affect the neutral spatial genetic structure of M. fijiensis. To achieve this objective, we analyzed the spatial genetic structure of M. fijiensis populations sampled within the so called ‘Mungo' production area in Cameroon, a 50 × 80-km area with several large commercial banana plantations (6000 Ha) surrounded by food-crop banana and plantains. Other large commercial rubber and oil palm plantations in which banana plants are practically nonexistent are present throughout the study site. Such ‘low host density areas' might also influence the spatial genetic structure of M. fijiensis populations. Because the disease appeared recently (1983) in this area and spread rapidly to all banana plants of the region, a prior analysis of the spatial genetic structure was conducted at this larger scale to detect possible genetic patterns resulting from range expansion of M. fijiensis. To further test the potential effect of agricultural practices, the distribution of genetic diversity was analyzed more precisely by comparing populations sampled inside a commercial banana plantation with those sampled in neighboring banana and plantain food-crops.
Materials and methods
Study site and isolate sampling
All isolates were collected in Southwest Cameroon in January 2009 using two independent strategies. First, we performed a ‘2D' sampling at a global scale of 50 × 80 km (that is, 40.106 Ha) in the Mungo area (see Figure 1). We collected five lesions on each of 150 untreated plantain banana plants sampled. Plants were sampled every 3.5 km along all accessible roads of the study site to construct the best bi-dimensional representation of the studied area. This area is situated just below the area sampled in 2006 (Rieux et al., 2011) and is ‘fragmented' by two potential natural barriers to gene flow: (i) various commercial banana plantations in which BLSD is controlled through the weekly application of multisite fungicides and the regular elimination of necrotic stages (ascospores are rarely produced in such areas), and (ii) large areas characterized by low host density (commercial plantations of palm oil or rubber) (Figure 1). The sampled lesions (stage 2, for more details, see Robert et al., 2010) were dried for 1 day at 55 °C and stored at –20 °C before genotyping. A second specific sampling was carried out at the local scale of a 1500-Ha single commercial banana plantation (Figure 1). We sampled 50 lesions on each of 12 plants. Six of these plants (Ins1–Ins6) were situated inside the commercial banana plantation, and six (Out1–Out6) were outside the commercial banana plantation (food-crop banana and plantain plantations). The distance between each sampled plant and the closest border of the commercial plantation was between 1 and 2 km in each case.
Figure 1.

Representation of the 50 × 80 km studied area localized in the Mungo area (region center of Cameroon) showing sampled sites. Left: distribution of the ‘2D' sampling sites (food-crop untreated plots) at a global scale (black dots) and of the sampling sites at the local scale of a single commercial plantation (black crosses). Right: enlargement of local scale sampling area.
Molecular analysis
Total genomic DNA was extracted directly from sampled lesions using the protocol described by Robert et al. (2010). Genotyping was carried out using four PCR multiplex sets comprising 20 microsatellite loci (Zapater et al., 2008; Robert et al., 2010). PCR reaction conditions are also detailed in Robert et al. (2010). Among the 20 markers, 10 were tetranucleotide, 2 were trinucleotide and 8 were dinucleotide microsatellite loci (Table 1). Finally, the amplified fragments were separated on a 16 capillary ABI Prism 3130 XL sequencer (Applied Biosystems, Foster City, CA, USA) and analyzed for length variation using GeneMapper Software (Applied Biosystems). We genotyped 32 lesions/plant for the local sampling (a total of 32 × 12=384 lesions genotyped) and 2 lesions/plant for the ‘2D' sampling (a total of 2 × 150=300 lesions genotyped). Any single lesion can be considered as one single isolate.
Table 1. Summary of basic information and global population genetics results obtained both at the global (2D) and local (commercial plantation) scales.
| Locus | Repeats | Global scale (N=300) | Local scale (N=378) | ||
|---|---|---|---|---|---|
| |
|
H
E
|
A
R
|
H
E
|
A
R
|
| MfSSR401(2) | 4 | 0.24 | 3 | 0.35 | 4 |
| MfSSR403(4) | 4 | 0.05 | 7 | 0.07 | 7 |
| MfSSR405(2) | 4 | 0.49 | 2 | 0.48 | 3 |
| MfSSR407(2) | 4 | 0.48 | 2 | 0.49 | 5 |
| MfSSR413(1) | 4 | 0.45 | 2 | 0.46 | 3 |
| MfSSR417(3) | 4 | 0.48 | 2 | 0.50 | 3 |
| MfSSR424(3) | 4 | 0.50 | 3 | 0.49 | 2 |
| MfSSR425(3) | 4 | 0.01 | 2 | 0.01 | 2 |
| MfSSR428(4) | 4 | 0.39 | 2 | 0.33 | 3 |
| MfSSR434(4) | 4 | 0.49 | 2 | 0.49 | 3 |
| MfSSR326(3) | 2 | 0.13 | 4 | 0.06 | 3 |
| MfSSR340(1) | 2 | 0.46 | 3 | 0.42 | 2 |
| MfSSR305b(2) | 2 | 0.47 | 2 | 0.46 | 2 |
| MfSSR322(1) | 2 | 0.61 | 3 | 0.56 | 3 |
| MfSSR324(1) | 2 | 0.44 | 5 | 0.30 | 6 |
| MfSSR350b(2) | 2 | 0.11 | 4 | 0.03 | 3 |
| MfSSR061(3) | 2 | 0.03 | 3 | 0.01 | 3 |
| MfSSR137(4) | 2 | 0.52 | 8 | 0.43 | 7 |
| MfSSR194b(2) | 3 | 0.36 | 3 | 0.46 | 4 |
| MfSSR203(1) | 3 | 0.57 | 4 | 0.54 | 3 |
| All loci | 0.36 | 3.28 | 0.35 | 3.55 | |
Abbreviations: AR, allelic richness are computed among the different loci; HE, unbiased estimate of gene diversity; N, sample size.
Repeats indicate the number of nucleotides corresponding to the repeat unit for each locus. The numbers in brackets indicate the multiplex panel to which each amplified locus belongs.
Analysis of genetic data
Basic population genetic analysis
Population gene diversity was estimated from the expected heterozygosity (HE, unbiased estimate calculated following Nei (1978)) and the allelic richness (AR). These two indices were calculated using FSTAT 2.9.3 (Goudet, 1995). We also used the rarefaction procedure implemented in the program HP-rare (Kalinowski, 2005) to estimate private allelic richness (PAR). The PAR of a population is the observed number of alleles that are detected in one population but absent in others. We calculated this index for each of the 12 populations sampled at the local scale (Ins1...Ins6 and Out1...Out6). In this case, the number of private alleles was calculated by comparing each population sampled inside or outside the commercial plantation with the six others belonging to the other group (inside or outside). We also calculated the PAR index by pooling together each population sampled inside vs outside the commercial plantation. The genotypic diversity, defined as the probability that two isolates taken at random have different multilocus genotypes, was measured using Multilocus (Agapow and Burt, 2001). We used the non-parametric Wilcoxon–Mann–Whitney test to look for significant differences in HE, AR, PAR and genotypic diversity between the populations sampled inside and outside the commercial plantation. Linkage disequilibrium between all pairs of loci was tested using Fisher's exact tests implemented in Genepop run on the web (http://genepop.curtin.edu.au/) (Rousset, 2008); (Markov chain parameters: dememorization number=2000, number of batches=250, number of iterations per batch=2000). We used the false discovery rate procedure (Benjamini and Hochberg, 1995) to control for multiple testing. The resulting adjusted P values are called Q values. This procedure is implemented in the R package QVALUE (Storey, 2002). We also calculated the multilocus linkage disequilibrium (r̂D) based on the index of association (Brown et al., 1980). This statistic is corrected for the number of loci. Departure from random association of alleles across loci was assessed by bootstrapping alleles among isolates 1000 times independently for each locus using Multilocus (Agapow and Burt, 2001).
