


Abstract
Amino acids form the building blocks of all proteins. Naturally occurring amino acids are restricted to a few tens of sidechains, even when considering post-translational modifications and rare amino acids such as selenocysteine and pyrrolysine. However, the potential chemical diversity of amino acid sidechains is nearly infinite. Exploiting this diversity by using non-natural sidechains to expand the building blocks of proteins and peptides has recently found widespread applications in biochemistry, protein engineering and drug design. Despite these applications, there is currently no unified online bioinformatics resource for non-natural sidechains. With the SwissSidechain database (http://www.swisssidechain.ch), we offer a central and curated platform about non-natural sidechains for researchers in biochemistry, medicinal chemistry, protein engineering and molecular modeling. SwissSidechain provides biophysical, structural and molecular data for hundreds of commercially available non-natural amino acid sidechains, both in l- and d-configurations. The database can be easily browsed by sidechain names, families or physico-chemical properties. We also provide plugins to seamlessly insert non-natural sidechains into peptides and proteins using molecular visualization software, as well as topologies and parameters compatible with molecular mechanics software.
INTRODUCTION
Amino acid sidechains confer their specific properties to all proteins. They govern the specific folding of polypeptide chains into distinct 3D structures, build complementary binding interfaces between members of macromolecular complexes or create enzyme catalytic sites by spatially arranging together different chemical groups (1). Despite the large diversity of existing proteins, it is clear that nature has not exhaustively sampled the set of macromolecules with specific functions that can be chemically synthesized. This is especially true because proteins are made out of a limited number (twenty in most cases) of genetically encoded amino acids. Although this limited number may confer advantages in terms of genetic encoding and synthesis of proteins in vivo, it clearly restricts protein’s functional diversity.
Introducing new chemistry into existing proteins is therefore an attractive strategy for biochemical studies, protein engineering and human therapeutics. A promising approach is to expand the repertoire of amino acids by introducing non-natural amino acids into existing proteins or peptides (2). Thereby, new regions of the chemical space can be explored that have not been already sampled along evolution. Non-natural amino acids, and especially l-alpha amino acids where the sidechain has only one single bond with the backbone (i.e. excluding amino acids with branching at Cα or bonds with other backbone atoms), are particularly interesting because they do not affect the chemical properties of the protein backbone. As such they are more likely to keep the global fold and secondary structure elements of a polypeptide chain unchanged. For instance, in a recent study, it was shown that non-natural sidechains dramatically increase the affinity of amyloid fiber inhibitors (3). Similarly, several cyclic and other kinds of modified peptides with non-natural amino acid sidechains have been developed for therapeutics use, such as cilengitide (4) or crafilzomib (5). Non-natural sidechains have also been used as independent ligands. For instance, l-3,4-dihydroxyphenylalanine is a non-natural amino acid used in Parkinson disease treatment (6), and 5-hydroxytryptophan (oxitriptan) has been used as an antidepressant (7). Another commonly used type of non-natural sidechains consists of d-amino acids. d-amino acids are enantiomers of l-amino acids where the sidechain and the hydrogen atom attached to the Cα atom have been switched. d-amino acids are known to increase resistance to protease degradation and are thus frequently used to enhance the biological stability of peptide-based drugs or biological tools (8,9).
In addition to therapeutics use, non-natural sidechains have found many other applications in biochemistry and protein studies (10). These include photo-crosslinking amino acids to probe in vivo protein interactions (11,12), fluorescent amino acids used as markers of specific proteins (13) or phosphorylated amino acid mimetics to probe the effect of post-translational modifications (14).
The importance of expanding the building blocks of proteins beyond the 20 natural ones is further emphasized by noting that a remarkably high degree of optimization is often observed in the choice of amino acid sequences for many proteins. Optimized sequences stand as an important hurdle to protein or peptide engineering based only on genetically encoded amino acids.
