

Abstract
Plasmodium falciparum thymidylate kinase (PfTMPK) is a key enzyme in pyrimidine nucleotide biosynthesis. 3-Trifluoromethyl-4-chloro-phenyl-urea-α-thymidine has been reported as an inhibitor of Mycobacterium tuberculosis TMPK (MtTMPK). Starting from this point, we designed, synthesized and evaluated a number of thymidine analogues as antimalarials. Both 5′-urea-α- and β-thymidine derivatives were moderate inhibitors of PfTMPK and furthermore showed moderate inhibition of parasite growth. The structure of several enzyme–inhibitor complexes provides a basis for improved inhibitor design. However, we found that certain 5′-urea-α-thymidine analogues had antimalarial activity where inhibition of PfTMPK is not the major mode of action. Optimization of this series resulted in a compound with potent antimalarial activity (EC50 = 28 nM; CC50 = 29 μM).
Introduction
There are approximately 300–500 million clinical cases of malaria each year.1−5 While there is significant progress in development of vaccines, they are still on trial and not available,3 so chemotherapy remains the mainstay for dealing with this enormous problem. Furthermore, owing to problems with resistance, there is a need for new drugs to treat the disease.
Analysis of the Plasmodium falciparum genome6 indicates that these parasites lack the enzymes required for pyrimidine salvage7 and are totally dependent on de novo pyrimidine nucleoside synthesis for DNA replication. In contrast, erythrocyte maturation is accompanied by the loss of the capacity to carry out de novo synthesis of pyrimidines8 reinforcing the importance of pyrimidine biosynthesis as a potential antimalarial drug target. Indeed, several enzymes in the pyrimidine metabolism pathway are validated antimalarial targets, such as dihydrofolate reductase9 and dihydroorotate dehydrogenase.10
Another enzyme involved in pyrimidine biosynthesis, P. falciparum thymidylate kinase (PfTMPK), which catalyzes phosphorylation of thymidine monophosphate (TMP) to thymidine diphosphate (TDP), represents a potentially attractive drug target for malaria.11,12 TMPK has been characterized in several organisms including human,13,14Escherichia coli,13,15Saccharomyces cerevisiae,13,15Mycobacterium tuberculosis,16Vaccinia virus,17 and P. falciparum,5,12 and is essential in most organisms. We have recently reported structural and kinetic studies of PfTMPK, which indicate significant differences from the human homologue.12
TMPK is a homodimer with a subunit fold consisting of a five-stranded parallel β-sheet surrounded by 7–11 α-helices.12 Two conserved sequence motifs (Figure 1) are found in the nucleotide substrate binding domain: the P-loop containing structural elements required for substrate recognition and catalysis and a LID domain that partially encloses the phosphate donor and is important for catalysis.12
Figure 1.
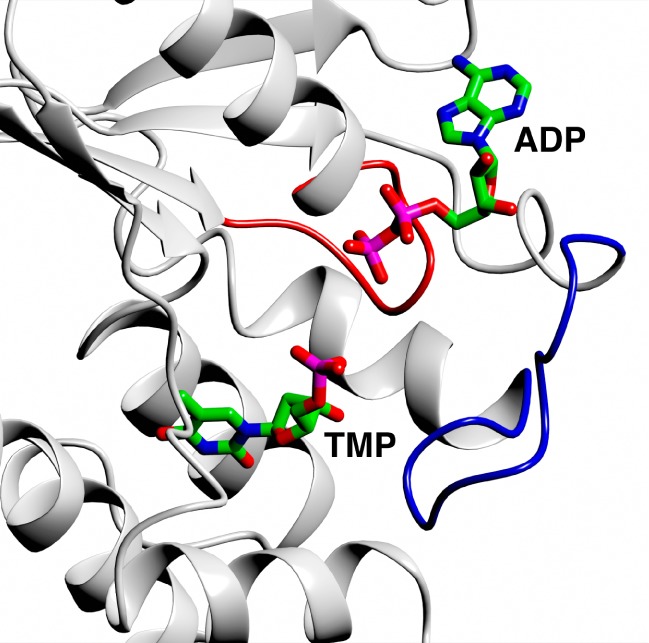
The crystal structure of the PfTMPK TMP-ADP complex showing the P-loop (red) and LID domain (blue), PDB code 2wwf, Whittingham et al.12 Figures 1 and 3 were made with CCP4mg.18
Superposition of the structure of PfTMPK-TMP-ADP with the equivalent complex of human TMPK (hTMPK) shows a highly conserved topology and mode of nucleotide binding, the TMP binding sites being essentially identical. However, several significant differences were observed in the P-loop and the LID domain.12 Therefore, reported TMPK inhibitors containing the thymidine base, but an optimized 5′-OH motif, will potentially target the P-loop and LID domain, and could lead to selective inhibition of PfTMPK compared to hTMPK.
TMPK has been investigated as a target against M. tuberculosis.16 Various series of inhibitors have been reported for MtTMPK.16,19,20 An interesting class is a series of 5′- substituted α-thymidine derivatives (Figure 2), some of which showed sub-micromolar activity against MtTMPK, and also good selectivity compared to hTMPK (Figure 2).16 A number of MtTMPK inhibitors were active against M. tuberculosis in cell culture, although others were inactive possibly due to issues concerning chemical solubility and cell permeability. Superimposition of the structures of hTMPK and MtTMPK revealed significant amino acid differences in the active site close to the 5′-position of the deoxyribose.21 Moreover, 5′-thiourea α-thymidine derivatives were completely inactive against human thymidine kinase 1 and only showed weak inhibition of human mitochondrial nucleoside kinase TK-2.22,23
Figure 2.

Example of a 5′- substituted α-thymidine inhibitor of MtTMPK.16
As a starting point we were interested to investigate if α-thymidine derivatives could act against PfTMPK, as these have been shown to inhibit MtTMPK, but not hTMPK.
Results and Discussion
Synthesis of α-Thymidine
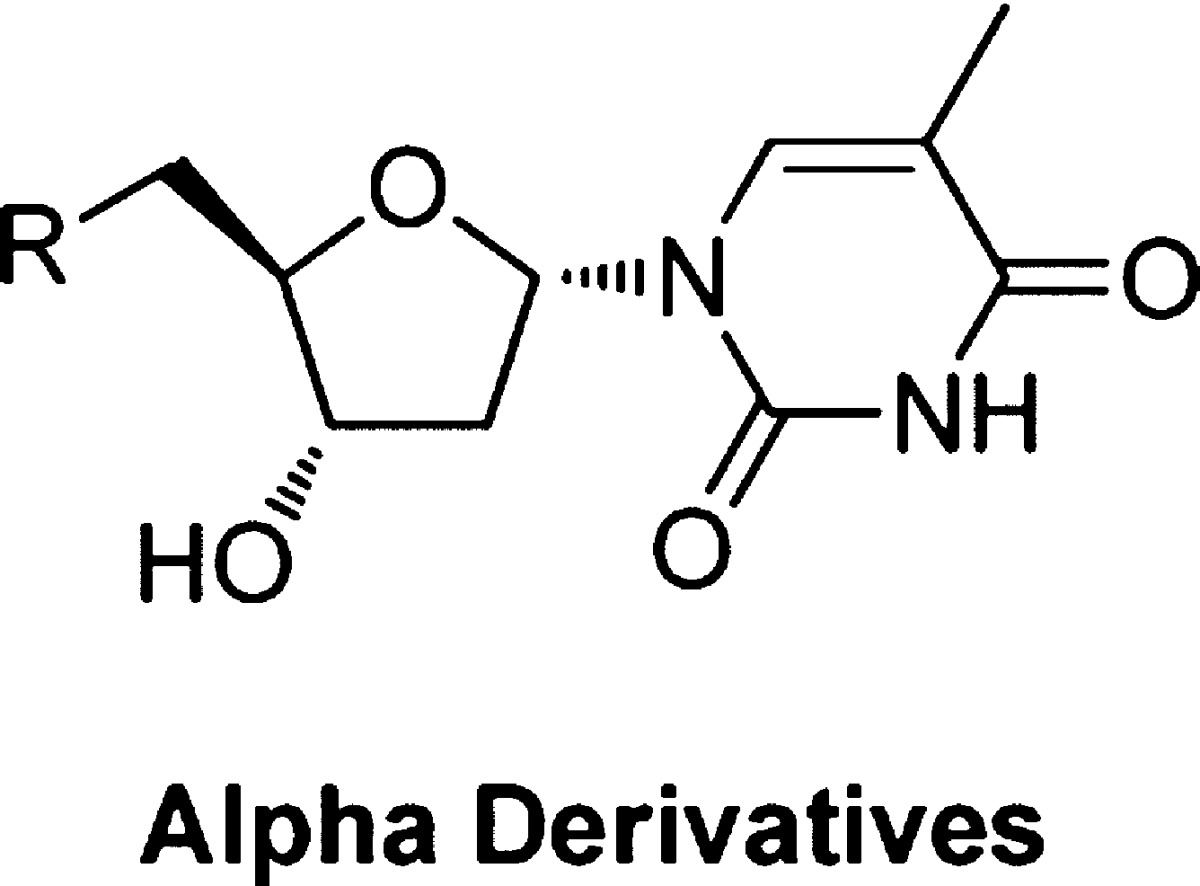
The synthesis of α-thymidine was based on literature methods.24,25 The key step was epimerization of the base. Several methods are reported for epimerization of β-thymidine including the use of TMSOTf reported by Sato25 and use of acetic anhydride/sulphuric acid as reported by Ward.24 Initially the method by Sato was followed. First, β-thymidine was selectively protected with diphenylacetyl chloride on the primary position in 50% yield (Scheme 1). The 3′-position was then protected with p-toluoyl chloride to give compound 3, as this substituent was reported to be selective for generation of the α-analogue on epimerization of the anomeric center.25 However, epimerization using TMSOTf was unsuccessful. It was decided to retain the 5′ and 3′ protecting groups suggested by Sato, since the compounds crystallized well, but epimerize the base using the acetic anhydride/sulphuric acid method of Ward.24 The ratio of α/β analogues after epimerization was around 3/1. The 3′,5′-O-diacylated α-thymidine derivative was readily separated by crystallization, giving the required product 4 in around 70% yield. Identification of the α/β-thymidine derivatives (3 and 4) was carried out by 1H NMR (Supporting Information, Figure S4), in particular, by looking at the chemical shifts of H1′ and H2′, which were affected by the stereochemistry of C1′. The two acyl groups were removed from compound 4 by sodium methoxide to give α-thymidine (5).