Analysis of population genetic structure at the global scale
Clustering analysis: We carried out Bayesian population structure analysis on the ‘2D' sampling data set using Structure v2.2 (Pritchard et al., 2000) and Geneland v3.1.4 (Guillot et al., 2005). The base units of these methods are individual multilocus genotypes. In these Bayesian methods, the data is subdivided into clusters by minimizing Hardy–Weinberg disequilibrium and gametic phase disequilibrium between loci within groups. Structure bases its inference on genetic data only, whereas Geneland takes into account explicitly spatial localization information for genotyped individuals. We followed the general recommendations of the authors of these methods to run the programs. Using Structure, we tested all values from K=1 to K=10 with a Markov chain MonteCarlo of 600 000 iterations following a burn-in of 60 000 iterations. Other parameters were set to default values (admixture model and correlated allele frequencies). For each K, we calculated the mean posterior probability over its runs, and we used this value to estimate the posterior probability of each K using the formula given by Pritchard and Wen (2003). Five independent runs were performed to check for the consistency of Markov chain convergence. Geneland analyses were conducted using the correlated model of allele frequencies and long Markov chain MonteCarlo runs to ensure convergence of the chain. We performed five independent runs with 1 000 000 iterations (thinning=100) allowing K to vary from 1 to 10. The maximum rate of the Poisson process was fixed at 500, the maximum number of nuclei in the Poisson–Voronoi tessellation at 300 and the potential error for spatial coordinates at 2 m. K was inferred from the modal value of the run highest likelihood. IBD was assessed using an ad hoc haploid version of the ‘IBD between individuals' suboption implemented in Genepop (version 4) (Rousset, 2008, see also Rieux et al., 2011 for more details on the haploid version). The Isolde procedure tests the correlation between genetic distance (ar) and the logarithm of geographical distance (as suggested for 2D populations). The significance of the regression slopes was evaluated from Mantel tests performed across 100 000 permutations. IBD was tested on the entire data set.
Evidence of recent population expansion
We used Bottleneck v. 1.2 (Cornuet and Luikart, 1996; Piry et al., 1999) to determine if there was an excess (expected in recently bottlenecked populations) or deficit (expected in the case of recent population expansion) in genetic diversity (H) relative to the number of alleles present in the population that includes all untreated strains sampled in 2D. The probability distribution was established using 1000 simulations. We used the Wilcoxon significance test to determine whether loci displayed a significant excess of H>HEQ (genetic diversity expected under mutation drift equilibrium) or deficit in gene diversity under a mutation drift equilibrium for loci evolving under the two-phase mutation model. Such a model is expected to best fit with microsatellite evolution (DiRienzo et al., 1994) and was parameterized using 70% of SMM (Stepwise Mutation Model) and 30% of IAM (Infinite Alleles Model).
Spatial genetic structure of populations sampled at the local scale
The existence of genetic structure between populations sampled inside and outside the commercial plantation was tested using analysis of molecular variance (AMOVA) (Excoffier et al., 1992) and a locus-by-locus AMOVA framework, as executed in Arlequin software. Total variance in gene frequencies was partitioned among groups (inside vs outside), among populations within groups and within populations by carrying out a hierarchical AMOVA on Euclidean pairwise distances among isolates. The significance of the fixation indices calculated at each level of analysis is tested using a non-parametric permutation approach described in Excoffier et al. (1992). Additionally, the level of genetic differentiation at microsatellite loci among populations and groups was estimated as FST (Weir and Cockerham, 1984) using Genepop run on the web. We used Fisher's exact test for population differentiation implemented in Genepop run on the web (Markov chain parameters: dememorization number=2000, number of batches=250, number of iterations per batch=2000) to test for significant values of FST between populations and groups.
Genotype assignments at the local scale
Geneclass 2.0 (Piry et al., 2004) software was used to generate the probability of exclusion of genotypes sampled in commercial plantations from food-crop plantation populations. The probability of isolates being excluded from populations was calculated using the standard criterion described by Rannala and Mountain (1997) and by simulating 1000 individuals per group of samples using the method of Paetkau et al. (2004).
Estimation of population sizes
Inoculum densities for the two types of propagules (ascospores and conidia) were estimated inside (site Ins2) and outside (site Out2) the commercial farm. Twenty banana plants were selected randomly at each site for these assessments.
Estimation of the number of ascospores per plant
For all 20 banana trees at each site, the number of leaves (n), their surface(s) and the severity index (SIj, the % of necrotic surface, see Stover and Dickson (1970)) of each leaf, j, were measured to calculate the necrotic surface of each banana tree (NSi):
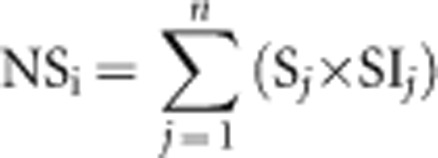 |
The average number of ascospores/banana tree (Nasc) was then estimated from its necrotic surface NSi (in cm2) according to the equation given by Burt et al. (1999):
Finally, we computed average values of Nasc among the different banana trees sampled both inside and outside the commercial plantation.
Estimation of the number of conidia per plant
For all 20 banana trees at each site, the number of lesions bearing conidia (that is, from stages 2, 3 and 4; Fouré, 1987) on each leaf were counted independently to determine the number of lesions at different stages present. From this number, we computed an average value of the number of lesions from the different stages among the 20 banana trees at each site. Then, for each leaf, a lesion of each stage (2, 3 and 4) was sampled (when present) and the lower face of each lesion was applied successively 10 times to the surface of an artificial medium in a Petri dish (20 g agar L-1) to remove all conidia from the lesion. The total number of conidia per lesion was counted under a microscope 24 h later to estimate the number of conidia produced by lesions of stages 2, 3 and 4. This procedure allowed us to calculate the expected total number of conidia produced by lesions at different stages among the 20 banana trees at each site, and therefore to estimate the average number of conidia on each plant at both sites.
Results
Genetic analysis at the global scale
Genetic diversity and linkage disequilibrium
Gene diversity for each locus ranged from 0.01 (locus MfSSR425) to 0.61 (locus MfSSR322), with an average value of 0.36, and mean AR ranged between 2 and 7 alleles/locus (3.20 on average) (Table 1). Genotypic diversity was very high (0.9999) (only two isolates out of 300 had the same multilocus genotype). We observed low (r̂D=0.002) and not significant values (P=0.211) of multilocus estimates of linkage disequilibrium. No pairs of loci showed significant linkage disequilibrium after false discovery rate control.
Spatial genetic structure
The results of Bayesian population structure analyses did not reveal any significant genetic differentiation across the study area. The estimated logarithm of likelihood for data analyzed with Structure was highest for K=1, and the posterior probability of having one population was equal to 1 (Table 2). For K>1, none of the 300 isolates could be assigned to a given cluster, as each isolate had a similar probability of belonging to each cluster. In Geneland, the mean posterior density distribution (in %) of the number of clusters estimated over the five replicates was higher for K=1. Second, we found no significant correlation between geographical and genetic distance (slope of regression b=0.005, confidence interval (–0.008; 0.018), P value=0.267).
Table 2. Results of individual-based Bayesian clustering analyses computed from K=1 to K=10.
| K |
Structure
a
|
Geneland b | |
| |
Mean lnP(D)
|
Pr(K|X) |
% Iterations (K) |
| 1 | −3712.5 | 1 | 58.48 |
| 2 | −3908.9 | 2.0E-113 | 15.75 |
| 3 | −3971.9 | 7.6E-206 | 9.28 |
| 4 | −4184.8 | 9.5E-238 | 4.04 |
| 5 | −5085.7 | 0 | 1.71 |
| 6 | −5742.5 | 0 | 0.49 |
| 7 | −6595.7 | 0 | 0.19 |
| 8 | −5215.3 | 0 | 0.06 |
| 9 | −4864 | 0 | 0 |
| 10 | −5000 | 0 | 9.91 |
Bold values correspond to the best K obtained in each analysis.
Structure analyses: mean values of likelihood (mean lnP(D)) over the 10 repetitions and posterior probability of having K clusters (Pr(K|X)).
Geneland analyses: mean posterior density distribution (in %) of the number of clusters estimated over the five replicates.
Evidence of recent population expansion
The possibility of population bottlenecks or expansions was tested using the program Bottleneck on the population that includes all untreated strains sampled in 2D. In the tested population, 13 loci out of 20 displayed a deficit in genetic diversity, and the multilocus probability of exhibing a deficiency in heterozygosity appeared to be significant (one tail Wilcoxon's test P-value=0.04174). This result is consistent with the recent history of M. fijiensis population expansion over the study area.
Genetic structure of populations sampled at the local scale
Genetic diversity and linkage disequilibrium
First, we calculated indices by pooling all populations together. In this case, gene diversity and mean AR were very similar to values obtained at the global scale (Table 1). Genotypic diversity was very high (0.9999), indicating that very few isolates (only 3 out of 384) had the same multilocus genotype. We observed low (r̂D=0.001) and not significant values (P=0.116) of multilocus estimates of linkage disequilibrium.
Second, we carried out the same calculations on each population independently (Table 3). We found no significant differences in HE (P=0.798) between populations sampled inside (commercial plantation sites) and outside (food-crop plantations sites) the commercial plantation. However, we detected a significantly higher value of multilocus AR (3.30>2.65; P=0.031) and PAR (18>5; P=0.006) in commercial plantation sites than in food-crop plantations. Importantly, all private alleles were present at very low frequencies (<0.05%, not shown). Genotypic diversity was very high in both commercial plantation (0.9998) and food-crop plantation (0.9999) plots—no significant difference between these populations was detected (P=0.987). Only two pairs of loci showed significant linkage disequilibrium after false discovery rate control, and this signal was detected only in two commercial plantation populations: MfSSR401 and MfSSR324 (population Ins5), and MfSSR401 and MfSSR407 (population Ins2).
Table 3. Summary of genetic diversity indices.
| Locus |
Food-crop plantations populations
|
Commercial plantation populations
|
Pooled populations
|
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Out1 | Out2 | Out3 | Out4 | Out5 | Out6 | Ins1 | Ins2 | Ins3 | Ins4 | Ins5 | Ins6 | Out | Ins | ||
| Heterozygosity | |||||||||||||||
| MfSSR401 | 0.452 | 0.186 | 0.061 | 0.246 | 0.473 | 0.226 | 0.349 | 0.39 | 0.409 | 0.398 | 0.437 | 0.422 | 0.293 | 0.394 | |
| MfSSR403 | 0.135 | 0 | 0.061 | 0 | 0.127 | 0.063 | 0.065 | 0.181 | 0 | 0 | 0 | 0.171 | 0.063 | 0.071 | |
| MfSSR405 | 0.515 | 0.514 | 0.409 | 0.443 | 0.508 | 0.37 | 0.516 | 0.516 | 0.522 | 0.396 | 0.494 | 0.458 | 0.466 | 0.499 | |
| MfSSR407 | 0.452 | 0.514 | 0.458 | 0.452 | 0.516 | 0.484 | 0.414 | 0.61 | 0.43 | 0.576 | 0.512 | 0.486 | 0.48 | 0.503 | |
| MfSSR413 | 0.396 | 0.426 | 0.498 | 0.473 | 0.508 | 0.49 | 0.503 | 0.417 | 0.486 | 0.452 | 0.428 | 0.484 | 0.46 | 0.454 | |
| MfSSR417 | 0.517 | 0.515 | 0.503 | 0.512 | 0.514 | 0.492 | 0.516 | 0.387 | 0.549 | 0.517 | 0.481 | 0.514 | 0.5 | 0.498 | |
| MfSSR424 | 0.49 | 0.49 | 0.515 | 0.514 | 0.515 | 0.514 | 0.473 | 0.514 | 0.484 | 0.49 | 0.514 | 0.428 | 0.496 | 0.494 | |
| MfSSR425 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0.057 | 0 | 0 | 0.01 | |
| MfSSR428 | 0.471 | 0.315 | 0.22 | 0.315 | 0.287 | 0.272 | 0.405 | 0.452 | 0.125 | 0.37 | 0.363 | 0.371 | 0.311 | 0.351 | |
| MfSSR434 | 0.512 | 0.508 | 0.511 | 0.466 | 0.473 | 0.484 | 0.503 | 0.433 | 0.444 | 0.452 | 0.481 | 0.499 | 0.484 | 0.497 | |
| MfSSR326 | 0.127 | 0.063 | 0 | 0 | 0 | 0.063 | 0 | 0.121 | 0.121 | 0.125 | 0.057 | 0 | 0.041 | 0.07 | |
| MfSSR340 | 0.488 | 0.434 | 0.361 | 0.49 | 0.503 | 0.476 | 0.508 | 0.331 | 0.452 | 0.331 | 0.254 | 0.379 | 0.454 | 0.385 | |
| MfSSR305b | 0.512 | 0.452 | 0.508 | 0.503 | 0.434 | 0.473 | 0.488 | 0.515 | 0.497 | 0.409 | 0.426 | 0.379 | 0.475 | 0.453 | |
| MfSSR322 | 0.637 | 0.6 | 0.492 | 0.571 | 0.471 | 0.514 | 0.602 | 0.623 | 0.579 | 0.568 | 0.541 | 0.528 | 0.545 | 0.568 | |
| MfSSR324 | 0.323 | 0.121 | 0.226 | 0.401 | 0.186 | 0.466 | 0.323 | 0.369 | 0.246 | 0.36 | 0.358 | 0.232 | 0.298 | 0.309 | |
| MfSSR350b | 0.067 | 0.063 | 0 | 0 | 0 | 0 | 0.065 | 0.125 | 0 | 0 | 0.057 | 0 | 0.021 | 0.041 | |
| MfSSR061 | 0 | 0 | 0.063 | 0 | 0 | 0 | 0 | 0.063 | 0 | 0 | 0 | 0 | 0.011 | 0.01 | |
| MfSSR137 | 0.561 | 0.448 | 0.55 | 0.236 | 0.31 | 0.609 | 0.52 | 0.391 | 0.179 | 0.391 | 0.477 | 0.494 | 0.456 | 0.411 | |
| MfSSR194b | 0.563 | 0.444 | 0.436 | 0.444 | 0.374 | 0.323 | 0.426 | 0.486 | 0.498 | 0.512 | 0.477 | 0.49 | 0.431 | 0.479 | |
| MfSSR203 | 0.516 | 0.605 | 0.558 | 0.563 | 0.516 | 0.577 | 0.54 | 0.587 | 0.556 | 0.497 | 0.54 | 0.528 | 0.549 | 0.53 | |
| N | 31 | 31 | 32 | 31 | 30 | 31 | 31 | 32 | 32 | 31 | 34 | 32 | 186 | 192 | |
| Mean | 0.39 | 0.33 | 0.32 | 0.33 | 0.34 | 0.34 | 0.36 | 0.38 | 0.33 | 0.34 | 0.35 | 0.34 | 0.342 | 0.351 | NS |
| Allelic richness | |||||||||||||||
| MfSSR401 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 4 | 3 | 3 | 3 | 3 | 2 | 4 | |
| MfSSR403 | 3 | 1 | 2 | 1 | 3 | 2 | 2 | 4 | 1 | 1 | 1 | 4 | 6 | 5 | |
| MfSSR405 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 3 | 2 | 2 | 2 | 2 | 3 | |
| MfSSR407 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 4 | 4 | 3 | 3 | 3 | 2 | 5 | |
| MfSSR413 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 3 | 2 | 2 | 2 | 2 | 3 | |
| MfSSR417 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 3 | 2 | 2 | 2 | 2 | 3 | |
| MfSSR424 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | |
| MfSSR425 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 2 | 1 | 1 | 2 | |
| MfSSR428 | 3 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 3 | 2 | |
| MfSSR434 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 3 | 2 | 2 | 2 | 2 | 2 | 3 | |
| MfSSR326 | 3 | 2 | 1 | 1 | 1 | 2 | 1 | 2 | 2 | 2 | 2 | 1 | 3 | 3 | |
| MfSSR340 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | |
| MfSSR305b | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | |
| MfSSR322 | 3 | 3 | 2 | 3 | 3 | 3 | 3 | 3 | 3 | 3 | 3 | 3 | 3 | 3 | |
| MfSSR324 | 2 | 2 | 2 | 3 | 2 | 2 | 2 | 5 | 3 | 3 | 3 | 3 | 3 | 5 | |
| MfSSR350b | 2 | 2 | 1 | 1 | 1 | 1 | 2 | 2 | 1 | 1 | 2 | 1 | 2 | 3 | |
| MfSSR061 | 1 | 1 | 2 | 1 | 1 | 1 | 1 | 2 | 1 | 1 | 1 | 1 | 2 | 2 | |
| MfSSR137 | 5 | 4 | 5 | 4 | 3 | 6 | 5 | 4 | 3 | 4 | 6 | 6 | 6 | 7 | |
| MfSSR194b | 3 | 2 | 2 | 2 | 3 | 2 | 2 | 3 | 2 | 2 | 3 | 3 | 3 | 4 | |
| MfSSR203 | 2 | 3 | 3 | 3 | 2 | 3 | 3 | 3 | 3 | 2 | 3 | 3 | 3 | 3 | |
| N | 31 | 31 | 32 | 31 | 30 | 31 | 31 | 32 | 32 | 31 | 34 | 32 | 186 | 192 | |
| Mean | 2.30 | 2.05 | 2.05 | 2.00 | 2.00 | 2.15 | 2.10 | 2.70 | 2.30 | 2.10 | 2.40 | 2.40 | 2.650 | 3.300 | * |
| Private allelic richness | |||||||||||||||
| MfSSR401 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 1 | 1 | 1 | 1 | 0 | 2 | |
| MfSSR403 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 2 | 1 | |
| MfSSR405 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 1 | |
| MfSSR407 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 2 | 1 | 1 | 1 | 0 | 3 | |
| MfSSR413 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 1 | |
| MfSSR417 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 1 | |
| MfSSR424 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| MfSSR425 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 1 | |
| MfSSR428 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | |
| MfSSR434 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 1 | |
| MfSSR326 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| MfSSR340 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| MfSSR305b | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| MfSSR322 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| MfSSR324 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 3 | 1 | 1 | 1 | 1 | 1 | 3 | |
| MfSSR350b | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 1 | |
| MfSSR061 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 1 | 1 | |
| MfSSR137 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 1 | |
| MfSSR194b | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | |
| MfSSR203 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| ∑ | 3 | 0 | 1 | 1 | 0 | 0 | 2 | 12 | 7 | 3 | 4 | 5 | 5 | 18 | |
| Mean | 0.16 | 0.00 | 0.05 | 0.05 | 0.00 | 0.00 | 0.10 | 0.62 | 0.35 | 0.16 | 0.20 | 0.25 | 0.26 | 0.94 | ** |
| N | 31 | 31 | 32 | 31 | 30 | 31 | 31 | 32 | 32 | 31 | 34 | 32 | 186 | 192 | |
Summary of genetic diversity indices calculated on (i) all populations independently and (ii) pools of food-crop (Out) vs commercial (Ins) plantation populations. Significance of the differences between treated and untreated populations calculated using Wilcoxon–Mann–Whitney test are computed among the different loci: (NS P>0.05 *P<0.05; **P<0.01).
Population genetic structure
The AMOVA indicated that most of the total genetic variation (99% P=0.027) was within populations (Table 4) but the analysis did not indicate any significant effect of either ‘populations within group' (0.21% P=0.237) or ‘group' (0.78% P=0.185), with ‘group' including populations sharing the same cultural practices (that is, commercial vs food-crop). FST values calculated with Genepop were consistent with AMOVA results. Indeed, multilocus values of FST were weak and not significant between groups and between each population (Table 5).
Table 4. Hierarchical AMOVA based on nuclear microsatellite DNA.
|
Source of variation
|
df
|
Sum of squares
|
Variance component
|
Percentage of variation
|
Fixation index (F)
|
P value |
| Among groups | 1 | 8.969 | 0.02734 Va | 0.78 | 0.00784 | 0.0045 |
| Among populations within groups | 10 | 36.933 | 0.00744 Vb | 0.21 | 0.00215 | 0.2375 |
| Within populations | 374 | 1291.844 | 3.45413 Vc | 99 | 0.00997 | 0.028 |
Abbreviation: AMOVA, analysis of molecular variance.
Percentage of variation explained by different source of variation (with groups reflecting the ‘inside vs outside the commercial plantation' populations partition), variance components and F statistics.
Table 5. Pairwise values of genetic differentiation (F ST) between food-crop (Out) vs commercial (Ins) plantation populations.
| Locus | FST | |
|---|---|---|
| MfSSR401 | 0.010 | ** |
| MfSSR403 | −0.004 | NS |
| MfSSR405 | 0.005 | NS |
| MfSSR407 | 0.005 | ** |
| MfSSR413 | −0.005 | NS |
| MfSSR417 | −0.004 | NS |
| MfSSR424 | −0.005 | NS |
| MfSSR425 | 0.000 | NS |
| MfSSR428 | −0.001 | NS |
| MfSSR434 | −0.004 | NS |
| MfSSR326 | −0.002 | NS |
| MfSSR340 | 0.012 | NS |
| MfSSR305b | −0.002 | NS |
| MfSSR322 | −0.004 | NS |
| MfSSR324 | 0.002 | * |
| MfSSR350b | 0.001 | NS |
| MfSSR061 | −0.003 | NS |
| MfSSR137 | −0.002 | NS |
| MfSSR194b | 0.001 | NS |
| MfSSR203 | −0.003 | NS |
| All loci | 0.0001 | NS |
Fisher's exact test: NS P>0.05; *P<0.05; **P<0.01.
Genotype assignment at the local scale
Considering a probability threshold of exclusion of <0.1, Geneclass indicated that a food-crop plantation population origin can be excluded in 112 out of 210 (53%) genotypes from the commercial population. When looking specifically at these 112 genotypes, we observed that 95% of them contain at least one rare (often private) allele. Among the remaining genotypes (that is, not excluded), 92 (43%) displayed a poor assignment probability (that is, <0.7) and only 6 (0.03%) displayed a good assignment probability (that is, >0.7) to a food-crop plantation population origin.
Estimation of population sizes in both commercial and food-crop sites
As expected, populations were very much larger in the food-crop site than in the commercial site (Table 6). In particular, in commercial plantations, the area of necrotic surface per plant (source of ascospores) and the number of young lesions per plant (stage 2 to 4, sources of conidia) was very low as a result of fungicide treatment and regular mechanical elimination of necrotic surfaces. Consequently, the estimated number of ascospores per plant was more than 300 000-fold higher in the food-crop site than in the commercial site (Table 6). The difference in the estimated amount of conidia between the two sites was less as the number of conidia per plant was 16-fold higher in the food-crop site than in the commercial site (Table 6).
Table 6. Average value and standard deviation for the estimation of M. fijiensis population sizes in both the food-crop plantations (site Out2) and the commercial plantation (site Ins2) populations.
| Number of lesions per plant | Necrotic surface per plant (m2) | Number of conidia per plant | Number of ascospores per plant | |
|---|---|---|---|---|
| Food-crop plantations | 5845±2637 | 5.3±2.3 | 84 172±40 493 | 1.6 × 108±8.1 × 107 |
| Commercial plantation | 164±124 | 0.00008±0.0002 | 5214±5456 | 489±950 |
Discussion
In a previous study aimed at assessing the influence of natural barriers on the spatial genetic structure of M. fijiensis populations in Cameroon, Rieux et al. (2011) detected a sharp break in allelic frequencies over the area studied. As the position of this genetic discontinuity was not correlated with any particular physical element, it was hypothesized that it might have been caused by a historical colonization event (for example, secondary contact between expanding populations). This would suggest that the historical signal caused by M. fijiensis range expansion in this area persists, and is still detectable today. In the present study, we investigated the spatial genetic structure of this plant pathogenic fungus over a slightly wider area (50 × 80 km) localized just south of that considered previously. Interestingly, this area encompassed new landscape features that could impact pathogen genetic diversity and structure. More specifically, instead of exhibiting natural and physical potential barriers to gene flow (that is, mountain, forest and others), this new area includes commercial banana plantations in which fungal populations are regularly reduced through fungicide applications, and other commercial rubber and oil palm plantations in which banana plants are practically nonexistent (low pathogen densities). To our knowledge, this is the first landscape genetics study in which the potential effect on gene flow of agricultural practices, including the use of chemicals, has been considered. In this context, it is important to note that considering commercial banana plantations (to test for either indirect barrier effects on food-crop plantation populations on a global scale or for direct effects on genetic diversity of commercial plantation populations on a local scale) does not allow us to test solely for an effect of fungicide. Indeed, commercial and food-producing banana plantations differ in several characteristics, including banana plant variety cultivated, plant density (higher in commercial areas), and the cultural practices for BLSD control used only in commercial plantations such as the intensive use of fungicides and the regular elimination of necrotic tissues producing ascospores (De Lapeyre de Bellaire et al., 2009). However, because all cultivated banana plants are fully sensitive to BLSD disease (Fouré et al., 1990), we hypothesized that these agronomic practices, which will influence population size and gene flow, might be the principal factors influencing the genetic diversity and structure of isolates in commercial banana plantations.
Homogeneous fungal population in an agricultural landscape following recent range expansion
Using two clustering algorithms on samples collected from untreated plants over the whole study area, we did not detect any spatial genetic structure in the M. fijiensis population. This absence of spatial genetic structure pattern can be informative regarding both (i) the dispersal processes that might have affected the spread of M. fijiensis over the area studied and (ii) the effects of potential barriers to gene flow.
M. fijiensis was first observed in 1983 in the Mungo banana-producing area (Fouré and Lescot, 1988), but there is no precise historical data indicating the spatial origin of this introduction and the direction of its expansion. In the current study, the characterization of a single homogeneous population suggests that colonization of the area probably arose from a single spatial introduction. Moreover, following recent advances in our understanding of the genetic consequences of range expansions (see Excoffier et al., 2009 for a review), we hypothesize two contrasting dispersal processes that could have effected M. fijiensis expansion over the study area. In the first, the absence of spatial genetic structure could result from the short-range dispersal of a large population. Indeed, it has been shown that such conditions limit gene surfing events and could lead to the emergence of homogeneous genetic sectors that can cover geographic zones as large as that studied here. Alternatively, it has also recently been demonstrated theoretically that frequent LDD events favor gene mixing and decrease the likelihood of a spatial break in allelic frequencies (due either to a stratified dispersal combination or to a gene surfing event) (Bialozyt et al., 2006; Fayard et al., 2009). Following such theoretical considerations, we argue that the second hypothesis, that is, that M. fijiensis colonized the Mungo area through the expansion of populations exhibiting frequent LDD events, is the most likely as it appears to be consistent with the biology of this fungus. Indeed, frequent long-distance (up to several kilometers) ascospore movement by wind dispersal has been suspected previously in this species (Burt et al., 1998; Amil et al., 2007). Thus, in contrast with our previously published work (Halkett et al., 2010; Rieux et al., 2011), we did not detect any genetic discontinuities over the area studied. However, this area exhibits some topographical differences in comparison to the areas previously considered. Indeed, the genetic discontinuity observed by Rieux et al., (2011) was mapped in a narrow valley, whereas the area considered in the present study appears uniformly flat (see Figure 1). This difference might be relevant because it has been shown theoretically that, during range expansion events of species exhibiting LDD, wide colonization corridors decreases the probability of observing a break in allelic frequencies (Fayard et al., 2009), which is consistent with the results obtained here.
The landscape elements present in the Mungo area also had no effect on the current spatial genetic structure of M. fijiensis populations. Two elements of the agricultural landscape are thought to have had an impact on M. fijiensis gene flow. The first is a ‘biological' barrier represented by wide areas in which host density is particularly weak. Such areas might decrease the connectivity between individuals at the global scale. The second element is represented by the four commercial banana plantations in the study site, which are thought to constrain gene movement because of BLSD control practices (plants are sprayed weekly with fungicides, and ascospores sources are eliminated regularly), expected to drastically reduce the probability of an individual being able to reproduce and disperse inside such areas. These ‘anthropical barriers' were already in place at the time of M. fijiensis colonization because another related pathogenic fungus (M. musicola) was already present in the area (Fouré and Lescot, 1988). Because of its high dispersal abilities, and because untreated plants are effectively distributed continuously (if scarcely) throughout the studied area, we hypothesize that M. fijiensis could easily by-pass ‘low host density' areas and commercial plantations during expansion.
BLSD control methods have no effect on M. fijiensis genetic diversity
We also asked whether BLSD control methods in commercial plantations, particularly the intensive use of fungicide, had any effect on current M. fijiensis genetic diversity and differentiation. We conducted a complementary analysis at the local scale of one particular banana commercial plantation, examining isolates sampled both within and outside the plantation. Combining an AMOVA with pairwise FST comparisons, we detected no significant influence of fungal control methods on genetic differentiation; however, we obtained contrasting results regarding their influence on genetic diversity. Indeed, although no significant difference in expected heterozygosity and genotypic diversity was found between the two groups, we observed a significantly higher number of alleles in the commercial plantation populations compared with the surrounding food-crop populations. This difference in allelic richness but not in heterozygosity can be explained by the excess of private alleles existing at very low frequencies observed in commercial plantation populations. We argue that this excess could be a consequence of the planting strategies adopted in commercial plantation. Due to fertility erosion in those monocultural agrosystems, commercial plots are effectively destroyed after 5–7 crop cycles and replanted with in vitro propagated banana plants. These in vitro plants are naturally contaminated with M. fijiensis during their bioacclimatization in nursery localities removed from the commercial plots. Private alleles present in nurseries might then be imported into commercial plantations during planting. The high probability of exclusion of genotypes from commercial plantations exhibiting rare/private alleles to the surroundings food-crop populations is consistent with this hypothesis.
Our findings contrast with the major decrease in genetic diversity observed previously for other pathogenic fungi in treated compared with untreated areas (Grunwald et al., 2006; Matasci et al., 2008). However, the fungal populations studied in the latter works were clonal, and directional selection at some loci certainly influenced the whole genome through selective sweep (Fisher, 1930). Such processes are not expected to occur in the present study as protectant fungicides have replaced systemic fungicides (which led to high levels of strain resistance) in recent years. No strains resistant to fungicides with a multisite mode action have been detected to date (De Lapeyre de Bellaire et al., 2010). Moreover, populations of M. fijiensis mate randomly at the scale considered, and present a low level of genetic disequilibrium due to recombination. Thus, the probability of detecting an indirect effect of selection on 20 neutral genetic markers chosen randomly is very low. Nevertheless, a loss of genetic diversity can also occur through random changes in allele frequency over the whole genome by genetic drift when population size decreases. This could have happened here; indeed, there was a drastic reduction in the size of the M. fijiensis population inside the commercial plantation due to fungal control methods. As no decrease in the expected heterozygosity and genotypic diversity was detected in the commercial area, we argue that the population size inside this area remains large enough to avoid the occurrence of a genetic drift effect. Alternatively, gene flow from the outside to the inside of the commercial plantation might continuously restore genetic diversity, as was suggested in the case of the mosquito Aedes rusticus when sites treated with bioinsecticides were compared with non-treated sites (Paris et al., 2010). The occurrence of sexual reproduction in M. fijiensis in commercial plantations is limited, mainly because of regular elimination of the necrotic tissues where sexual ascospores are produced. As a result, reproduction in commercial plantations is mainly via production of asexual spores (that is, conidia), which, in contrast to ascospores, are thought to disperse over very short distances (Gauhl, 1994). Consequently, we argue that an asymmetric migration rate from surrounding food-crop plantation to commercial plantation populations might exist due to ascospores dispersal.
Conclusion
In summary, we showed that the historical expansion of a plant pathogenic fungus in an agricultural landscape has resulted in the establishment of a single homogeneous population. This result suggests that colonization of the area probably arose from a single introduction, with subsequent expansion of populations through frequent LDD events. Landscape elements representing potential barriers to the spread of the pathogen had no effect on the spatial genetic structure. Moreover, we found no influence of agricultural control methods such as the intensive use of fungicides or the elimination of ascospores sources on neutral genetic diversity and spatial genetic structure. We hypothesize that large population size and/or high migration rate from the outside to the inside of the commercial plantation may have prevented the detection of any influence of genetic drift on genetic diversity and structure. Further work is needed to disentangle the relative contributions of both evolutionary forces. Finally, this study illustrates the fact that surrounding food-crop populations might have an effect on the evolution of fungicide resistance in commercial plantations and should be considered explicitly in theoretical models aiming at defining efficient management strategies.
Data Archiving
Data have been deposited at Dryad:doi:10.5061/dryad.64p8t.
Acknowledgments
We thank CARBAP and particularly Josué Essoh Ngando and Oscar Nguidjo for their technical and logistical support during sampling in Cameroon; PHP company for giving us the access to their banana plantation; Luc Pignolet for help with the molecular work; Renaud Vitalis, Thomas Lenormand, Etienne Klein, Denis Bourguet, Anne-Sophie Walker, Elisabeth Fournier, Didier Tharreau, Benois Barrès and Eric Bazin for their helpful discussions; three anonymous referees for useful comments on a previous version of this manuscript; Helen Rothnie for her attentive reading and careful language checking. Data used in this work were produced partly through the molecular genetic analysis technical facilities of the IFR119 ‘Montpellier Environnement Biodiversité'. Adrien Rieux was supported by a CIFRE doctoral fellowship from Bayer CropScience and the Agence Nationale de la Recherche et de la Technologie(ANRT-123/2008). This work was also supported by a Europaid contract ATF/UE- N°146-762/786/798/801.
The authors declare no conflict of interest.
References
- Agapow PM, Burt A. Indices of multilocus linkage disequilibrium. Mol Ecol Notes. 2001;1:101–102. [Google Scholar]
- Amil AF, Heaney SP, Stanger C, Shaw MW. Dynamics of QoI sensitivity in Mycosphaerella fijiensis in Costa Rica during 2000 to 2003. Phytopathology. 2007;97:1451–1457. doi: 10.1094/PHYTO-97-11-1451. [DOI] [PubMed] [Google Scholar]
- Anderson PK, Cunningham AA, Patel NG, Morales FJ, Epstein PR, Daszak P. Emerging infectious diseases of plants: pathogen pollution, climate change and agrotechnology drivers. Trend Ecol Evolut. 2004;19:535–544. doi: 10.1016/j.tree.2004.07.021. [DOI] [PubMed] [Google Scholar]
- Archie EA, Luikart G, Ezenwa VO. Infecting epidemiology with genetics: a new frontier in disease ecology. Trend Ecol Evolut. 2009;24:21–30. doi: 10.1016/j.tree.2008.08.008. [DOI] [PubMed] [Google Scholar]
- Balkenhol N, Gugerli F, Cushman SA, Waits LP, Coulon A, Arntzen JW, et al. Identifying future research needs in landscape genetics: where to from here. Landscape Ecol. 2009;24:455–463. [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J R Stat Soc Ser B Met. 1995;57:289–300. [Google Scholar]
- Bialozyt R, Ziegenhagen B, Petit RJ. Contrasting effects of long distance seed dispersal on genetic diversity during range expansion. J Evol Biol. 2006;19:12–20. doi: 10.1111/j.1420-9101.2005.00995.x. [DOI] [PubMed] [Google Scholar]
- Biek R, Real LA. The landscape genetics of infectious disease emergence and spread. Mol Ecol. 2010;19:3515–3531. doi: 10.1111/j.1365-294X.2010.04679.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown AHD, Feldman MW, Nevo E. Multilocus structure of natural populations of Hordeum spontaneum. Genetics. 1980;96:523–536. doi: 10.1093/genetics/96.2.523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JKM, Hovmoller MS. Epidemiology - Aerial dispersal of pathogens on the global and continental scales and its impact on plant disease. Science. 2002;297:537–541. doi: 10.1126/science.1072678. [DOI] [PubMed] [Google Scholar]
- Burt PJA, Rosenberg LJ, Rutter J, Ramirez F, Gonzales H. Forecasting the airborne spread of Mycosphaerella fijiensis, a cause of black Sigatoka disease on banana: estimations of numbers of perithecia and ascospores. Ann Appl Biol. 1999;135:369–377. [Google Scholar]
- Burt PJA, Rutter J, Ramirez F. Airborne spore loads and mesoscale dispersal of the fungal pathogens causing Sigatoka diseases in banana and plantain. Aerobiologia. 1998;14:209–214. [Google Scholar]
- Cornuet JM, Luikart G. Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genetics. 1996;144:2001–2014. doi: 10.1093/genetics/144.4.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Lapeyre de Bellaire L, Essoh Ngando J, Abadie C, Chabrier C, Blanco R, Lescot T, et al. Is chemical control of Mycosphaerella foliar diseases of bananas sustainable. Acta Hortic. 2009;828:161–170. [Google Scholar]
- De Lapeyre de Bellaire L, Fouré E, Abadie CJC, Carlier J. Black leaf streak disease is challenging the banana industry. Fruits. 2010;65:327–342. [Google Scholar]
- Dionne M, Caron F, Dodson JJ, Bernatchez L. Landscape genetics and hierarchical genetic structure in Atlantic salmon: the interaction of gene flow and local adaptation. Mol Ecol. 2008;17:2382–2396. doi: 10.1111/j.1365-294X.2008.03771.x. [DOI] [PubMed] [Google Scholar]
- DiRienzo A, Peterson AC, Garza JC, Valdes AM, Slatkin M, Freimer NB. Mutational processes of simple-sequence repeat loci in human populations. Proc Natl Acad Sci USA. 1994;91:3166–3170. doi: 10.1073/pnas.91.8.3166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Excoffier L, Foll M, Petit RJ. Genetic consequences of range expansions. Annu Rev Ecol Evol Syst. 2009;40:481–501. [Google Scholar]
- Excoffier L, Smouse PE, Quattro JM. Analysis of molecular variance from metric distances among DNA haplotypes: Application to human mitochondrial DNA restriction data. Genetics. 1992;131:479–491. doi: 10.1093/genetics/131.2.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fayard J, Klein EK, Lefevre F. Long distance dispersal and the fate of a gene from the colonization front. J Evol Biol. 2009;22:2171–2182. doi: 10.1111/j.1420-9101.2009.01832.x. [DOI] [PubMed] [Google Scholar]
- Fisher RA. The Genetical Theory of Natural Selection. Clarendon Press: Oxford; 1930. [Google Scholar]
- Fouré E.1987Varietal reactions of bananas and plantains to black leaf streak disease Banana and Plantain Breeding Strategies, Proceedings of an International WorkshopPersley GJaDL, E.A. ed)13–17.Cairns: Australia; 1986. [Google Scholar]
- Fouré E, Lescot T. Variabilité génétique des Mycosphaerella inféodés au genre Musa. Mise en évidence de la présence au Cameroun sur bananiers et plantains d'une cercosporiose (Mycosphaerella musicola) au comportement pathogène atypique. Fruits. 1988;43:407–415. [Google Scholar]
- Fouré E, Mouliom-Pefoura A, Mourichon X. Etude de la sensibilité variétale des bananiers et des plantains à M. fijiensis Morelet au Cameroun. Cractérisation de la résistance au champ de bananiers appartenant à divers groupes génétiques. Fruits. 1990;45:339–345. [Google Scholar]
- Gauhl F. Epidemilogy and Ecology of Black Sigatoka (PhD Thesis) INIBAP: Montpellier; 1994. p. 120. [Google Scholar]
- Goudet J. FSTAT (Version 1.2): A computer program to calculate F-statistics. J Hered. 1995;86:485–486. [Google Scholar]
- Grunwald NJ, Sturbaum AK, Montes GR, Serrano EG, Lozoya-Saldaña H, Fry WE. Selection for fungicide resistance within a growing season in field populations of Phytophthora infestans at the center of origin. Phytopathology. 2006;96:1397–1403. doi: 10.1094/PHYTO-96-1397. [DOI] [PubMed] [Google Scholar]
- Guillot G, Estoup A, Mortier F, Cosson JF. A spatial statistical model for landscape genetics. Genetics. 2005;170:1261–1280. doi: 10.1534/genetics.104.033803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halkett F, Coste D, Rivas Platero GG, Zapater MF, Abadie C, Carlier J. Isolation by distance and genetic discontinuities in recently established populations of the plant pathogenic fungus Mycosphaerella fijiensis. Mol Ecol. 2010;19:3909–3923. doi: 10.1111/j.1365-294X.2010.04774.x. [DOI] [PubMed] [Google Scholar]
- Hoffmann AA, Willi Y. Detecting genetic responses to environmental change. Nat Rev Genet. 2008;9:421–432. doi: 10.1038/nrg2339. [DOI] [PubMed] [Google Scholar]
- Holderegger R, Wagner HH. Landscape genetics. Bioscience. 2008;58:199–207. [Google Scholar]
- Jones DR. Diseases of Banana, Abaca and Enset. CABI Publishing: Oxon; 2000. [Google Scholar]
- Kalinowski ST. HP-RARE 1.0: a computer program for performing rarefaction on measures of allelic richness. Mol Ecol Notes. 2005;5:187–189. [Google Scholar]
- Manel S, Schwartz MK, Luikart G, Taberlet P. Landscape genetics: combining landscape ecology and population genetics. Trend Ecol Evolut. 2003;18:189–197. [Google Scholar]
- Matasci CL, Gobbin D, Scharer HJ, Tamm L, Gessler C. Selection for fungicide resistance throughout a growing season in populations of Plasmopara viticola. Eur J Plant Pathol. 2008;120:79–83. [Google Scholar]
- McRae BH, Beier P. Circuit theory predicts gene flow in plant and animal populations. Proc Natl Acad Sci USA. 2007;104:19885–19890. doi: 10.1073/pnas.0706568104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nei M. Estimation of average heterozygosity and genetic distance from a small number of individuals. Genetics. 1978;89:583–590. doi: 10.1093/genetics/89.3.583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols RA, Hewitt GM. The genetic consequences of long-distance dispersal during colonization. Heredity. 1994;72:312–317. [Google Scholar]
- Paetkau D, Slade R, Burden M, Estoup H. Genetic assignment methods for the direct, real-time estimation of migration rate: a simulation-based exploration of accuracy and power. Mol Ecol. 2004;13:55–65. doi: 10.1046/j.1365-294x.2004.02008.x. [DOI] [PubMed] [Google Scholar]
- Paris M, Boyer S, Bonin A, Collado A, David JP, Despres L. Genome scan in the mosquito Aedes rusticus: population structure and detection of positive selection after insecticide treatment. Mol Ecol. 2010;19:325–337. doi: 10.1111/j.1365-294X.2009.04437.x. [DOI] [PubMed] [Google Scholar]
- Parisod C, Bonvin G. Fine-scale genetic structure and marginal processes in an expanding population of Biscutella laevigata L. (Brassicaceae) Heredity. 2008;101:536–542. doi: 10.1038/hdy.2008.95. [DOI] [PubMed] [Google Scholar]
- Piry S, Alapetite A, Cornuet JM, Paetkau D, Baudouin L, Estoup A. GENECLASS2: a software for genetic assignment and first-generation migrant detection. J Hered. 2004;95:536–539. doi: 10.1093/jhered/esh074. [DOI] [PubMed] [Google Scholar]
- Piry S, Luikart G, Cornuet JM. Bottleneck: a computer program for detecting recent reductions in the effective population size using allele frequency data. J Hered. 1999;90:502–503. [Google Scholar]
- Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155:945–959. doi: 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard JK, Wen W.2003. Documentation for STRUCTURE Software, Ver. 2Department of Human Genetics, the University of Chicago [Google Scholar]
- Rannala B, Mountain JL. Detecting immigration by using multilocus genotypes. Proc Natl Acad Sci USA. 1997;94:9197–9201. doi: 10.1073/pnas.94.17.9197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieux A, Halkett F, de Lapeyre de Bellaire L, Zapater MF, Rousset F, Ravigne V, et al. Inferences on pathogenic fungus population structures from microsatellite data: new insights from spatial genetics approaches. Mol Ecol. 2011;20:1661–1674. doi: 10.1111/j.1365-294X.2011.05053.x. [DOI] [PubMed] [Google Scholar]
- Robert S, Rieux A, Argout X, Carlier J, Zapater MF. Optimized genotyping with microsatellite markers in the fungal banana pathogen Mycosphaerella Fijiensis. Am J Bot. 2010;97:e130–e132. doi: 10.3732/ajb.1000295. [DOI] [PubMed] [Google Scholar]
- Robert S, Ravigne V, Zapater MF, Abadie C, Carlier J. Contrasting introduction scenarios among continents in the worldwide invasion of the banana fungal pathogen Mycosphaerella fijiensis. Mol Ecol. 2012;21:1098–1114. doi: 10.1111/j.1365-294X.2011.05432.x. [DOI] [PubMed] [Google Scholar]
- Rousset F. Genepop'007: a complete re-implementation of the Genepop software for Windows and Linux. Mol Ecol Resour. 2008;8:103–106. doi: 10.1111/j.1471-8286.2007.01931.x. [DOI] [PubMed] [Google Scholar]
- Schmidt T, Arens P, Smulders MJM, Billeter R, Liira J, Augenstein I, et al. Effects of landscape structure on genetic diversity of Geum urbanum L. populations in agricultural landscapes. Flora. 2009;204:549–559. [Google Scholar]
- Sork VL, Waits L. Contributions of landscape genetics - approaches, insights and future potential. Mol Ecol. 2010;19:3489–3495. doi: 10.1111/j.1365-294X.2010.04786.x. [DOI] [PubMed] [Google Scholar]
- Storey JD. A direct approach to false discovery rates. J R Stat Soc Ser B Stat Met. 2002;64:479–498. [Google Scholar]
- Stover RH, Dickson JD. Leaf spot of bananas caused by Mycosphaerella musicola: methods of measuring spotting prevalence and severity. Trop Agric (Trinidad) 1970;47:289–302. [Google Scholar]
- Stukenbrock EH, McDonald BA. The origins of plant pathogens in agro-ecosystems. Annu Rev Phytopathol. 2008;46:75–100. doi: 10.1146/annurev.phyto.010708.154114. [DOI] [PubMed] [Google Scholar]
- Weir BS, Cockerham CC. Estimating F-statistics for the analysis of population structure. Evolution. 1984;38:1358–1370. doi: 10.1111/j.1558-5646.1984.tb05657.x. [DOI] [PubMed] [Google Scholar]
- Zapater MF, Duchemin M, Dussart JF, Coste D, Brottier P, Carlier J. Microsatellite markers for the fungal banana pathogens Mycosphaerella fijiensis, Mycosphaerella musicola and Mycosphaerella eumusae. Mol Ecol Resour. 2008;8:1121–1125. doi: 10.1111/j.1755-0998.2008.02180.x. [DOI] [PubMed] [Google Scholar]