Experimental research on non-natural sidechains is based mainly on solid-phase synthesis technology (15) and genetically encoding non-natural sidechains into micro-organisms or cell cultures (2). The former technique uses in vitro chemical peptide synthesis to assemble polypeptide chains with both natural and non-natural residues. This technology is routinely used to synthesize non-natural peptides and can nowadays be applied to small proteins of up to hundred amino acids (16). The latter is typically based on generating novel unique codon–tRNA pairs, together with the corresponding aminoacyl tRNA synthetases in a given organism (2). This approach enables production of non-natural sidechain containing polypeptide chains in micro-organisms that can be used as in vivo biological tools (13) or as modified peptide libraries for ligand screening (17). Recent progresses currently enable encoding simultaneously hundreds of different non-natural sidechains in the same organism (18).
Bio- and chemo-informatics, structural modeling and visualization tools are powerful to guide and interpret experimental studies with both natural and non-natural amino acids. Recently, we and others developed different computational tools to better characterize non-natural sidechains and predict the effect of introducing them into proteins or peptides (19–22). This includes sidechain 3D coordinates, biomolecular parameters describing properties such as bond length flexibility, as well characterization of conformational diversity in terms of rotamers (23). For the latter, different strategies have been elaborated to predict rotamer probabilities, using either solely free energy–based calculations (20,22), or combining physics-based calculations together with statistical analysis of conserved properties in natural sidechain rotamer libraries (19).
Here, we introduce the SwissSidechain database, a unified and integrated resource providing access to curated biochemical and structural information for 210 non-natural sidechains. Many of the data are unique to SwissSidechain (e.g. rotamers, biomolecular parameters), whereas the rest has been collected from existing resources to provide a rapid access to this information. The SwissSidechain database further includes visualization and molecular modeling tools. Different options to browse the non-natural sidechains based on their chemical properties or their families are also provided. The database can be accessed at http://www.swisssidechain.ch.
Content of the SwissSidechain database
The SwissSidechain database contains molecular and structural data for 210 non-natural alpha amino acid sidechains, both in l- and d-configurations, in addition to the 20 natural ones. These amino acids have been selected based on two main criteria: first, the presence of non-natural sidechains in publicly available protein structures in the PDB (Protein Data Bank) (24) and second, the commercial availability. Each amino acid in SwissSidechain has been given a three- or four-letter code. For all sidechains present in the PDB, we kept the existing three-letter code. For other sidechains, a four-letter code was created. These include many d-amino acids whose l-configuration is present in the PDB. For them, a ‘d’ was simply added in front of the three-letter code (e.g. NLE stands for l-Norleucine and DNLE for d-Norleucine). For the rest, four-letter codes were chosen so as to recall as much as possible the full chemical name (e.g. AZDA for l-azido-alanine), and always starting with a ‘d’ for non-natural sidechains in d-configuration (e.g. DZDA for d-azido-alanine).
SwissSidechain data are available in different files describing the chemical structure (SMILES and 2D chemical drawing) and 3D coordinates (PDB, Mol2) of these sidechains, files describing their physico-chemical properties (molecular weight, volume, logP, pKa, partial charges, bond/angle/torsion constants), as well as files describing the probability of their possible conformations (rotamers). Among the different experimentally measurable chemical properties of non-natural sidechains in SwissSidechain, volumes have been calculated with CHARMM (Chemistry at HARvard Molecular Mechanics) (25) and molecular weights with OpenBabel (26). LogP values give information about the hydrophobicity. Experimental LogP for both the full amino acid and the sidechain only have been manually collected from literature when available. For the rest of the sidechains, LogP values have been predicted with XlogP3 (27). PKa values for each protonatable group of the full amino acids have been retrieved from literature when available and computed with MarvinSketch (ChemAxon, http://www.chemaxon.com) otherwise. Model parameters, such as bond, angle and torsion constants, which provide information about sidechain flexibility and dynamics, are provided as parameter files compatible with the CHARMM force field (28) and can be readily used in the CHARMM (25) and GROMACS (GROningen MAchine for Chemical Simulations) (29) molecular mechanics software. Similarly, partial atomic charges, which are useful to predict possible interactions (e.g. H-bond, electrostatic) of a sidechain with its surrounding atoms, are provided as topology files in the CHARMM (25) and GROMACS (29) format. The computation of these model parameters is described in detail elsewhere (19).
Finally, rotamers have been generated as described in Gfeller et al. (19). To include information about sidechain conformations in recent crystal structures, we developed the current version of SwissSidechain based on an up-to-date rotamer library for natural sidechains, instead of using rotamer libraries published in 2002 (23). In practice, we performed a global analysis of natural sidechain conformations in all crystallographic structures available in the PDB (as on May 2012) with resolution lower or equal to 1.75Å. This updated rotamer library of natural sidechains, and especially the conserved probabilities of the first dihedral angles of long sidechains, was used in all our analysis to predict rotamers for non-natural sidechains, as described in Gfeller et al. (19). We further expand rotamer libraries to d-amino acids, both natural and non-natural. For non-chiral sidechains, rotamer probabilities can be readily generated using the properties of mirror images. In particular, a d-configuration characterized by backbone dihedral angles 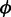 and
and 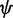 and sidechain dihedral angles χi is equivalent to the l-configuration with dihedral angles −
and sidechain dihedral angles χi is equivalent to the l-configuration with dihedral angles −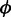 , −
, − and −χi (i = 1, … N, where N stands for the number of freely rotating dihedral angles along the sidechain). In other words,
and −χi (i = 1, … N, where N stands for the number of freely rotating dihedral angles along the sidechain). In other words, 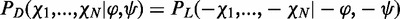 , where PD stands for the conformational probability of a d-amino acid and PL stands for the conformational probability of the same amino acid in l-configuration (30). Note that this does not consider interactions with other residues, neither whether the previous and the next residues are in l- or d-configuration. However, such information is typically not explicitly considered in rotamer libraries. For chiral sidechains, the previous relation does not hold because of the additional asymmetric centers. Therefore, we used the same strategy based on molecular dynamics simulations and renormalization that was used for l-sidechains in Gfeller et al. (19) to predict the rotamer libraries for chiral d-sidechains.
, where PD stands for the conformational probability of a d-amino acid and PL stands for the conformational probability of the same amino acid in l-configuration (30). Note that this does not consider interactions with other residues, neither whether the previous and the next residues are in l- or d-configuration. However, such information is typically not explicitly considered in rotamer libraries. For chiral sidechains, the previous relation does not hold because of the additional asymmetric centers. Therefore, we used the same strategy based on molecular dynamics simulations and renormalization that was used for l-sidechains in Gfeller et al. (19) to predict the rotamer libraries for chiral d-sidechains.
To help exploring the structural environment of non-natural sidechains incorporated in polypeptide chains, we developed visualization plugins for PyMOL and UCSF Chimera (31). These plugins enable users to mutate residues in protein structures to any sidechain (natural and non-natural, in both l- and d-configurations) present in the SwissSidechain database. This is particularly useful to evaluate the structural environment of a new residue and detect possible clashes that may arise on mutation. Moreover, the resulting structures can be readily used for future investigations with molecular mechanics software. Detailed instructions for installing and running these plugins are provided on the SwissSidechain web site, together with short movies describing standard use for mutating a residue to a non-natural sidechain in a protein structure.
Web interface
For each sidechain, all data are collected in dedicated web pages containing 2D and 3D structures, the different names, biochemical properties, links to existing databases and download options. An example is shown in Figure 1. The biochemical parameters listed on the right have been computed as described above. Links to existing databases include the PDB (24), the PDB Ligand Expo (32) and PubChem (33). Files to be downloaded consist of structure files containing 3D coordinates (PDB and Mol2 formats), 2D structures, rotamers (backbone dependent and independent) and topology files for molecular mechanics software such as CHARMM (25) and GROMACS (29) Parameter files are available at the Molecular Dynamics (MD) simulations pages. These data are provided for both l- and d-configurations. The amino acid 3D structures for the l-configuration can be visualized online thanks to the open-source Java viewer for chemical structures Jmol (http://www.jmol.org). In addition we provide bulk download options for all data at the download page of SwissSidechain.
Figure 1.

Sidechain general information page. 2D and 3D views of the full amino acids are provided. Names, SMILES and several biochemical parameters are listed on the right part of the page, as well as links to other structural or chemical databases. Download options are provided in the lower part.
Browsing options
SwissSidechain data can be browsed in large alphabetical tables containing the three- or four-letter codes and the sidechain full names, 2D structures, internal links to the sidechain pages, external links to the PDB, as well as download links for structural files. Both a full table and tables restricted to sub-families of natural sidechain derivatives (e.g. methionine derivatives) are provided.
Another alternative is to query data based on sidechain physico-chemical properties. Two particularly important parameters are the volume and the hydrophobicity (logP) of the sidechains. In SwissSidechain, we provide an interactive 2D plot of these parameters. This enables users to zoom, visualize and select specific subsets of non-natural sidechains (see Figure 2). Sidechains can be selected by clicking on the corresponding circles or selecting a region of the graph and using the ‘select all’ button. Selected sidechains appear in the box below the graph and links to sidechain pages can be followed from there. This approach is especially useful for protein engineering and ligand design, to select a subset of sidechains with specific properties.
Figure 2.

Sidechain chemical properties browsing plot. Each sidechain is represented by a yellow circle positioned according to the sidechain volume and logP. On mouseover, the amino acid 2D structure and full name appear. Sidechains can be selected by clicking on the circles or using the ‘select all’ button to select all sidechains in the plot. Selected sidechains appear in the box below the plot, where users can follow the links to the sidechain web page (see Figure 1) or download all structural and molecular files. Users can zoom on the graph by selecting subregions (see red rectangle in the overview panel). Specific families of sidechain derivatives can be selected in the menu on the left.
CONCLUSION AND OUTLOOK
Exploiting naturally evolved, and often well optimized, proteins or peptides while not being restricted to the limited set of 20 natural amino acids is a promising approach for protein engineering with applications in biochemistry, protein structure and function analysis, and for drug design (3,10). Toward this goal, non-natural sidechains are being increasingly used in experimental studies. The SwissSidechain database provides a unified web resource for hundreds of non-natural sidechains. The biochemical parameters allow researchers to rapidly retrieve sidechains with specific properties, such as volume or hydrophobicity. The different visualization tools enable seamless introduction of non-natural sidechains in protein and peptide structures. The model parameters, such as bond and angle flexibility or partial atomic charges are useful to investigate non-natural sidechains with molecular mechanics software. Most of the non-natural sidechains are commercially available, so that researchers interested in using the predictions obtained with the SwissSidechain data can easily test them experimentally. The naming is consistent with the PDB so that published experimental structures can be readily analyzed with the tools provided in SwissSidechain.
In the current version of SwissSidechain, we focus on alpha amino acid sidechains that do not modify the backbone atoms (i.e. without branching at Cα, modification of the nitrogen atom or longer backbones such as beta amino acids). These amino acids in their l-configuration are less likely to lead to large structural remodeling when incorporated into proteins or peptides. Therefore they are often used in protein engineering experiments, and predictions of their effect on protein structure and function are in general more accurate. The same amino acids in d-configuration have found many applications in biochemistry and drug discovery. Better characterizing d-amino acid properties is also useful to study d-retro-inverso peptides that can provide mimetics of l-peptides (34), or d-peptides obtained by screening l-peptides against d-targets and using the mirror-image property of d-enantiomers (35). Structural modeling of these types of non-natural sidechains is becoming feasible, although it is still more challenging. This is for instance the case with peptides binding to peptide recognition domains, such as Post synaptic density protein, Drosophila disc large tumor suppressor, and Zonula occludens-1 protein or Src Homology 3 domains (36,37). As the binding interface is often relatively short, one may be able to probe in silico the effect of alpha-amino acids in d-configuration (38). For these different reasons, we have included data for the d-configuration of all amino acid sidechains present in the current version of SwissSidechain.
As experimental techniques keep progressing, more and more non-natural sidechains will become commercially available or be encountered in protein structures. We plan to include these new sidechains in future updates of the SwissSidechain database. Other developments may include focusing on different families of non-natural sidechains, such as fluorescent or photo-cleavable amino acids.
FUNDING
European Molecular Biology Organization long-term fellowship [ALTF 241-2010 to D.G.]; Swiss Institute of Bioinformatics. Funding for open access charge: Swiss Institute of Bioinformatics.
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
The authors would like to thank Eric Pettersen for valuable help in building the UCSF Chimera plugin, and Aurélien Grosdidier for insightful comments on the database.
REFERENCES
- 1.Alberts B, Johnson A, Lewis J, Raff M, Roberts K, Walter P. Molecular Biology of the Cell. 5th edn. New York: Garland Science; 2007. [Google Scholar]
- 2.Wang L, Xie J, Schultz PG. Expanding the genetic code. Annu. Rev. Biophys. Biomol. Struct. 2006;35:225–249. doi: 10.1146/annurev.biophys.35.101105.121507. [DOI] [PubMed] [Google Scholar]
- 3.Sievers SA, Karanicolas J, Chang HW, Zhao A, Jiang L, Zirafi O, Stevens JT, Munch J, Baker D, Eisenberg D. Structure-based design of non-natural amino-acid inhibitors of amyloid fibril formation. Nature. 2011;475:96–100. doi: 10.1038/nature10154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Burke PA, DeNardo SJ, Miers LA, Lamborn KR, Matzku S, DeNardo GL. Cilengitide targeting of alpha(v)beta(3) integrin receptor synergizes with radioimmunotherapy to increase efficacy and apoptosis in breast cancer xenografts. Cancer Res. 2002;62:4263–4272. [PubMed] [Google Scholar]
- 5.Parlati F, Lee SJ, Aujay M, Suzuki E, Levitsky K, Lorens JB, Micklem DR, Ruurs P, Sylvain C, Lu Y, et al. Carfilzomib can induce tumor cell death through selective inhibition of the chymotrypsin-like activity of the proteasome. Blood. 2009;114:3439–3447. doi: 10.1182/blood-2009-05-223677. [DOI] [PubMed] [Google Scholar]
- 6.Smith Y, Wichmann T, Factor SA, DeLong MR. Parkinson's disease therapeutics: new developments and challenges since the introduction of levodopa. Neuropsychopharmacology. 2012;37:213–246. doi: 10.1038/npp.2011.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Turner EH, Blackwell AD. 5-Hydroxytryptophan plus SSRIs for interferon-induced depression: synergistic mechanisms for normalizing synaptic serotonin. Med. Hypotheses. 2005;65:138–144. doi: 10.1016/j.mehy.2005.01.026. [DOI] [PubMed] [Google Scholar]
- 8.Welch BD, VanDemark AP, Heroux A, Hill CP, Kay MS. Potent D-peptide inhibitors of HIV-1 entry. Proc. Natl Acad. Sci. USA. 2007;104:16828–16833. doi: 10.1073/pnas.0708109104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schorderet DF, Manzi V, Canola K, Bonny C, Arsenijevic Y, Munier FL, Maurer F. D-TAT transporter as an ocular peptide delivery system. Clin. Experiment. Ophthalmol. 2005;33:628–635. doi: 10.1111/j.1442-9071.2005.01108.x. [DOI] [PubMed] [Google Scholar]
- 10.Wang Q, Parrish AR, Wang L. Expanding the genetic code for biological studies. Chem. Biol. 2009;16:323–336. doi: 10.1016/j.chembiol.2009.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ai HW, Shen W, Sagi A, Chen PR, Schultz PG. Probing protein-protein interactions with a genetically encoded photo-crosslinking amino acid. Chembiochem. 2011;12:1854–1857. doi: 10.1002/cbic.201100194. [DOI] [PubMed] [Google Scholar]
- 12.Kessler B, Michielin O, Blanchard CL, Apostolou I, Delarbre C, Gachelin G, Gregoire C, Malissen B, Cerottini JC, Wurm F, et al. T cell recognition of hapten. Anatomy of T cell receptor binding of a H-2kd-associated photoreactive peptide derivative. J. Biol. Chem. 1999;274:3622–3631. doi: 10.1074/jbc.274.6.3622. [DOI] [PubMed] [Google Scholar]
- 13.Wang J, Xie J, Schultz PG. A genetically encoded fluorescent amino acid. J. Am. Chem. Soc. 2006;128:8738–8739. doi: 10.1021/ja062666k. [DOI] [PubMed] [Google Scholar]
- 14.Lemke EA, Summerer D, Geierstanger BH, Brittain SM, Schultz PG. Control of protein phosphorylation with a genetically encoded photocaged amino acid. Nat. Chem. Biol. 2007;3:769–772. doi: 10.1038/nchembio.2007.44. [DOI] [PubMed] [Google Scholar]
- 15.Kent SB. Chemical synthesis of peptides and proteins. Annu. Rev. Biochem. 1988;57:957–989. doi: 10.1146/annurev.bi.57.070188.004521. [DOI] [PubMed] [Google Scholar]
- 16.Mandal K, Kent SB. Total chemical synthesis of biologically active vascular endothelial growth factor. Angew. Chem. Int. Ed. Engl. 2011;50:8029–8033. doi: 10.1002/anie.201103237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tian F, Tsao ML, Schultz PG. A phage display system with unnatural amino acids. J. Am. Chem. Soc. 2004;126:15962–15963. doi: 10.1021/ja045673m. [DOI] [PubMed] [Google Scholar]
- 18.Neumann H, Wang K, Davis L, Garcia-Alai M, Chin JW. Encoding multiple unnatural amino acids via evolution of a quadruplet-decoding ribosome. Nature. 2010;464:441–444. doi: 10.1038/nature08817. [DOI] [PubMed] [Google Scholar]
- 19.Gfeller D, Michielin O, Zoete V. Expanding molecular modeling and design tools to non-natural sidechains. J. Comput. Chem. 2012;55:1525–1535. doi: 10.1002/jcc.22982. [DOI] [PubMed] [Google Scholar]
- 20.Renfrew PD, Choi EJ, Bonneau R, Kuhlman B. Incorporation of noncanonical amino acids into Rosetta and use in computational protein-peptide interface design. PLoS One. 2012;7:e32637. doi: 10.1371/journal.pone.0032637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Revilla-Lopez G, Rodriguez-Ropero F, Curco D, Torras J, Isabel Calaza M, Zanuy D, Jimenez AI, Cativiela C, Nussinov R, Aleman C. Integrating the intrinsic conformational preferences of noncoded alpha-amino acids modified at the peptide bond into the noncoded amino acids database. Proteins. 2011;79:1841–1852. doi: 10.1002/prot.23009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Revilla-Lopez G, Torras J, Curco D, Casanovas J, Calaza MI, Zanuy D, Jimenez AI, Cativiela C, Nussinov R, Grodzinski P, et al. NCAD, a database integrating the intrinsic conformational preferences of non-coded amino acids. J. Phys. Chem. B. 2010;114:7413–7422. doi: 10.1021/jp102092m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dunbrack RL., Jr Rotamer libraries in the 21st century. Curr. Opin. Struct. Biol. 2002;12:431–440. doi: 10.1016/s0959-440x(02)00344-5. [DOI] [PubMed] [Google Scholar]
- 24.Rose PW, Beran B, Bi C, Bluhm WF, Dimitropoulos D, Goodsell DS, Prlic A, Quesada M, Quinn GB, Westbrook JD, et al. The RCSB Protein Data Bank: redesigned web site and web services. Nucleic Acids Res. 2011;39:D392–D401. doi: 10.1093/nar/gkq1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brooks BR, Brooks CL, 3rd, Mackerell AD, Jr, Nilsson L, Petrella RJ, Roux B, Won Y, Archontis G, Bartels C, Boresch S, et al. CHARMM: the biomolecular simulation program. J. Comput. Chem. 2009;30:1545–1614. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.O'Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, Hutchison GR. Open Babel: An open chemical toolbox. J. Cheminform. 2011;3:33. doi: 10.1186/1758-2946-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang R, Fu Y, Lai L. A new atom-additive method for calculating partition coefficients. J. Chem. Inf. Model. 1997;37:615–621. [Google Scholar]
- 28.MacKerell JAD, Bashford D, Bellott M, Dunbrack RL, Jr, Evanseck JD, Field MJ, Fischer S, Gao J, Guo H, Ha S, et al. All-atom empirical potential for molecular modeling and dynamics Studies of proteins. J. Phys. Chem. G. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 29.Van Der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJ. GROMACS: fast, flexible, and free. J. Comput. Chem. 2005;26:1701–1718. doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- 30.Mitchell JB, Smith J. D-amino acid residues in peptides and proteins. Proteins. 2003;50:563–571. doi: 10.1002/prot.10320. [DOI] [PubMed] [Google Scholar]
- 31.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 32.Feng Z, