

Abstract
Kinases play central roles in signaling pathways and are promising therapeutic targets for many diseases. Designing selective kinase inhibitors is an emergent and challenging task, because kinases share an evolutionary conserved ATP-binding site. KIDFamMap (http://gemdock.life.nctu.edu.tw/KIDFamMap/) is the first database to explore kinase-inhibitor families (KIFs) and kinase-inhibitor-disease (KID) relationships for kinase inhibitor selectivity and mechanisms. This database includes 1208 KIFs, 962 KIDs, 55 603 kinase-inhibitor interactions (KIIs), 35 788 kinase inhibitors, 399 human protein kinases, 339 diseases and 638 disease allelic variants. Here, a KIF can be defined as follows: (i) the kinases in the KIF with significant sequence similarity, (ii) the inhibitors in the KIF with significant topology similarity and (iii) the KIIs in the KIF with significant interaction similarity. The KIIs within a KIF are often conserved on some consensus KIDFamMap anchors, which represent conserved interactions between the kinase subsites and consensus moieties of their inhibitors. Our experimental results reveal that the members of a KIF often possess similar inhibition profiles. The KIDFamMap anchors can reflect kinase conformations types, kinase functions and kinase inhibitor selectivity. We believe that KIDFamMap provides biological insights into kinase inhibitor selectivity and binding mechanisms.
INTRODUCTION
Protein kinases play central roles in signaling pathways and cell cycle regulation (1,2). Protein kinases are one of the most important classes of drug targets, because the deregulation of kinase functions is often implicated in many diseases, such as cancers and neurological and metabolic diseases (2–4). Therefore, inhibition of protein kinases has been considered as a promising therapeutic strategy for the treatment of the diseases. Although many kinase inhibitors have been developed, most of them lack selectivity and interact with multiple protein kinases, resulting in unexpected side effects (5–7). The major factor is that the protein kinases share an evolutionary conserved ATP-binding site (8). Therefore, understanding of kinase-inhibitor binding mechanisms and selectivity, as well as kinase-inhibitor-disease (KID) relationships will be helpful for designing selective kinase inhibitors.
As increasing numbers of reliable kinase-inhibitor assays and complex structures become available, and as high-throughput binding assays provide systematic identification of kinase-inhibitor interactions (KIIs), there is a growing need for the establishment of a comprehensive database to describe kinase-inhibitor and KID relationships for studying protein kinase inhibitor selectivity and binding mechanisms. Kinase-inhibitor structures provide the atomic details of KIIs, kinase conformations and inhibitor types. Large-scale kinase profiling of known inhibitors has proven useful for studying the selectivity of protein kinases and inhibitors, with various reports elucidating the inhibition assays of 38 compounds against 317 kinases (5), 178 compounds against 300 kinases (6) and 72 compounds against 442 kinases (7). Moreover, some databases such as Protein Data Bank (PDB) (9) and BindingDB (10) have accumulated kinase inhibition assays. ChEMBL kinase SARfari incorporates and links kinase sequences, structures, compounds and screening data (11). As the number of these databases and binding assays continues to grow, they will become increasingly useful for analyzing kinase inhibitor selectivity and binding mechanisms. In addition, many methods have been proposed to design selective kinase inhibitors for minimizing adverse effects (12–15) by comparing the sequence and structure diversity and conservation. However, most of these methods are often unable to provide the large-scale subsite–moiety interactions of kinase subsites and compound moieties for reflecting kinase inhibitor selectivity and binding mechanisms.
We have recently reported site-moiety maps (SiMMaps) for elucidating protein-inhibitor binding mechanisms and discovering new inhibitors (16,17). A SiMMap represents physicochemical properties and interaction preferences of a protein-binding site by several anchors. A SiMMap anchor consists of three essential elements: the binding pocket (a part of the binding site) with conserved interacting residues; the compound moiety preferences of the pocket; and the pocket–moiety interaction type (electrostatic, hydrogenbonding or van der Waals). The consensus anchor, the subpocket–moiety interactions with statistical significance sharing by some particular protein kinases, can be regarded as a ‘hot spot’ that represents the conserved binding environments involved in inhibitor bindings and biological functions. As a result, a group of KIIs with consensus anchors can constitute a kinase-inhibitor family (KIF), which is analogous to a protein sequence family (18,19), a structure family (20) and a protein–protein interaction family (21).
To elucidate protein kinase inhibitor selectivity and binding mechanisms, we have developed the KIDFamMap database to explore KIFs and KID relationships. The KIIs exhibited in a KIF are often conserved on a number of consensus anchors, the conserved structural subsites interacting with consensus moieties of their inhibitors. These anchors are situated in the ATP-binding site, N-terminal lobe (N-lobe), head of activation loop (A-loop) pocket, C-terminal lobe (C-lobe) and substrate site. We evaluated 1208 KIFs in this database by comparing the results of large-scale kinase profiling assays. Our experimental results reveal that the members of a KIF often possess similar inhibition profiles. In this database, we also collected 962 kinase-disease relationships and 638 disease allelic variants from public databases to provide KID relationships. In addition, the anchors of KIFs can reflect several major kinase conformation types [e.g. DFG-in (22), DFG-out (22), A-loop-out and A-loop-in], kinase functions (638 disease allelic variants are often conserved interacting residues) and kinase inhibitor types [e.g. type I, II and III inhibitors (23,24)]. Our results show that the KIDFamMap database can provide further insights in the elucidation of protein kinase inhibitor selectivity and binding mechanisms. We believe that this database can be further applied to design selective kinase inhibitors.
MATERIALS AND METHODS
Data collection and preparation
KIDFamMap contains 1208 KIFs, 962 kinase-disease relationships, 186 985 kinase-inhibitor assays, 339 kinase-related diseases and 638 disease allelic variants (Table 1) collected from the following sources, such as ChEMBL kinase SARfari, PDB, BindingDB, PubChem (25), KinBase (8), UniProt (26), KEGG (27), OMIM (28) and large-scale kinase profiling assays (5–7). First, the annotations of 518 human protein kinases were obtained from the KinBase and UniProt databases. Among these 518 kinases, 172 kinases with 1208 X-ray structures were obtained from PDB. In addition, we used the in-house protein structure prediction server, (PS)2 (29,30), to model 227 protein kinases whether both the sequence similarity (BLASTP E-value ≤e−40) and interface interacting residues (sequence identity ≥60%) are significantly similar between the structure template and the modelled kinases. Furthermore, we collected non-redundant 35 788 kinase inhibitors and 55 603 KIIs (binding affinity or inhibitory activity ≤10 μM) by eliminating the redundant compounds and interactions.
Table 1.
A summary of the contents of KIDFamMap
| Content type | Number |
|---|---|
| Kinases | 399 |
| Kinases with PDB structures | 172 |
| Kinases with predicted models | 227 |
| Kinase inhibitors | 35 788 |
| Kinase-inhibitor assays | 186 985 |
| KIIs (≤10 μM) | 55 603 |
| Diseases | 339 |
| Disease allelic variants | 638 |
| KIFs | 1208 |
| Conformation types of 1208 KIFs | |
| Type A structures | 669 |
| Type B structures | 381 |
| Type C structures | 34 |
| Type D structures | 16 |
| Type E structures | 80 |
| Type F structures | 28 |
| KID relationships | 962 |
Identification of a KIF
Figure 1A and Supplementary Figure S1 show the details of the KIDFamMap database to identify the KIF and KID relationships of a kinase-inhibitor crystal structure. For a query kinase or compound, KIDFamMap first identifies the structure template candidate (kinase K and inhibitor I) of the query using BLASTP (31) [or compound topology similarity tools (32–34)] to search the structural template database (Figure 1B). For this structure template (K–I), the KIDFamMap then searches the kinase candidates (K′) with significant sequence similarity (E-values ≤e−10) using BLASTP and also searches the compound candidates (I′) with significant topology similarity (≥0.6) using atom pairs and moiety composition from the annotated KII database (≤10 μM) (Figure 1C). Our template-based scoring function was utilized to statistically evaluate the interaction similarity between the kinase-inhibitor candidates (K′–I′) and the template (K–I) according to the KIDFamMap anchors derived from our in-house tool SiMMap (16,17). Those kinase-inhibitor candidates with significant interaction similarity (Z-value ≥1.645) were considered as the KIF members (Figure 1D) of the template K–I. For each KIF, we inferred its consensus anchors, the subsite–moiety interactions with statistical significance. A consensus anchor (e.g. anchors ATP1-4 and NLP1-2 in Figure 1D) often represents the conserved binding environment that is involved in inhibitor binding mechanisms, biological functions and kinase inhibitor selectivity (Supplementary Figure S1D). In addition, the anchors of a KIF can reflect the kinase conformations and inhibitor types. Based on the members in a KIF, KIDFamMap provides the KID and anchor-mutation-inhibitor relationships (Figure 1E and F; Supplementary Figure S1F). Thus, for a given query, this database finally provides the KIFs, KID relationships, graphic visualization of binding models, KIDFamMap anchors (conserved interacting residues and moiety preferences), inhibitor-anchor map, kinase profiling assays, kinase conformation types and inhibitor types.
Figure 1.

Overview of the KIDFamMap database’s process and workflow for identifying KIF and KID relationships using tyrosine-protein kinase ABL1 as the query. (A) Main procedure. (B) Identification of the template candidate ABL1 (PDB code 3CS9) with inhibitor nilotinib of the query using BLASTP to scan the structural template database. The KIDFamMap anchors of ABL1 are shown. (C) Identification of the kinase and compound candidates of the template using BLASTP and compound topology similarity tools, respectively, for the subsequent search of the annotated KII database. The anchors occupied by nilotinib are labeled with red dots. (D) Identification of the kinase-inhibitor candidates with significant interaction similarity (Z-value ≥1.645) of the template ABL1-nilotinib using interaction similarity scores. (E) Identification of the KID relationships of ABL1-nilotinib family. (F) The relationships between KIDFamMap anchors and drug resistance mutations. The anchor NLP1 is formed by three residues (T315, K271 and F382) and the residue T315 forms a hydrogen bond with nilotinib. The mutation, T315I (threonine to isoleucine), reduces inhibitory activity of nilotinib against ABL1 by ∼150 folds (IC50 value from 13 to >2000 nM).
The kinase-inhibitor family
The concept of a KIF forms the core of the KIDFamMap database to explore the binding mechanisms, inhibition selectivity and KID relationships of a query kinase or compound. Here, we used a structural template (kinase K and inhibitor I) as a simple case to define the KIF as follows: (i) the kinases (e.g. K and K′) in a KIF with significant sequence similarity (BLASTP E-value ≤e−10); (ii) the inhibitors (e.g. I and I′) in a KIF with significant topology similarity (≥0.6); (iii) the KIIs in a KIF with significant interaction similarity (Z-value ≥1.645). The interaction similarity Z-value is defined as Z = (S − μ)/σ, where S is the interaction similarity score between the KIIs K′–I′ and K–I. S is calculated as S = EX × IR × MP × CI, where EX is the similarity score of anchor patterns; IR and MP are the similarity scores of the conserved interacting residues and moiety preferences of the aligned anchors, respectively; CI is the similarity score of the KII profiles. The four similarity scores range between 0 and 1. μ and σ are the mean and the standard deviation of the interaction similarity scores of 12 090 pairs of complex comparisons derived from non-redundant 156 X-ray kinase-inhibitor complexes in PDB.
KIDFamMap anchors
To study the binding mechanisms of KIIs and evaluate the interaction similarity between any two KIIs, we used SiMMap to describe the conserved kinase structural subsites interacting with consensus moieties of the inhibitors (Figure 2D; Supplementary Figure S1D). We constructed a SiMMap for each X-ray kinase-inhibitor complex using the in-house tools GEMDOCK (35) and SiMMap. We selected 5844 diverse kinase inhibitors from a total of 35 788 inhibitors by discarding the inhibitors with similar topology through the use of atom pairs and moiety compositions. For the kinase structure of each of the 1208 complexes selected from PDB, these 5844 inhibitors were docked into the binding site of each structure using GEMDOCK, which yielded similar performance to that reported with other docking tools (36–38). Among these 5844 kinase inhibitors, the docked poses of the top-ranked 2000 inhibitors, as determined by using the piecewise linear potentials of GEMDOCK, were used to construct the site-moiety map by considering the interaction profiles between the inhibitors and the kinase. Figure 1B and Supplementary Figure S1B show the site-moiety map of the ABL1 kinase.
Figure 2.

KIDFamMap search results using compound staurosporine as the query. (A) The query interface for inputting a kinase, compound or disease name. (B) The ‘Template’ page shows summarized query results, related diseases, the available KIFs (such as CKD2-staurosporine and ITK-staurosporine families), kinase conformation types and inhibitor types. (C) The members of the CKD2-staurosporine family (PDB code 1AQ1) share similar interactions and atomic interactions, as seen on the ‘Kinase-Inhibitor Family’ page. KIDFamMap also offers the analysis of family kinases and kinase profiling assays. (D) The ‘Family Anchors’ page provides anchor patterns, interacting residues and moiety preferences of each anchor. (E) The ‘Inhibitor-Anchor Map’ offers the anchor pattern distributions (consensus anchors) of KIIs and the docked poses of family inhibitors. (F) The family related diseases, the associated family kinases and disease allelic variants are provided on the ‘Diseases’ page.
After constructing 1208 site-moiety maps for all the kinase structures, the global protein structures were aligned using a structural alignment tool (39). Among the residues of the kinase domain, 14 residues (i.e. A179-K181 of β3-strand, M229-N233 of hinge and K277-M282 of catalytic loop in AKT2 kinase numbering) are common to 399 human protein kinases, the conformations of these 14 residues does not change substantially upon kinase activation (40). We then manually refined the structural alignments by visual inspection (41) to minimize the root-mean-square deviation of these 14 residues between the kinases and the template structure AKT2 structure (PDB code 1O6K). The superimposed SiMMap anchors of these kinases can be roughly clustered into 14 groups, called KIDFamMap anchors (Supplementary Figure S2), based on spatial distances and domain knowledge. According to the conformations and functions of the kinases, these 14 KIDFamMap anchors were divided into five pockets, namely, ATP site (anchors ATP1-5), N-lobe pocket (anchors NLP1-3), head of A-loop pocket (HAP pocket, anchors HAP1-3), C-lobe pocket (anchors CLP1-2) and substrate pocket (anchor SP). To facilitate the observation, we constructed a pseudostructure with 14 anchors by combining six representative kinase structures (i.e. PDB codes 1AQ1 [CDK2], 3EQH [MAP2K1], 1O6K [AKT2], 2WGJ [MET], 3CS9 [ABL1] and 2HZ0 [ABL1]) and by eliminating the collision residues located near the bound inhibitors. In addition, we deleted the phosphate-binding loop and A-loop occupying the ATP and HAP pockets, respectively, for clarity of the structure. Furthermore, to identify the consensus anchors (e.g. anchors NLP1-2, HAP1-2 and CLP1 in Figure 1D) of a KIF, we utilized the structural alignment tool and an automatic procedure to align these structures and anchors of these kinase members. The consensus anchors of the KIF revealed the conserved binding environments which are often involved in inhibitor binding mechanisms, biological functions and kinase inhibitor selectivity for these kinases.
Kinase conformation types
Kinase conformations are highly correlated to catalytic activity and inhibitor selectivity (kinase inhibitor types) (24,42). The 14 KIDFamMap anchors identified in this study can reflect kinase conformations and inhibitor types (24,43). An inhibitor occupying mainly the anchors ATP1-5 is assigned as a type I inhibitor (e.g. staurosporine in Figure 2), whereas an inhibitor occupying both ATP site (anchors ATP1-4) and the HAP pocket (anchors HAP1-3) is assigned as a type II inhibitor (e.g. nilotinib in Figure 1). The type III inhibitors [e.g. U0126 (44)], that bind outside the ATP-binding site, occupy both N-lobe pocket (anchors NLP1-3) and HAP pocket (anchors HAP1-3).
Based on the 14 KIDFamMap anchors, we can divide kinase conformations into the following six types (Supplementary Figure S3): (A) DFG-in and A-loop-out; (B) N-lobe pocket presented and HAP pocket presented; (C) C-lobe pocket absent; (D) DFG-in and A-loop-in; (E) DFG-out and A-loop-in; (F) DFG-out and A-loop-out. Here, the A-loop is regarded as an ‘in’ conformation (A-loop-in) when A-loop is close to the γ-phosphate group of ATP; otherwise, the A-loop is regarded as an ‘out’ conformation (A-loop-out). The anchors NLP2, NLP3 and HAP2 appear for binding type III inhibitors when the αC-helix in the ‘out’ conformation (Supplementary Figure S3B) (24). The anchors CLP1 and CLP2 are absent in the AGC group kinases which consist of the AGC C-terminal domain occupying these two anchors (Supplementary Figure S3C). The anchors HAP1-3 are present or absent when the DFG motif is ‘out’ or ‘in’ conformation, respectively (Supplementary Figure S3A and E). The ATP5 anchor, which is close to the γ-phosphate group of ATP and which forms electrostatic interactions with the DFG motif, is often occupied by the A-loop which adopts an ‘in’ conformation (Supplementary Figure S3E). The A-loop would be inferred as ‘in’ or ‘out’ conformation when the anchor ATP5 is absent or present, respectively.
DATABASE ACCESS
User interface
KIDFamMap is an easy-to-use database in which the users give an input in the form of a kinase, compound or disease name. The typical workflow of KIDFamMap is shown in Figure 2 and Supplementary Figure S1. This database provides KIF candidates and KID relationships in the query result pages, which are termed ‘Template’, ‘Kinase-Inhibitor Family’, ‘Family Anchors’, ‘Inhibitor-Anchor Map’ and ‘Diseases’. Here, we show the steps in accessing information by querying the compound staurosporine (Figure 2A).
Template: A quick overview includes the molecular formula, molecular weight, the conformation type of the target kinases of the query compound (Figure 2B). For each conformation type, the 3D visualization of the KIDFamMap anchor pattern is presented dynamically using Jmol (45). Users can select the interested template to examine its KIF and KID relationships in the following results pages. KIDFamMap shows the KIF statistics of the selected template, including the kinases, inhibitors and diseases associated with the query compound.
Kinase-Inhibitor Family: KIDFamMap indicates the binding interfaces, binding models and binding affinities of KIIs for the selected KIF on this page (Figure 2C). For the family kinases, the multiple sequence alignment, which was obtained from Pfam (19), is used to present conserved residue positions. Recently, large-scale kinase profiling assays have provided new insights for the kinase inhibitor selectivity. KIDFamMap uses two sets of large-scale profiling assays (6,7) to present the inhibition patterns between the family kinases and their inhibitors.
Family Anchors: This page first lists the anchors with different interacting forces (electrostatic in red, hydrogen bonding in green and van der Waals in gray) (Figure 2D; Supplementary Figure S1D). For each anchor, the interacting residues, pocket patterns, moiety preferences and reached compounds are shown for studying the binding mechanisms. Moreover, disease allelic variants often provide the clues for revealing KID relationships. The conserved interacting residues, which are recorded as disease allelic variants (e.g. T315I), are labeled (Supplementary Figure S1D).
Inhibitor-Anchor Map: This page offers an overview of the relationships between the anchor patterns and inhibitory activities of KIIs (Figure 2E), which provide useful clues to design inhibitors. For example, the anchor patterns with potent binding affinities can guide the direction to design potency inhibitors. Moreover, the inhibitor-anchor map would be helpful for exploring kinase inhibitor selectivity.
Diseases: The related diseases and disease allelic variants of the selected KIF are presented on this page (Figure 2F). Based on these diseases and associated mutations, KIDFamMap provides the KID relationships (Figure 1E).
Examples
ABL1-nilotinib family
Chronic myeloid leukemia (CML), a malignant condition of white blood cells with an annual incidence of ∼10 cases per million, is caused by a defect in the BCR-ABL1 fusion gene coding for an aberrant tyrosine kinase (46,47). Currently, the ABL1 kinase is the main pharmaceutical target for the treatment of CML (48). After querying for the ABL1 kinase, KIDFamMap found 18 structure template candidates with three conformation types (A, E and F), six diseases (i.e. three types of leukemia and three infectious diseases) and six allelic variants (e.g. T315I and Y253H) (Supplementary Figure S1B). The template ABL1-nilotinib complex (PDB code 3CS9) with conformation type E (DFG-out and A-loop-in), which binds to the type II inhibitor nilotinib approved for the treatment of drug-resistant CML (49,50), was selected as an example. The ABL1-nilotinib family consists of 195 KIIs with 125 similar inhibitors (40 known inhibitors for ABL1) and 12 similar kinases, including 9 kinases (e.g. ABL2 and KIT) in the TK group, 2 kinases (MAPK11 and MAPK14 with E-values of e−17 and e−18, respectively) in the CMGC group, and the BRAF kinase in the TKL group (Figures 1D and 3). Conversely, the FES kinase, which shares a high-sequence similarity but different anchor patterns with ABL1 (E-value = e−63), is not a member of the ABL1-nilotinib family because FES is in the ‘DFG-in and A-loop-out’ conformation (Figure 3B). KIDFamMap also provides additional information about this KIF, such as the docked poses of ABL1 inhibitors and the multiple sequence alignment of these 12 kinases. Excluding KDR kinase, the remaining 11 kinases are inhibited by nilotinib according to the kinase profiling assays (6,7).
Figure 3.

The anchor patterns and interacting residues of the kinases ABL1, MAPK14, EPHA7 and FES. (A) The partial anchor patterns of the ABL1-nilotinib family. The kinase groups are labeled in parentheses with the corresponding kinase names. (B) The partial anchor patterns of EPHA7 and FES, which are not members of the ABL1-nilotinib family. (C) Residues, pocket surfaces and nilotinib reference poses of ABL1, MAPK14, EPHA7 and FES using the ABL1-nilotinib structure (PDB code 3CS9). (D) Superimposed structures. (E) Residues of ABL1, MAPK14, EPHA7 and FES.
For the ABL1-nilotinib family, the KIDFamMap database infers nine consensus anchors (i.e. ATP1-4, NLP1-2, HAP1-2 and CLP1) to represent the conserved binding environments (Supplementary Figure S1C and D). For each anchor, the database shows the conserved interacting residues, pocket patterns and moiety preferences. The anchor NLP1 consists of three interacting residues (T315, K271 and F382) to form a van der Waals environment that prefers aromatic and heterocyclic moieties. The gatekeeper residue T315 often forms hydrogen bonds with inhibitors (e.g. nilotinib) (Figures 1F and 3C). The conserved residue K271 located on the β3-strand binds to the phosphate groups of ATP, and the residue F382 situated in the DFG motif is also conserved. The gatekeeper residue T106 and corresponding residues of MAPK14 are also involved in hydrogen bond and van der Waals interactions, respectively. Experimental results show that nilotinib inhibits both ABL1 and MAPK14 (7), which suggests that kinases in a KIF can be inhibited by similar inhibitors due to the similar binding environments. In contrast, the EPHA7 kinase, which shares a high-sequence similarity with ABL1 (E-value = e−55), is not a member of ABL1-nilotinib family and has the different binding environment at the anchor NLP1. The corresponding residues of EPHA7 (I711) and FES (M636) located at the gatekeeper form a longer side chain than the threonine of ABL1, thus generating a steric clash with the methylphenyl group of nilotinib and disrupting the hydrogen bonding with nilotinib (Figure 3C–E).
Among the six allelic variants of ABL1, two (Y253 and T315) of them are located at the ligand binding site and are identified as conserved interacting residues. Three approved drugs (nilotinib, imatinib and dasatinib) targeting ABL1 form hydrogen bonds with T315. The mutation T315I disrupts the hydrogen bonding and the inhibitory effects of three drugs are drastically reduced more than 140-fold (Figure 1F) on average (48). Conversely, the moiety of CHEMBL1171836 has been modified to overcome this mutation is responsible for drug resistance (51). Based on the relationships between the KIF and diseases (Figure 1E), KIDFamMap can help in the understanding of adverse effects of drugs and in the identification of new uses for existing drugs. For instance, nilotinib is a drug for treating CML by inhibiting ABL1 originally. Applying the relationships between the KIF and diseases, nilotinib can be potentially used to some indications, such as amyotrophic lateral sclerosis, non-small cell lung cancer and gastrointestinal stromal tumor (GIST), as revealed by the inhibition of MAPK14, BRAF and KIT, respectively. For example, nilotinib was previously used to inhibit KIT for the treatment of GIST in a phase III study (52–54).
CDK2-staurosporine family
We used the staurosporine, a microbial alkaloid widely used kinase inhibitor, as the second example (Figure 2). The KIDFamMap database found cyclin-dependent kinase 2 (CDK2)-staurosporine structure as the template of the query. CDK2 is essential for the G1/S phase transition of the cell cycle. Supplementary Figure S4A shows the relationship between interaction similarity and sequence similarity in the CDK2-staurosporine family, revealing that more than 189 kinases in eight kinase groups have similar interfaces for staurosporine binding in the conformation type A (e.g. DFG-in and A-loop-out). This result is consistent with the previous kinase profiling studies that report staurosporine as one of the most non-selective inhibitor (24,43). Supplementary Figure S4B presents the similar docked poses of staurosporine in these diverse kinases. These poses generally match the anchors ATP1-ATP4 and CLP1, all of which are low selectivity anchors.
To understand the inhibitor selectivity and binding mechanisms of staurosporine, we selected the following kinases to compare the structures with KIDFamMap anchors (Supplementary Figure S5A): CDK2 (CMGC group), IRAK4 (TKL group), MAPKAPK3 (CAMK group), PRKCQ (AGC group), AURKA (other group), STK25 (STE group) and MAPK11 (CMGC group). The selected kinases are good representatives due to the fact that their interfaces are significantly similar, and their sequences are diverse in this KIF. We found that the selected kinases have similar kinase conformations and anchor patterns; in particular, they contain the ATP1-ATP4 and CLP1 anchors, which match staurosporine. Moreover, these anchors have similar interaction types, the conserved interacting residues and moiety preferences (Supplementary Figure S5B).
The anchors and their properties within a KIF can be applied to explore kinase inhibitor selectivity and to understand binding mechanisms. For example, in the CDK2-staurosporine family, the hydrophobic residues (V18, F80 and L134) of the ATP2 anchor are involved in van der Waals interactions with the planar ring of staurosporine (Supplementary Figure S6A). In this family, the RPS6KB1 kinase has a similar binding environment formed by the three hydrophobic residues (V105, L172 and M225) interacting with staurosporine (Supplementary Figure S6B). Previous studies showed that staurosporine inhibits CDK2 and RPS6KB1 (24,43), suggesting that kinases in a KIF can be inhibited by the similar inhibitors and can serve as new uses of existing drugs. In contrast, the RAF1 and BRAF kinases, which are not the members of the CDK2-staurosporine family, have a different binding environment at the ATP2 anchor from that of CDK2. The ATP2 pockets of these two kinases consists of two hydrophobic residues (V363 and F475 in RAF1 numbering) and one polar residue (T421) (Supplementary Figure S6C). The gatekeeper residue T421 has a shorter and a polar side chain than the phenylalanine residue (F80) of CDK2 and the leucine residue (L172) of RPS6KB1. These three interacting residues of the anchor ATP2 of RAF1 and BRAF cannot provide stable van der Waals interactions with the planar ring of staurosporine (Supplementary Figure S6D and E). In addition, the ATP2 pockets of RAF1 and BRAF contain a phenylalanine residue that has a relatively long side chain compared with the leucine (L134) of CDK2 and the methionine (M225) of RPS6KB1, resulting in the reduction of the volume of the pocket. Therefore, the pocket cannot accommodate the planar ring of staurosporine, and staurosporine fails to exert an inhibitory effect on RAF1 and BRAF (24,43). The ATP2 anchor can be further applied to design selective inhibitors for RAF1 and BRAF by forming hydrogen bonds with the threonine residue. These results reveal that the kinases belonging to a KIF often interact with similar inhibitors and the anchor properties are useful for studying the binding mechanisms and kinase selectivity of inhibitors.
EVALUATION
To evaluate the KIDFamMap database for KIFs, we collected two sets with the available large-scale kinase profiling assay (7), term PDB156 and LIG72. In total, 12 090 pairs of comparisons between protein interfaces in the PDB156 set, which consists of 156 kinase-inhibitor structures with binding affinities, were used to assess the correlation between inhibition similarities and interaction similarity Z-values (Figure 4). Pearson’s correlation coefficient (PCC) was calculated as 0.97. The distribution of Z-values is shown in Supplementary Figure S7. These 12 090 comparisons were also used to calculate the correlation between inhibition similarities and kinase sequence similarities (PCC = 0.93; Supplementary Figure S8A). Furthermore, we evaluated the correlation between inhibition similarities and compound topology similarities (PCC = 0.9) by generating 2556 pairs of comparisons between 72 inhibitors in the LIG72 set, which are diverse inhibitors that have been previously tested against 370 human protein kinases in large-scale kinase profiling assays (Supplementary Figure S8B). These results show that compounds with similar topologies possess similar inhibition profiles and inhibit similar kinases. Similarly, kinases with significant sequence similarity are inhibited by similar inhibitors and KIIs with significant Z-values have similar inhibition profiles.
Figure 4.
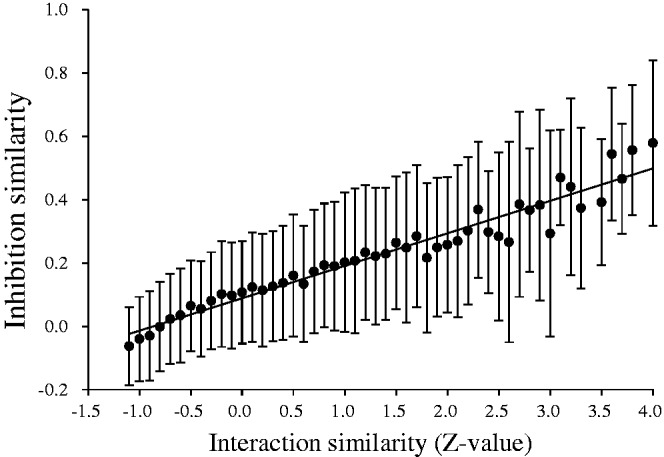
The correlation between inhibition similarity and interaction similarity (Z-value). Z-values of interaction similarity scores are highly correlated with inhibition similarities and the PCC is 0.97.
We collected 3240 inhibition assays of 72 inhibitors against 45 mutant variants (belonging to nine kinases) from large-scale profiling inhibition assays (7) to validate that conserved interacting residues (Supplementary Tables S1 and S2) are indeed causally implicated in the inhibitions. Among these 45 mutant variants, 27 have mutations in the conserved interacting residues. Moreover, 1663 (inhibitory activity ≤10 μM; Supplementary Dataset S1) of these 3240 inhibition assays were used to calculate the average fold changes between the wild-type kinases and mutant variants. For example, the phosphorylated ABL1 kinase is inhibited by 41 (inhibitory activity ≤10 μM) of 72 inhibitors (Supplementary Table S2) and the average fold change of T315I-mutant/wild-type ABL1 is 122.62 (Supplementary Figure S9A). For T315I mutant of ABL1, the top three inhibitors with the highest fold changes are dasatinib (from 0.046 to 120 nM, 2608 folds), PD-173955 (from 0.58 to 480 nM, 827 folds) and nilotinib (from 13 to ≥10 000 nM, 769 folds). Among 27 mutant variants with mutations in the conserved interacting residues, 55.6% (15/27) have high average fold changes (≥10). Conversely, the average fold changes of 100% (18/18) mutant variants with mutations in the other residues are low (<4). In addition, we collected 638 disease allelic variants from OMIM (28) and 1041 kinase mutations in cancer from MoKCa (55). Among 51 conserved interacting residues, 42 (82.4%) of them were disease related mutations (Supplementary Table S1). These results suggested that conserved interacting residues often implicated in the inhibitory activity and diseases.
CONCLUSIONS
The KIDFamMap database provides the utility and feasibility to explore KIFs and KID relationships for kinase inhibitor selectivity and binding mechanisms. To our knowledge, KIDFamMap is unique in exploring comprehensive ‘family-selective’ inhibitors based on KIDFamMap anchors and KIFs. This database includes 1208 KIFs, 962 KIDs, 55 603 KIIs, 35 788 kinase inhibitors, 399 human protein kinases, 339 diseases and 638 disease allelic variants. Our experimental results indicate that the KIIs of a KIF often possess similar inhibition profiles and our interaction similarity Z-values are highly consistent with those obtained from large-scale kinase profiling assays. In addition, the KIDFamMap anchors of a KIF often represent the conserved binding environments involved in the structural conformations of kinases and can reliably guide the processes of discovering selective kinase inhibitors. We believe that KIDFamMap is able to provide valuable insights for elucidating kinase inhibitor selectivity and binding mechanisms.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Tables 1–2, Supplementary Figures 1–9 and Supplementary Dataset 1.
ACKNOWLEDGEMENTS
J.-M.Y. thanks Core Facility for Protein Structural Analysis supported by National Core Facility Program for Biotechnology.
FUNDING
Funding for open access charge: National Science Council, partial supports of Ministry of Education and National Health Research Institutes [NHRI-EX100-10009PI]; ‘Center for Bioinformatics Research of Aiming for the Top University Program’ of the National Chiao Tung University and Ministry of Education, Taiwan.
Conflict of interest statement. None declared.
REFERENCES
- 1.Blume-Jensen P, Hunter T. Oncogenic kinase signalling. Nature. 2001;411:355–365. doi: 10.1038/35077225. [DOI] [PubMed] [Google Scholar]
- 2.Cohen P. Protein kinases—the major drug targets of the twenty-first century? Nat. Rev. Drug Discov. 2002;1:309–315. doi: 10.1038/nrd773. [DOI] [PubMed] [Google Scholar]
- 3.Noble MEM, Endicott JA, Johnson LN. Protein kinase inhibitors: insights into drug design from structure. Science. 2004;303:1800–1805. doi: 10.1126/science.1095920. [DOI] [PubMed] [Google Scholar]
- 4.Weinmann H, Metternich R. Drug discovery process for kinase inhibitors. Chembiochem. 2005;6:455–459. doi: 10.1002/cbic.200500034. [DOI] [PubMed] [Google Scholar]
- 5.Karaman MW, Herrgard S, Treiber DK, Gallant P, Atteridge CE, Campbell BT, Chan KW, Ciceri P, Davis MI, Edeen PT, et al. A quantitative analysis of kinase inhibitor selectivity. Nat. Biotechnol. 2008;26:127–132. doi: 10.1038/nbt1358. [DOI] [PubMed] [Google Scholar]
- 6.Anastassiadis T, Deacon SW, Devarajan K, Ma HC, Peterson JR. Comprehensive assay of kinase catalytic activity reveals features of kinase inhibitor selectivity. Nat. Biotechnol. 2011;29:1039–1045. doi: 10.1038/nbt.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Davis MI, Hunt JP, Herrgard S, Ciceri P, Wodicka LM, Pallares G, Hocker M, Treiber DK, Zarrinkar PP. Comprehensive analysis of kinase inhibitor selectivity. Nat. Biotechnol. 2011;29:1046–1051. doi: 10.1038/nbt.1990. [DOI] [PubMed] [Google Scholar]
- 8.Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298:1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- 9.Rose PW, Beran B, Bi CX, Bluhm WF, Dimitropoulos D, Goodsell DS, Prlic A, Quesada M, Quinn GB, Westbrook JD, et al. The RCSB Protein Data Bank: redesigned web site and web services. Nucleic Acids Res. 2011;39:D392–D401. doi: 10.1093/nar/gkq1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu TQ, Lin YM, Wen X, Jorissen RN, Gilson MK. BindingDB: a web-accessible database of experimentally determined protein-ligand binding affinities. Nucleic Acids Res. 2007;35:D198–D201. doi: 10.1093/nar/gkl999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gaulton A, Bellis LJ, Bento AP, Chambers J, Davies M, Hersey A, Light Y, McGlinchey S, Michalovich D, Al-Lazikani B, et al. ChEMBL: a large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2012;40:D1100–D1107. doi: 10.1093/nar/gkr777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Traxler P, Furet P. Strategies toward the design of novel and selective protein tyrosine kinase inhibitors. Pharmacol. Ther. 1999;82:195–206. doi: 10.1016/s0163-7258(98)00044-8. [DOI] [PubMed] [Google Scholar]
- 13.Liu Y, Gray NS. Rational design of inhibitors that bind to inactive kinase conformations. Nat. Chem. Biol. 2006;2:358–364. doi: 10.1038/nchembio799. [DOI] [PubMed] [Google Scholar]
- 14.McGregor MJ. A pharmacophore map of small molecule protein kinase inhibitors. J. Chem. Info. Model. 2007;47:2374–2382. doi: 10.1021/ci700244t. [DOI] [PubMed] [Google Scholar]
- 15.Liao JJL. Molecular recognition of protein kinase binding pockets for design of potent and selective kinase inhibitors. J. Med. Chem. 2007;50:409–424. doi: 10.1021/jm0608107. [DOI] [PubMed] [Google Scholar]
- 16.Chen YF, Hsu KC, Lin SR, Wang WC, Huang YC, Yang JM. SiMMap: a web server for inferring site-moiety map to recognize interaction preferences between protein pockets and compound moieties. Nucleic Acids Res. 2010;38:W424–W430. doi: 10.1093/nar/gkq480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hsu KC, Cheng WC, Chen YF, Wang HJ, Li LT, Wang WC, Yang JM. Core site-moiety maps reveal inhibitors and binding mechanisms of orthologous proteins by screening compound libraries. PLoS One. 2012;7:e32142. doi: 10.1371/journal.pone.0032142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hunter S, Apweiler R, Attwood TK, Bairoch A, Bateman A, Binns D, Bork P, Das U, Daugherty L, Duquenne L, et al. InterPro: the integrative protein signature database. Nucleic Acids Res. 2009;37:D211–D215. doi: 10.1093/nar/gkn785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Punta M, Coggill PC, Eberhardt RY, Mistry J, Tate J, Boursnell C, Pang N, Forslund K, Ceric G, Clements J, et al. The Pfam protein families database. Nucleic Acids Res. 2012;40:D290–D301. doi: 10.1093/nar/gkr1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Andreeva A, Howorth D, Brenner SE, Hubbard TJP, Chothia C, Murzin AG. SCOP database in 2004: refinements integrate structure and sequence family data. Nucleic Acids Res. 2004;32:D226–D229. doi: 10.1093/nar/gkh039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen CC, Lin CY, Lo YS, Yang JM. PPISearch: a web server for searching homologous protein-protein interactions across multiple species. Nucleic Acids Res. 2009;37:W369–W375. doi: 10.1093/nar/gkp309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pargellis C, Tong L, Churchill L, Cirillo PF, Gilmore T, Graham AG, Grob PM, Hickey ER, Moss N, Pav S, et al. Inhibition of p38 MAP kinase by utilizing a novel allosteric binding site. Nat. Struct. Biol. 2002;9:268–272. doi: 10.1038/nsb770. [DOI] [PubMed] [Google Scholar]
- 23.Dar AC, Shokat KM. The evolution of protein kinase inhibitors from antagonists to agonists of cellular signaling. Annu. Rev. Biochem. 2011;80:769–795. doi: 10.1146/annurev-biochem-090308-173656. [DOI] [PubMed] [Google Scholar]
- 24.Norman RA, Toader D, Ferguson AD. Structural approaches to obtain kinase selectivity. Trends Pharmacol. Sci. 2012;33:273–278. doi: 10.1016/j.tips.2012.03.005. [DOI] [PubMed] [Google Scholar]
- 25.Wang YL, Xiao JW, Suzek TO, Zhang J, Wang JY, Zhou ZG, Han LY, Karapetyan K, Dracheva S, Shoemaker BA, et al. PubChem's BioAssay database. Nucleic Acids Res. 2012;40:D400–D412. doi: 10.1093/nar/gkr1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Magrane M, Consortium U. UniProt Knowledgebase: a hub of integrated protein data. Database. 2011;2011:bar009. doi: 10.1093/database/bar009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kanehisa M, Goto S, Sato Y, Furumichi M, Tanabe M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 2012;40:D109–D114. doi: 10.1093/nar/gkr988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Amberger J, Bocchini CA, Scott AF, Hamosh A. McKusick's Online Mendelian Inheritance in Man (OMIM®) Nucleic Acids Res. 2009;37:D793–D796. doi: 10.1093/nar/gkn665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen CC, Hwang JK, Yang JM. (PS)(2): protein structure prediction server. Nucleic Acids Res. 2006;34:W152–W157. doi: 10.1093/nar/gkl187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen CC, Hwang JK, Yang JM. (PS)(2)-v2: template-based protein structure prediction server. BMC Bioinform. 2009;10:366. doi: 10.1186/1471-2105-10-366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Altschul SF, Madden TL, Schaffer AA, Zhang JH, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Carhart RE, Smith DH, Venkataraghavan R. Atom pairs as molecular-features in structure activity studies—definition and applications. J. Chem. Info. Comput. Sci. 1985;25:64–73. [Google Scholar]
- 33.Willett P, Barnard JM, Downs GM. Chemical similarity searching. J. Chem. Info. Comput. Sci. 1998;38:983–996. [Google Scholar]
- 34.Haider N. Functionality pattern matching as an efficient complementary structure/reaction search tool: an open-source approach. Molecules. 2010;15:5079–5092. doi: 10.3390/molecules15085079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang JM, Chen CC. GEMDOCK: a generic evolutionary method for molecular docking. Proteins. 2004;55:288–304. doi: 10.1002/prot.20035. [DOI] [PubMed] [Google Scholar]
- 36.Jones G, Willett P, Glen RC, Leach AR, Taylor R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997;267:727–748. doi: 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- 37.Kramer B, Rarey M, Lengauer T. Evaluation of the FLEXX incremental construction algorithm for protein-ligand docking. Proteins. 1999;37:228–241. doi: 10.1002/(sici)1097-0134(19991101)37:2<228::aid-prot8>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 38.Ewing TJA, Makino S, Skillman AG, Kuntz ID. DOCK 4.0: search strategies for automated molecular docking of flexible molecule databases. J. Comput. Aided Mol. Des. 2001;15:411–428. doi: 10.1023/a:1011115820450. [DOI] [PubMed] [Google Scholar]
- 39.Shindyalov IN, Bourne PE. Protein structure alignment by incremental combinatorial extension (CE) of the optimal path. Protein Eng. 1998;11:739–747. doi: 10.1093/protein/11.9.739. [DOI] [PubMed] [Google Scholar]
- 40.Hubbard SR. Crystal structure of the activated insulin receptor tyrosine kinase in complex with peptide substrate and ATP analog. EMBO J. 1997;16:5572–5581. doi: 10.1093/emboj/16.18.5572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Guex N, Peitsch MC. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis. 1997;18:2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- 42.Huse M, Kuriyan J. The conformational plasticity of protein kinases. Cell. 2002;109:275–282. doi: 10.1016/s0092-8674(02)00741-9. [DOI] [PubMed] [Google Scholar]
- 43.Zhang JM, Yang PL, Gray NS. Targeting cancer with small molecule kinase inhibitors. Nat. Rev. Cancer. 2009;9:28–39. doi: 10.1038/nrc2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fischmann TO, Smith CK, Mayhood TW, Myers JE, Reichert P, Mannarino A, Carr D, Zhu H, Wong J, Yang RS, et al. Crystal structures of MEK1 binary and ternary complexes with nucleotides and inhibitors. Biochemistry. 2009;48:2661–2674. doi: 10.1021/bi801898e. [DOI] [PubMed] [Google Scholar]
- 45.Herraez A. Biomolecules in the computer—Jmol to the rescue. Biochem. Mol. Biol. Educ. 2006;34:255–261. doi: 10.1002/bmb.2006.494034042644. [DOI] [PubMed] [Google Scholar]
- 46.Chissoe SL, Bodenteich A, Wang YF, Wang YP, Burian D, Clifton SW, Crabtree J, Freeman A, Iyer K, Li JA, et al. Sequence and analysis of the human Abl gene, the Bcr gene, and regions involved in the philadelphia chromosomal translocation. Genomics. 1995;27:67–82. doi: 10.1006/geno.1995.1008. [DOI] [PubMed] [Google Scholar]
- 47.Faderl S, Talpaz M, Estrov Z, Kantarjian HM. Chronic myelogenous leukemia: biology and therapy. Ann. Intern. Med. 1999;131:207–219. doi: 10.7326/0003-4819-131-3-199908030-00008. [DOI] [PubMed] [Google Scholar]
- 48.Radich JP. Measuring response to BCR-ABL inhibitors in chronic myeloid leukemia. Cancer. 2012;118:300–311. doi: 10.1002/cncr.26280. [DOI] [PubMed] [Google Scholar]
- 49.Cortes JE, Jones D, O'Brien S, Jabbour E, Konopleva M, Ferrajoli A, Kadia T, Borthakur G, Stigliano D, Shan JQ, et al. Nilotinib as front-line treatment for patients with chronic myeloid leukemia in early chronic phase. J. Clin. Oncol. 2010;28:392–397. doi: 10.1200/JCO.2009.25.4896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Weisberg E, Choi HG, Ray A, Barrett R, Zhang JM, Sim T, Zhou WJ, Seeliger M, Cameron M, Azam M, et al. Discovery of a small-molecule type II inhibitor of wild-type and gatekeeper mutants of BCR-ABL, PDGFR alpha, Kit, and Src kinases: novel type II inhibitor of gatekeeper mutants. Blood. 2010;115:4206–4216. doi: 10.1182/blood-2009-11-251751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Huang WS, Metcalf CA, Sundaramoorthi R, Wang YH, Zou D, Thomas RM, Zhu XT, Cai LS, Wen D, Liu SY, et al. Discovery of 3-[2-(Imidazo[1,2-b]pyridazin-3-yl)ethynyl]-4-methyl-N-{4-[(4-methylpiperazin-1-yl)methyl]-3-(trifluoromethyl)phenyl}benzamide (AP24534), a potent, orally active pan-inhibitor of breakpoint cluster region-Abelson (BCR-ABL) kinase including the T315I Gatekeeper Mutant. J. Med. Chem. 2010;53:4701–4719. doi: 10.1021/jm100395q. [DOI] [PubMed] [Google Scholar]
- 52.Brose MS, Volpe P, Feldman M, Kumar M, Rishi I, Gerrero R, Einhorn E, Herlyn M, Minna J, Nicholson A, et al. BRAF and RAS mutations in human lung cancer and melanoma. Cancer Res. 2002;62:6997–7000. [PubMed] [Google Scholar]
- 53.Ackerley S, Grierson AJ, Banner S, Perkinton MS, Brownlees J, Byers HL, Ward M, Thornhill P, Hussain K, Waby JS, et al. p38 alpha stress-activated protein kinase phosphorylates neurofilaments and is associated with neurofilament pathology in amyotrophic lateral sclerosis. Mol. Cell. Neurosci. 2004;26:354–364. doi: 10.1016/j.mcn.2004.02.009. [DOI] [PubMed] [Google Scholar]
- 54.Reichardt P, Blay JY, Gelderblom H, Schlemmer M, Demetri GD, Bui-Nguyen B, McArthur GA, Yazji S, Hsu Y, Galetic I, et al. Phase III study of nilotinib versus best supportive care with or without a TKI in patients with gastrointestinal stromal tumors resistant to or intolerant of imatinib and sunitinib. Ann. Oncol. 2012;23:1680–1687. doi: 10.1093/annonc/mdr598. [DOI] [PubMed] [Google Scholar]
- 55.Richardson CJ, Gao Q, Mitsopoulous C, Zvelebil M, Pearl LH, Pearl FMG. MoKCa database—mutations of kinases in cancer. Nucleic Acids Res. 2009;37:D824–D831. doi: 10.1093/nar/gkn832. [DOI] [PMC free article] [PubMed] [Google Scholar]