Scheme 1. Synthesis of α-Thymidine.

(a) Diphenylacetyl chloride, pyridine, 0 °C, 50%; (b) p-toluoyl chloride, pyridine, RT, 56%; (c) acetic anhydride, H2SO4, CH3CN, 51%; (d), NaOMe, MeOH, 90%.
Preparation of 5′ Substituted α- and β-Thymidine
Synthesis of 5′-amino substituted α-thymidine was carried out as shown in Scheme 2, by sulfonation, displacement with sodium azide and then hydrogenation to give the amine 8. Amine 8 was then coupled with thioisocyanate to give thioureas. The ureas, sulphonamides, amides and amines were also prepared to explore the SAR.
Scheme 2. Synthesis of 5′-Substituted α- and β-Thymidine Derivatives.

(a) Methanesulfonyl chloride, pyridine, −38 °C, [α: 43%; β: 50%]; (b) sodium azide, DMF, 60°C, [α: 43%; β 64%]; (c) 10% Pd/C, MeOH, [α: 95%; β 95%]; (d) sulfonyl chloride, DMF, RT; (e) carbonyl chloride, DMF, RT; (f) aryl chloride, DMF, RT; (g) (thio)isocyanate, DMF, RT.
For comparison, some 5′-substituted β-thymidine analogues were prepared (Scheme 2) using similar conditions as for the preparation of 5′-substituted α-thymidine. The β-thymidine amine 33 was coupled to give the urea, sulfonamide, amide and amine.
PfTMPK Inhibition Assay
Compounds were screened against recombinant PfTMPK using a coupled assay with pyruvate kinase and lactate dehydrogenase (Figure S1).12 Activity was measured spectrophotometrically by following the change in absorption at 340 nm due to the oxidation of NADH. The assay was carried out using TMP as substrate (Km = 11 μM). Control spectrophotometric assays were performed to verify that the compounds were inhibitors of PfTMPK and not of the coupling enzymes, by using ADP as substrate and the two coupling enzymes pyruvate kinase and lactate dehydrogenase, but no PfTMPK or TMP.12
The 5′-urea α-thymidine derivatives (14–30) showed moderate inhibition of PfTMPK, with most compounds having Ki values between 80 and 200 μM (Table 1). The most active were the 3-trifluoromethyl-4-chlorophenyl derivative 28 (Ki = 31 μM) and the 4-nitrophenyl derivative 30 (Ki = 11 μM). There were no significant differences between urea and thio-urea derivatives. 5′-(Thio)urea α-thymidine derivatives have been previously reported to be inhibitors of MtTMPK but are less active against hTMPK,16 human thymidine kinase23 and human mitochondrial thymidine kinase.23 They have also been shown to inhibit growth of M. tuberculosis and are selective compared to human cells.16
Table 1. Evaluation of 5′-α and β-Thymidine Derivatives against PfTMPK, P. falciparum and MRC5 Cellsc.

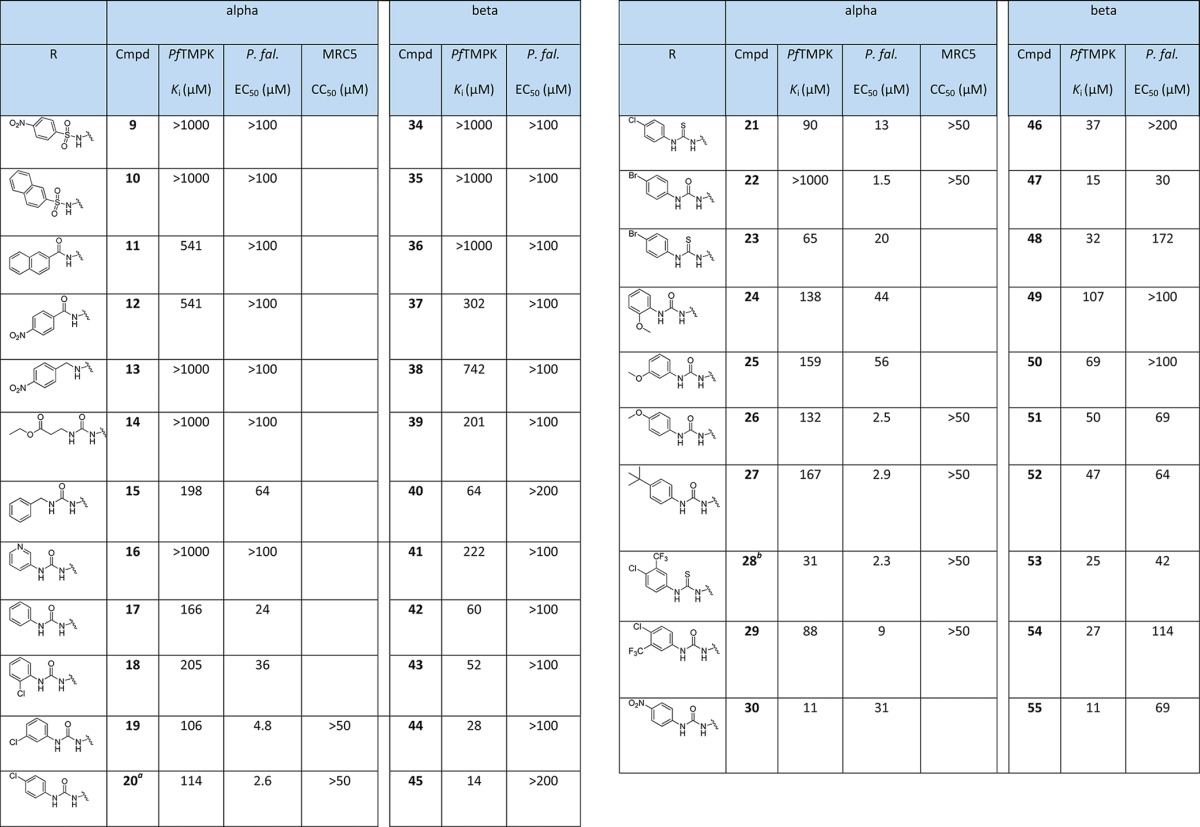
The 5′-urea β-thymidine derivatives (39–55) (Table 1) exhibited moderate inhibitory activity against PfTMPK in a similar fashion to the α-derivatives. Most of them inhibited in a similar range (50–100 μM), with again the 3-trifluoromethyl-4-chlorophenyl derivatives 53 (Ki = 25 μM) and 54 (Ki = 27 μM) and the 4-nitrophenyl derivative 55 (Ki = 11 μM) showing slightly higher potency.
There was no or very low inhibitory activity with the other 5′ substituents in either the α- or the β- series: this includes the sulphonamides (9, 10, 34, 35), the amides (11, 12, 36, 37), and the amines (13, 38). Interestingly, some 5′-substituted β-thymidine derivatives, including 5′-sulfonamides, 5′-(thio) ureas and 5′-amides, have been reported as moderate inhibitors of Bacillus anthracis TMPK but do not inhibit bacterial growth.26
Since the 5′-urea α-thymidine derivatives and the 5′-urea β-thymidine derivatives showed moderate inhibitory activity against PfTMPK, they were further investigated by cocrystallizing them with PfTMPK to assist in possible optimization.
Crystal Structure of Ligand Complexes
Co-crystallization with PfTMPK was successful with three of our active PfTMPK inhibitors (28, 30 and 53) (Figure 3). Crystallization conditions of the three inhibitors are listed in Supplementary Table S1, Supporting Information. Statistics for the X-ray data and refinement are summarized in Supplementary Table S2, and data for the three complexes have been deposited in the PDB with codes, 2YOG (28), 2YOH (30) and 2YOF (53). Compounds 28 and 53 are pairs of α- and β-thymidine derivatives (3-trifluoromethyl-4-chlorophenyl) that allow for comparison. The structures of all three complexes show that the enzyme is well ordered, with an essentially identical fold to that reported for the complexes with nucleotides and nucleotide derivatives.12 There was clear electron density for most of the ligands in all three complexes. A summary of some key features of the structural data on the complexes is given in Supporting Information Table S3. One substantial change in the protein is that the extended loop, residues 141–152, is disordered with no visible density in all three ligand structures, with the exception of Chain C in the complex with 53 where it packs against one of the two alternate conformations of the ligand.
Figure 3.

(A) Superposition of three ligand complexes 53 (blue), 30 (coral) and 28 (tan) on the TMP–ADP complex (colored by atom type) determined previously (pdb: 1wwf). The chain was selected in which the ligand was best ordered. The two sodium atoms in the TMP–ADP complex are shown as spheres. The peptide backbone is shown for the 53 complex (pale blue). (B) The binding pocket for 28, showing interactions with the protein. (C) The binding pocket for 28. The surface of the protein is colored by electrostatic potential. The four waters that form H-bond bridges to the protein are numbered. (D) The two conformations for 53 in Chain C, the least well-ordered ligand site.
The thymidine ring of the synthetic ligands mimics that of the natural TMP substrate (Figure 3B). The deoxyribose ring in contrast shows its typical variation in pucker and orientation relative to the thymidine allowing the end of the synthetic ligands to take up optimum interactions with the enzyme. The thymidine and deoxyribose are buried deep within the pocket, which also accommodates two water molecules, W27 and W303, that form bridges between ligand and protein (Figure 3C). Two additional waters form bridging H-bonds between ligand and protein. The H-bonds between the ligand and the protein and surrounding ordered water molecules (red spheres) are shown. The aromatic ring with its fluoro substituents extends out of the pocket and packs against the protein surface.
Chain C of the 53 complex displays the least well-ordered ligand (Figure 3D), and we have modeled this in two conformations. There is reasonable density for the thymidine and deoxyribose, though the deoxyribose is already showing disorder over two alternative puckers. There is essentially no density for the aromatic ring, and its positions are solely based on density for the fluorine groups. This ligand is well ordered in Chain B. A summary of the experimental electron density for the ligands is given in Supporting Table S3.
Anti-Malarial Activity
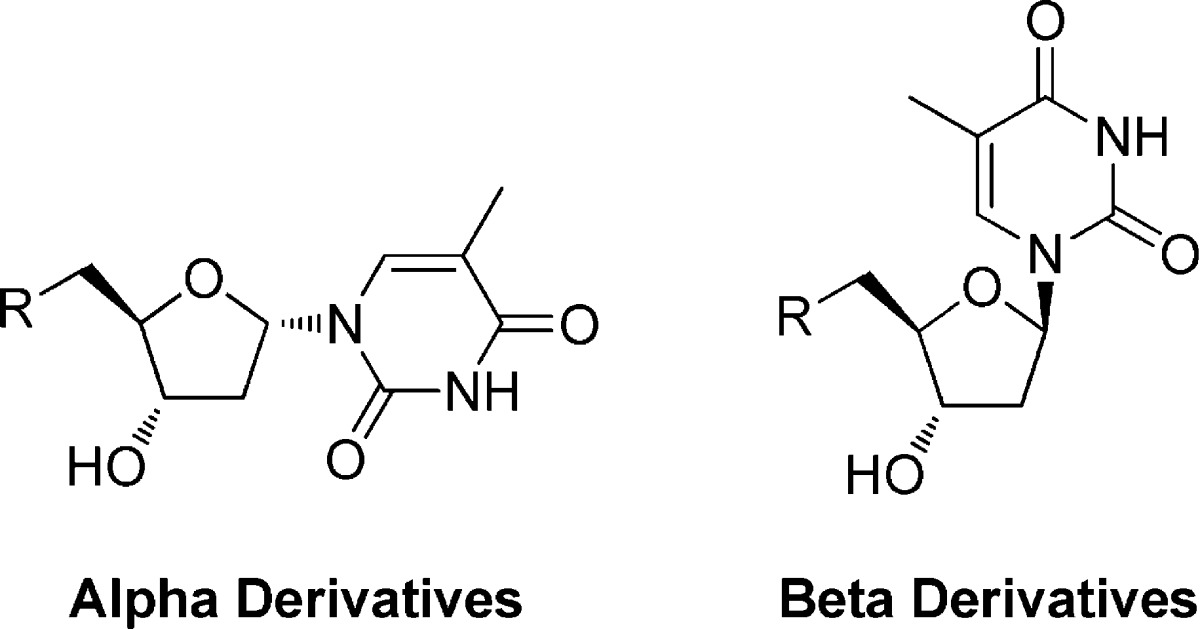
All of the compounds were evaluated for growth inhibition activity against P. falciparum using a SYBR green assay as reported in the literature.27 Most compounds from both series with Ki values for PfTMPK below 50 μM inhibited parasite growth. A number of compounds gave low micromolar inhibition of growth, the actives belonging almost exclusively to the α-thymidine analogue series (Table 1). The most potent compounds were 20, 26, 27 and 28, which had EC50 values of approximately 2 μM. They were all 4′-substituted phenyl compounds, which was the most active subset of this series of inhibitors in producing inhibition of parasite growth. All of the tested inhibitors showed good selectivity between malaria parasites and human MRC5 cells (Table 1). The β-thymidine derivatives had very low or no activity against the parasite (Table 1), and we therefore focused our attention on the α-derivatives.
There was only a low correlation between inhibition of PfTMPK and inhibition of the growth of P. falciparum. Most compounds were more potent inhibitors of parasite growth than inhibitors of the enzyme. For example, compound 17 (Figure 4) has a Ki of 166 μM against PfTMPK, but an EC50 of 23 μM against the parasite suggesting that the principal mode of action is not through inhibition of PfTMPK. These compounds showed good selectivity between the malaria parasite and human MRC5 cells. For instance, compound 26 has an EC50 of 2.5 μM, but a CC50 > 50 μM. Additional experiments were performed to probe the antimalarial activity of these derivatives.
Figure 4.

Two 5′-phenyl urea α-thymidine derivatives.
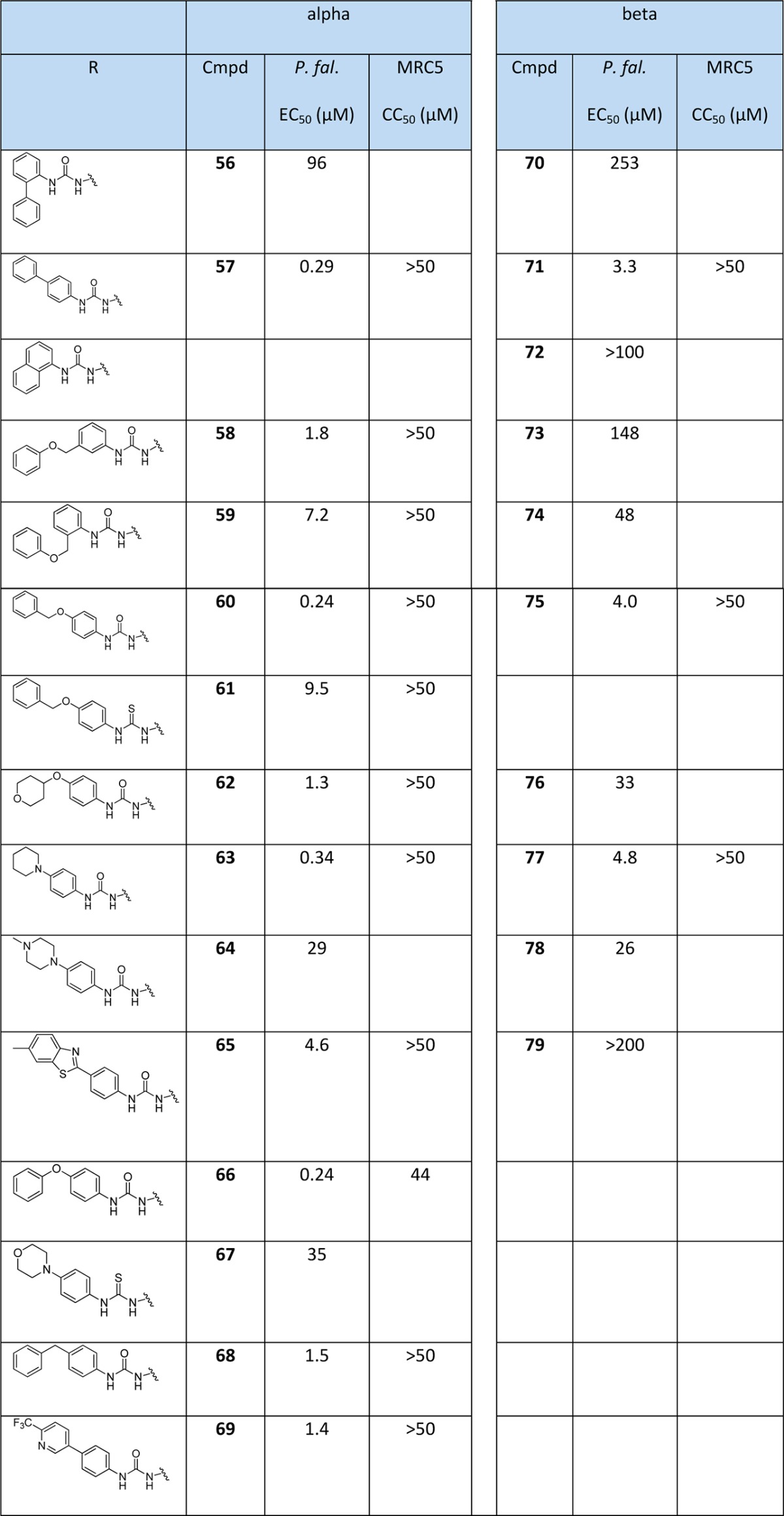
Compound 17 is a representative of the 5′-phenyl urea α-thymidine derivatives (Figure 4). Compound 17 can be divided into three components: the α-anomeric thymidine base, the deoxyribose ring and the substituted phenyl urea. The initial SAR showed that a substitution of the 5′-phenyl urea gave a significant increase in the activity against the parasite. For example, a para-methoxy group on the phenyl increased potency against the parasite 10-fold (compound 26, Figure 4). Work was carried out to optimize the phenyl group in the 5′-position, and the corresponding β-thymidine analogues were also evaluated for comparison purposes. Compounds were prepared as shown in Scheme 2 and tested against the parasite (Table 2).
Table 2. Evaluation of 5′-Phenyl (thio)urea α- and β-Thymidine Derivatives against P. falciparum and MRC5 Cellsa.
Reference compound: chloroquine EC50 = 0.007 μM.
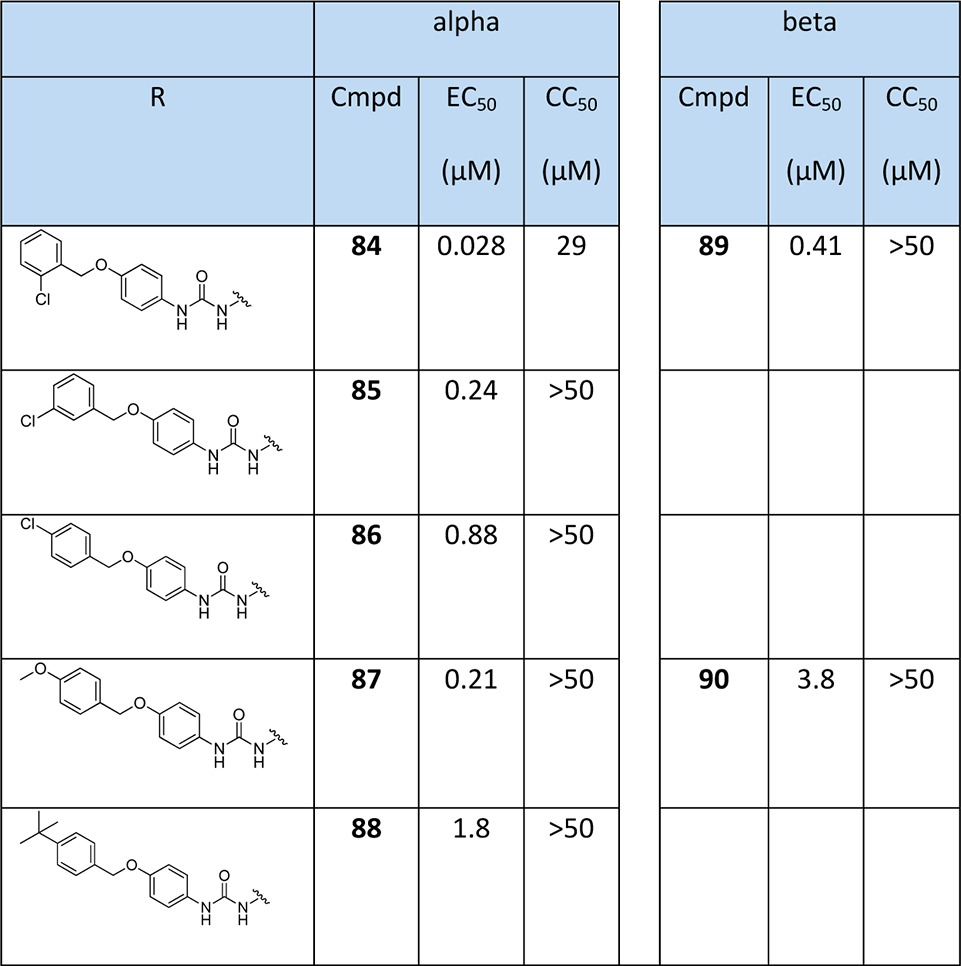
Since the 4-benzyloxy derivatives appeared to give very potent activity (compound 60), this series was expanded by preparing 4-benzyloxy phenyl isocyanates using the conditions reported by Knaggs et al.28 with triphosgene. The isocyanates were rapidly passed through a column for purification and then reacted with α- or β-thymidine amine to give the final compounds 84–90 (Scheme 3 and Table 3).
Scheme 3. Preparation of 4-Benzyloxy-phenyl Urea α- and β-Thymidine Derivatives.

(a) NaH, N-Boc protected 4-aminophenol, DMF; (b) TFA, DCM; (c) Et3N, triphosgene, EtOAC; (d) DMF, RT.
Table 3. Biological Evaluation of 4-Benzyloxy-phenyl Urea α- and β-Thymidine Derivativesa.

Reference compound: chloroquine EC50 = 0.007 μM.
Anti-Malarial Structure–Activity Relationship
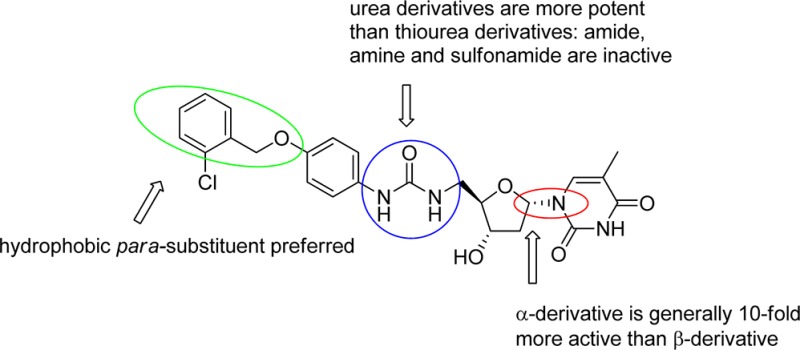
Data for the antiparasite assays are given in Tables 1–3. Compounds 57, 60, 63, 66, 84, 85, 86, 87 and 89 showed sub-micromolar activity. Most notably, compound 84 had an EC50 of 28 nM, which represents an increase in inhibition of approximately 1000-fold compared to the starting compound 17 (EC50 = 23 μM). We can summarize the key trends as follows:
-
•
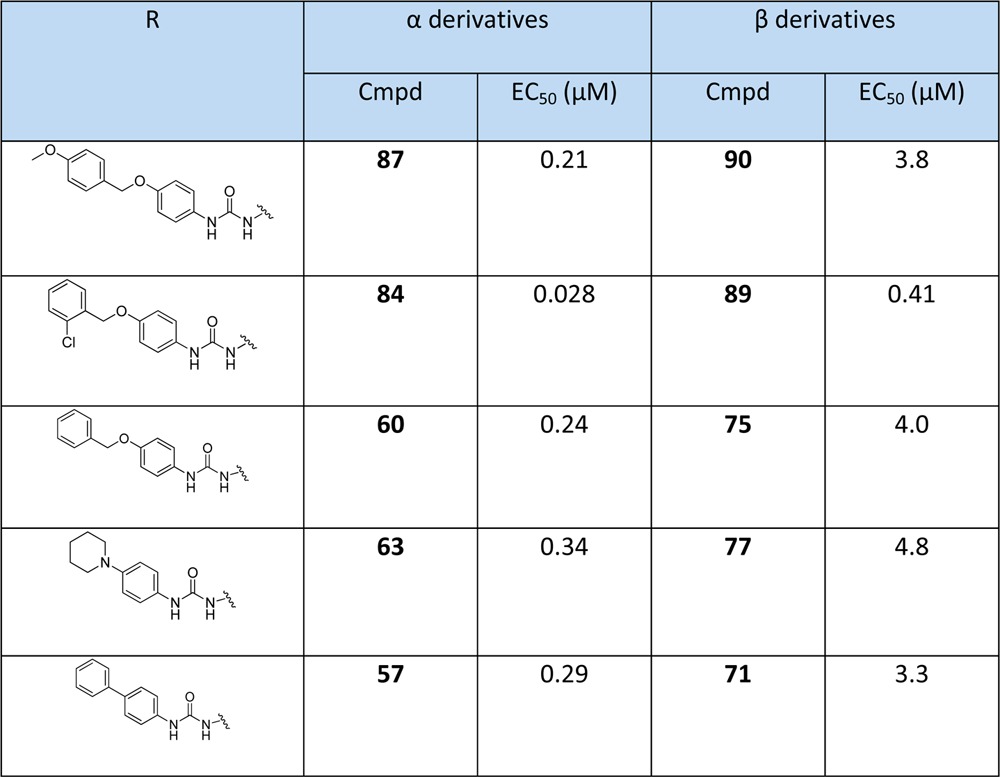
α-Anomers are much more potent than β-anomers. Five of the most active pairs of derivatives are compared in Table 4 which shows that α- had at least 10 times more activity than β-derivatives, presumably due to the α-anomer constraining the base in a much more favorable orientation. Interestingly compound 84, which has an ortho substitution in the benzyl group, gave the best antimalarial activity with an EC50 of 28 nM, and the related β-derivative 89 also gave the best inhibition activity of the β-derivatives, albeit with a 20-fold drop in activity.
-
•
A para-substitution gives enhanced activity. The phenyl urea is optimally substituted in the para-position rather than the ortho or meta positions for both the α- and β-anomers. For example, 56 (2-phenyl, EC50 = 96 μM) is much less active than 57 (4-phenyl, EC50 = 0.29 μM) (Table 2). Most of the active compounds in this study are para-substituted derivatives, for both α- and β- derivatives of the same pairs.
-
•
Ureas are more potent than thiourea. In the α-derivatives, thiourea and urea derivatives were investigated for their contribution to the inhibitory activity. Four pairs of inhibitors were chosen (Table 5). Urea derivatives are more active than thioureas except for the pair containing the 3-trifluoromethyl-4-chloro substituent, which is slightly more active for the thio-urea (28 and 29).
-
•
The para-substituent of the phenyl urea should be preferably a hydrophobic group. For the α-derivatives, improved activity is observed when the para-subsituents are hydrophobic groups. This is evident in comparisons of 66 (4-phenyloxy, EC50 = 0.24 μM) versus 62 (4-tetrahydropyran-oxy, EC50 = 1.3 μM); 63 (4-piperidine, EC50 = 0.34 μM) versus 67 (4-morpholino, EC50 = 35 μM) or 64 (4-(4-methyl-piperazine), EC50 = 29 μM).
Table 4. Comparison in Parallel of the α- and β-Urea Derivativesa.

Reference compound: chloroquine EC50 = 0.007 μM.
Table 5. Comparison in Parallel of the Thiourea and Urea α-Derivativesa.
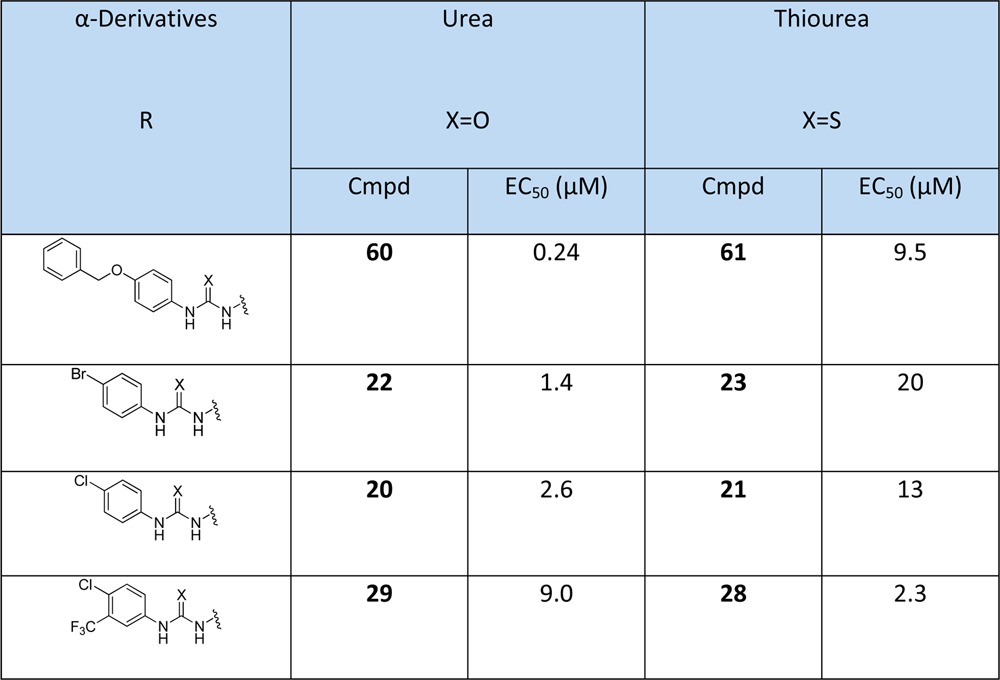
Reference compound: chloroquine EC50 = 0.007 μM.
DMPK Studies
To assess the further developability of the compounds, we carried out in vitro DMPK studies on five key compounds (Table 6).29 All showed reasonable microsomal stability (CLi < 5 mL/min/g) except for compound 84, which exhibited rapid turnover. Plasma protein binding was less than 95% in most cases, with compound 63 showing a particularly large unbound fraction. Unfortunately the most potent compound 84 was the most unstable when incubated with hepatic microsomes. However, related compounds showed good in vitro DMPK properties (57, 60, 63, 66), suggesting that there is nothing inherently problematic associated with the scaffold in terms of microsomal stability and protein binding.
Table 6. The Stability and Plasma Protein Binding Data of Five Selected Compoundsa.
| cmpd | microsomal stability (mL/min/g) | plasma protein binding (% bound) | EC50 (μM) | CC50 (μM) |
|---|---|---|---|---|
| 84 | 6 | 94.1 | 0.028 | 29 |
| 57 | 1.9 | 92.0 | 0.29 | >50 |
| 60 | 3 | 81.2 | 0.24 | >50 |
| 63 | 0.5 | 52.3 | 0.34 | >50 |
| 66 | 1.8 | 90.5 | 0.24 | 43.6 |
Reference compound: chloroquine EC50 = 0.007 μM.
Conclusion
In this study, we optimized a series of 5′-para substituted phenyl urea α-thymidine derivatives to produce compounds with improved antimalarial activity. Initially different series of compounds were designed as inhibitors of PfTMPK. While inhibition of the enzyme and parasite growth was moderate, the elucidation here of the crystal structure of several enzyme–inhibitor complexes provides the basis for a future structure-based drug discovery program.
Interestingly, while poorly active against the enzyme, the α-thymidine derivatives showed promising antimalarial activity and optimization was performed in order to increase potency. Several features were found to contribute to antiplasmodial action. The α-thymidine derivatives with para substituted phenyl groups (preferably hydrophobic substitutents) and ureas (better than thiourea) exhibited increased growth inhibition. Testing of the inhibitors gave activities in the nanomolar range and compounds showed a good selectivity between P. falciparum and human MRC5 cells. The most potent inhibitor from this series is compound 84 with an EC50 of 28 nM and CC50 of 29 μM, an increase in potency of nearly 1000 times compared to the starting compound 17 (EC50 = 23 μM). Furthermore some of the most active compounds have reasonable microsomal stability and free fractions. The resulting SAR information obtained for this series of inhibitors is shown in Figure 5.
Figure 5.
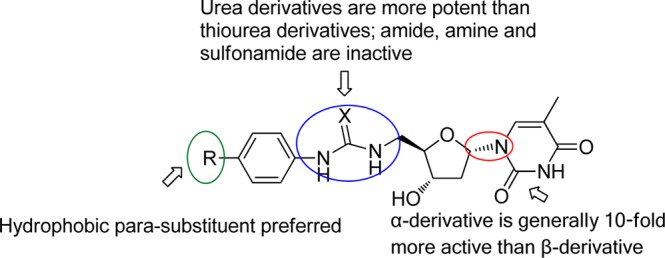
Summary of the SAR data for the thymidine-derived inhibitors.
Experimental Section
Chemistry
General
Chemicals and solvents were purchased from the Sigma-Aldrich Chemical Co., Fluka, VWR, Acros, Fisher Chemicals and Alfa Aesar. 1H NMR and 13C NMR were recorded on a Bruker Avance DPX 500 spectrometer (1H at 500.1 MHz and 13C at 125.8 MHz). Chemical shifts (δ) are expressed in ppm. Signal splitting patters are described as singlet (s), double (d), double doublet (dd), triplet (t), quarter (qt), multiplet (m). Low resolution electrospray (ES) mass spectra were recorded either on an Agilent Technologies 1200 series HPLC connected to an Agilent Technologies 6130 quadrupole spectrometer and to an Agilent diode array detector or on a Bruker MicroTof mass spectrometer, run in a positive ion mode, using either methanol, methanol/water (95:5), or water/acetonitrile (1:1) + 0.2% formic acid as the mobile phase. High resolution electrospray measurements were performed on a Bruker Daltonics MicrOTOF mass spectrometer. Column chromatography was carried out using silica gel 60 from Fluka. Thin layer chromatography (TLC) was carried out on Merck silica gel 60 F254 plates using UV light or PMA for visualization.
Purity was determined using both LCMS and NMR spectroscopy. Compounds had a purity of >95%.
General Procedure for Compounds 84–90
For the synthesis of compounds 84–90, amine 8 or 33 (1 equiv) was dissolved in DMF at 0 °C. The coupling reagents (1.1 equiv) were added, and the reaction mixture was allowed to stir at room temperature for 3 h. After the completion of the reaction, the reaction mixture was evaporated to dry (ethanol and toluene were used to coevaporate), and the residue was purified by column chromatography to yield the compounds 84–90 as a solid with the yields ranging from 67% to 83%.
N-(5′-Deoxy-α-thymidin-5′-yl)-N′-(4-(2-chlorobenzyloxy)phenyl)urea 84
4-(2-Chlorobenzyloxy)phenyl isocyanate reacted with amine 8 to yield compound 84 as a solid; 1H NMR (500 MHz, DMSO): δ 11.28 (s, 1H, NH), 8.40 (s, 1H, NH), 7.78 (d, J = 1.20 Hz, 1H, H6), 7.57–7.60, 7.50–7.53, 7.37–7.40, 7.29–7.31, 6.90–6.94 (m, 8H, H-Ph), 6.16–6.19 (m, 2H, H1′ and NH), 5.45 (d, J = 3.20 Hz, 1H, OH), 5.09 (s, 2H, CH2), 4.18–4.21 (m, 2H, H3′ and H4′), 3.21–3.28, 3.09–3.13 (m, 2H, H5′), 2.53–2.60, 1.91–1.97 (m, 2H, H2′), 1.78 (s, 3H, CH3); 13C NMR (125 MHz, DMSO): δ 163.8 (C4), 155.4 (CO), 150.5 (C2), 136.9 (C6), 152.8, 134.6, 132.5, 130.0, 129.7, 129.3, 127.3, 119.3, 114.9 (C-Ph), 108.8 (C5), 86.9 (C1′), 84.7 (C4′), 70.8 (C3′), 67.0 (CH2), 55.0 (C5′), 41.0 (C5′), 39.0 (C2′), 12.3 (CH3); LCMS (ES+): m/z (%) 501 (100) [M + H]+; HRMS (ES+): calcd for C24H26Cl1N4O6 [M + H]+ 501.1535 m/z, found 501.1533 m/z (0.54 ppm).
N-(5′-Deoxy-α-thymidin-5′-yl)-N′-(4-(3-chlorobenzyloxy)phenyl)urea 85
4-(3-Chlorobenzyloxy)phenyl isocyanate reacted with amine 8 to yield compound 85 as a solid; 1H NMR (500 MHz, DMSO): δ 11.28 (s, 1H, NH), 8.38 (s, 1H, NH), 7.78 (d, J = 1.20 Hz, 1H, H6), 7.37–7.42, 7.28–7.30, 6.89–6.91 (m, 8H, H-Ph), 6.16–6.19 (m, 2H, H1′ and NH), 5.45 (d, J = 3.25 Hz, 1H, OH), 5.06 (s, 2H, CH2), 4.16–4.20 (m, 2H, H3′ and H4′), 3.12–3.22 (m, 1H, H5′), 2.57 (m, 1H, H2′), 1.92–1.95 (m, 1H, H2′), 1.78 (s, 3H, CH3); 13C NMR (125 MHz, DMSO): δ 163.8 (C4), 155.3 (CO), 150.5 (C2), 136.9 (C6), 152.7, 136.9, 133.9, 133.0, 130.0, 127.6, 127.2, 126.1, 119.3, 115.0 (C-Ph), 108.8 (C5), 86.9 (C1′), 84.7 (C4′), 70.8 (C3′), 68.4 (CH2), 41.0 (C5′), 39.0 (C2′), 12.3 (CH3); LCMS (ES+): m/z (%) 501 (100) [M + H]+; HRMS (ES+): calcd for C24H26Cl1N4O6 [M + H]+ 501.1535 m/z, found 501.1533 m/z (0.54 ppm).
N-(5′-Deoxy-α-thymidin-5′-yl)-N′-(4-(4-chlorobenzyloxy)phenyl)urea 86
4-(4-Chlorobenzyloxy)phenyl isocyanate reacted with amine 8 to yield compound 86 as a solid; 1H NMR (500 MHz, DMSO): δ 11.28 (s, 1H, NH), 8.37 (s, 1H, NH), 7.77 (d, J = 1.20 Hz, 1H, H6), 7.44–7.45, 7.27–7.29, 6.88–6.90 (m, 8H, H-Ph), 6.16–6.19 (m, 2H, H1′ and NH), 5.45 (d, J = 3.30 Hz, 1H, OH), 5.03 (s, 2H, CH2), 4.16–4.18 (m, 2H, H3′ and H4′), 3.75 (s, 3H, CH3), 3.22–3.32 (m, 1H, H5′), 3.15–3.27 (m, 1H, H5′), 2.57 (m, 1H, H2′), 1.92–1.94 (m, 1H, H2′), 1.78 (s, 3H, CH3); 13C NMR (125 MHz, DMSO): δ 163.8 (C4), 155.3 (CO), 150.5 (C2), 136.9 (C6), 152.7, 136.4, 133.8, 132.2, 129.4, 128.4, 119.2, 115.0 (C-Ph), 108.8 (C5), 86.9 (C1′), 84.6 (C4′), 70.8 (C3′), 68.9 (CH2), 41.0 (C5′), 39.0 (C2′), 12.3 (CH3); LCMS (ES+): m/z (%) 501 (100) [M + H]+; HRMS (ES+): calcd for C24H26Cl1N4O6 [M + H]+ 501.1535 m/z, found 501.1519 m/z (3.33 ppm).
N-(5′-Deoxy-α-thymidin-5′-yl)-N′-(4-(4-methyoxybenzyloxy)phenyl)urea 87
4-(4-Methyoxybenzyloxy)phenyl isocyanate reacted with amine 8 to yield compound 87 as a solid; 1H NMR (500 MHz, DMSO): δ 11.28 (s, 1H, NH), 8.37 (s, 1H, NH), 7.78 (d, J = 1.20 Hz, 1H, H6), 7.27–7.31, 6.99–7.00, 6.87–6.90 (m, 8H, H-Ph), 6.16–6.19 (m, 2H, H1′ and NH), 5.45 (d, J = 3.25 Hz, 1H, OH), 5.01 (s, 2H, CH2), 4.16–4.18 (m, 2H, H3′ and H4′), 3.75 (s, 3H, CH3), 3.22–3.32 (m, 1H, H5′), 3.15–3.27 (m, 1H, H5′), 2.57 (m, 1H, H2′), 1.94 (m, 1H, H2′), 1.78 (s, 3H, CH3); 13C NMR (125 MHz, DMSO): δ 163.8 (C4), 155.4 (CO), 150.5 (C2), 136.9 (C6), 159.3, 152.9, 138.9, 129.5, 119.2, 114.9, 113.1, 113.0 (C-Ph), 108.8 (C5), 87.0 (C1′), 84.6 (C4′), 70.8 (C3′), 69.2 (CH2), 55.0 (CH3), 41.0 (C5′), 39.0 (C2′), 12.3 (CH3); LCMS (ES+): m/z (%) 497 (100) [M + H]+; HRMS (ES+): calcd for C25H29N4O7 [M + H]+ 497.2031 m/z, found 497.2031 m/z (−0.12 ppm).
N-(5′-Deoxy-α-thymidin-5′-yl)-N′-(4-(4-tertbutylbenzyloxy)phenyl)urea 88
4-(4-tert-Butylbenzyloxy)phenyl isocyanate reacted with amine 8 to yield compound 88 as a solid; 1H NMR (500 MHz, DMSO): δ 11.23 (s, 1H, NH), 8.37 (s, 1H, NH), 7.77 (d, J = 1.20 Hz, 1H, H6), 7.67–7.76, 7.26–7.41, 6.77–6.89 (m, 8H, H-Ph), 6.16–6.19 (m, 2H, H1′ and NH), 5.45 (d, J = 3.20 Hz, 1H, OH), 4.99 (s, 2H, CH2), 4.18–4.21 (m, 2H, H3′ and H4′), 3.21–3.28, 3.09–3.13 (m, 2H, H5′), 2.52–2.59, 1.91–1.98 (m, 2H, H2′), 1.78 (s, 3H, CH3), 1.28 (s, 9H, CH3); LCMS (ES+): m/z (%) 523 (100) [M + H]+; HRMS (ES+): calcd for C28H35N4O6 [M + H]+ 523.2551 m/z, found 523.2554 m/z (−0.57 ppm).
Acknowledgments
The research leading to these results has received funding from AntiMal, an FP6-funded integrated project under contract number LSHP-CT-2005-0188, the RICET FIS Network (RD06/0021) and the Junta de Andalucía (BIO-199, P09-CVI-5367). We would also like to acknowledge the support of the Wellcome Trust (Grant 083481). We would like to thank Suzanne Norval for the DMPK study. The authors thank the ESRF for access to beamlines and support by staff during visits.
Glossary
Abbreviations Used
- CC50
concentration required to reduce growth of MRC5 cells by 50%
- CLi
intrinsic clearance
- TDP
thymidine diphosphate
- TMP
thymidine monophosphate
- hTMPK
human thymidylate kinase
- Pf
Plasmodium falciparum
- Mt
Mycobacterium tuberculosis
- TMPK
thymidylate kinase
- TMSOTf
trimethylsilyl trifluoromethanesulfonate
Supporting Information Available
Methodology for enzyme assays, parasite assays, crystallography, DMPK and chemistry. Stereo views of several protein–ligand complexes. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Kilama W.; Ntoumi F. Malaria: a research agenda for the eradication era. Lancet 2009, 374, 1480–1482. [DOI] [PubMed] [Google Scholar]
- Gelb M. H.; Hol W. G. J. Parasitology - Drugs to combat tropical protozoan parasites. Science 2002, 297, 343–344. [DOI] [PubMed] [Google Scholar]
- Geels M. J.; Imoukhuede E. B.; Imbault N.; van Schooten H.; McWade T.; Troye-Blomberg M.; Dobbelaer R.; Craig A. G.; Leroy O. European Vaccine Initiative: lessons from developing malaria vaccines. Expert Rev. Vaccines 2011, 10, 1697–1708. [DOI] [PubMed] [Google Scholar]
- Cui H. Q.; Ruda G. F.; Carrero-Lerida J.; Ruiz-Perez L. M.; Gilbert I. H.; Gonzalez-Pacanowska D. Exploring new inhibitors of Plasmodium falciparum purine nucleoside phosphorylase. Eur. J. Med. Chem. 2010, 45, 5140–5149. [DOI] [PubMed] [Google Scholar]
- Cui H. Q.; Ruiz-Perez L. M.; Gonzalez-Pacanowska D.; Gilbert I. H. Potential application of thymidylate kinase in nucleoside analogue activation in Plasmodium falciparum. Bioorg. Med. Chem. 2010, 18, 7302–7309. [DOI] [PubMed] [Google Scholar]
- Gardner M. J.; Hall N.; Fung E.; White O.; Berriman M.; Hyman R. W.; Carlton J. M.; Pain A.; Nelson K. E.; Bowman S.; Paulsen I. T.; James K.; Eisen J. A.; Rutherford K.; Salzberg S. L.; Craig A.; Kyes S.; Chan M. S.; Nene V.; Shallom S. J.; Suh B.; Peterson J.; Angiuoli S.; Pertea M.; Allen J.; Selengut J.; Haft D.; Mather M. W.; Vaidya A. B.; Martin D. M. A.; Fairlamb A. H.; Fraunholz M. J.; Roos D. S.; Ralph S. A.; McFadden G. I.; Cummings L. M.; Subramanian G. M.; Mungall C.; Venter J. C.; Carucci D. J.; Hoffman S. L.; Newbold C.; Davis R. W.; Fraser C. M.; Barrell B. Genome sequence of the human malaria parasite Plasmodium falciparum. Nature 2002, 419, 498–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes P.; Rathod P. K.; Sanchez D. J.; Mrema J. E. K.; Rieckmann K. H.; Heidrich H. G. Enzymes of purine and pyrimidine metabolism from the human malaria, Plasmodium-falciparum. Mol. Biochem. Parasitol. 1982, 5, 275–290. [DOI] [PubMed] [Google Scholar]
- Sherman I. W.Malaria: Parasite Biology, Pathogenesis and Protection; ASM Press: Washington, DC, 1998. [Google Scholar]
- Anderson A. C. Targeting DHFR in parasitic protozoa. Drug Discovery Today 2005, 10, 121–128. [DOI] [PubMed] [Google Scholar]
- Booker M. L.; Bastos C. M.; Kramer M. L.; Barker R. H.; Skerlj R.; Sidhu A. B.; Deng X. Y.; Celatka C.; Cortese J. F.; Bravo J. E. G.; Llado K. N. C.; Serrano A. E.; Angulo-Barturen I.; Jimenez-Diaz M. B.; Viera S.; Garuti H.; Wittlin S.; Papastogiannidis P.; Lin J. W.; Janse C. J.; Khan S. M.; Duraisingh M.; Coleman B.; Goldsmith E. J.; Phillips M. A.; Munoz B.; Wirth D. F.; Klinger J. D.; Wiegand R.; Sybertz E. Novel inhibitors of Plasmodium falciparum dihydroorotate dDehydrogenase with Aanti-malarial activity in the mouse model. J. Biol. Chem. 2010, 285, 33054–33064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandeel M.; Ando T.; Kitamura Y.; Abdel-Aziz M.; Kitade Y. Mutational, inhibitory and microcalorimetric analyses of Plasmodium falciparum TMP kinase. Implications for drug discovery. Parasitology 2009, 136, 11–25. [DOI] [PubMed] [Google Scholar]
- Whittingham J. L.; Carrero-Lerida J.; Brannigan J. A.; Ruiz-Perez L. M.; Silva A. P. G.; Fogg M. J.; Wilkinson A. J.; Gilbert I. H.; Wilson K. S.; Gonzalez-Pacanowska D. Structural basis for the efficient phosphorylation of AZT-MP (3′-azido-3′-deoxythymidine monophosphate) and dGMP by Plasmodium falciparum type I thymidylate kinase. Biochem. J. 2010, 428, 499–509. [DOI] [PubMed] [Google Scholar]
- Lavie A.; Ostermann N.; Brundiers R.; Goody R. S.; Reinstein J.; Konrad M.; Schlichting I. Structural basis for efficient phosphorylation of 3 ′-azidothymidine monophosphate by Escherichia coli thymidylate kinase. Proc. Natl. Acad. Sci. U.S.A. 1998, 95, 14045–14050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostermann N.; Schlichting I.; Brundiers R.; Konrad M.; Reinstein J.; Veit T.; Goody R. S.; Lavie A. Insights into the phosphoryltransfer mechanism of human thymidylate kinase gained from crystal structures of enzyme complexes along the reaction coordinate. Structure 2000, 8, 629–642. [DOI] [PubMed] [Google Scholar]
- Lavie A.; Konrad M.; Brundiers R.; Goody R. S.; Schlichting I.; Reinstein J. Crystal structure of yeast thymidylate kinase complexed with the bisubstrate inhibitor P-1-(5′-adenosyl) P-5-(5′-thymidyl) pentaphosphate (TP(5)A) at 2.0 Å resolution: Implications for catalysis and AZT activation. Biochemistry 1998, 37, 3677–3686. [DOI] [PubMed] [Google Scholar]
- Van Daele I.; Munier-Lehmann H.; Froeyen M.; Balzarini J.; Van Calenbergh S. Rational design of 5′-thiourea-substituted alpha-thymidine analogues as thymidine monophosphate kinase inhibitors capable of inhibiting mycobacterial growth. J. Med. Chem. 2007, 50, 5281–5292. [DOI] [PubMed] [Google Scholar]
- Topalis D.; Collinet B.; Gasse C.; Dugue L.; Balzarini J.; Pochet S.; Deville-Bonne D. Substrate specificity of vaccinia virus thymidylate kinase. FEBS J. 2005, 272, 6254–6265. [DOI] [PubMed] [Google Scholar]
- McNicholas S.; Potterton E.; Wilson K. S.; Noble M. E. M. Presenting your structures: the CCP4mg molecular-graphics software. Acta Crystallogr. D 2011, 67, 386–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Daele I.; Munier-Lehmann H.; Hendrickx P. M. S.; Marchal G.; Chavarot P.; Froeyen M.; Qing L.; Martins J. C.; Van Calenbergh S. Synthesis and biological evaluation of bicyclic nucleosides as inhibitors of M-tuberculosis thymidylate kinase. ChemMedChem 2006, 1, 1081–1090. [DOI] [PubMed] [Google Scholar]
- Familiar O.; Munier-Lehmann H.; Negri A.; Gago F.; Douguet D.; Rigouts L.; Hernandez A. I.; Camarasa M. J.; Perez-Perez M. J. Exploring acyclic nucleoside analogues as inhibitors of Mycobacterium tuberculosis thymidylate kinase. ChemMedChem 2008, 3, 1083–1093. [DOI] [PubMed] [Google Scholar]
- Pochet S.; Dugue L.; Labesse G.; Delepierre M.; Munier-Lehmann H. Comparative study of purine and pyrimidine nucleoside analogues acting on the thymidylate kinases of Mycobacterium tuberculosis and of humans. ChemBiochem 2003, 4, 742–747. [DOI] [PubMed] [Google Scholar]
- Van Poecke S.; Munier-Lehmann H.; Helynck O.; Froeyen M.; Van Calenbergh S. Synthesis and inhibitory activity of thymidine analogues targeting Mycobacterium tuberculosis thymidine monophosphate kinase. Bioorg. Med. Chem. 2011, 19, 7603–7611. [DOI] [PubMed] [Google Scholar]
- Balzarini J.; Van Daele I.; Negri A.; Solaroli N.; Karlsson A.; Liekens S.; Gago F.; Van Calenbergh S. Human mitochondrial thymidine kinase is selectively inhibited by 3 ′-thiourea derivatives of beta-thymidine: identification of residues crucial for both inhibition and catalytic activity. Mol. Pharmacol. 2009, 75, 1127–1136. [DOI] [PubMed] [Google Scholar]
- Ward D. I.; Jeffs S. M.; Coe P. L.; Walker R. T. A Mild Procedure for the Anomerization of 2′-Deoxynucleosides. Tetrahedron Lett. 1993, 34, 6779–6782. [Google Scholar]
- Sato Y.; Tateno G.; Seio K.; Sekine M. A convenient method for the conversion of beta-thymidine to alpha-thymidine based on TMSOTf-mediated C1′-epimerization. Tetrahedron Lett. 2002, 43, 3251–3254. [Google Scholar]
- Byun Y.; Vogel S. R.; Phipps A. J.; Camrot C.; Eriksson S.; Tiwari R.; Tjarks W. Synthesis and biological evaluation of inhibitors of thymidine monophosphate kinase from Bacillus anthracis. Nucleosides, Nucleotides Nucleic Acids 2008, 27, 244–260. [DOI] [PubMed] [Google Scholar]
- Smilkstein M.; Sriwilaijaroen N.; Kelly J. X.; Wilairat P.; Riscoe M. Simple and inexpensive fluorescence-based technique for high-throughput antimalarial drug screening. Antimicrob. Agents Chemother. 2004, 48, 1803–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knaggs S.; Malkin H.; Osborn H. M. I.; Williams N. A. O.; Yaqoob P. New prodrugs derived from 6-aminodopamine and 4-aminophenol as candidates for melanocyte-directed enzyme prodrug therapy (MDEPT). Org. Biomol. Chem. 2005, 3, 4002–4010. [DOI] [PubMed] [Google Scholar]
- Ruda G. F.; Wong P. E.; Alibu V. P.; Norval S.; Read K. D.; Barrett M. P.; Gilbert I. H. Aryl phosphoramidates of 5-phospho erythronohydroxamic acid, a new class of potent trypanocidal compounds. J. Med. Chem. 2010, 53, 6071–6078. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.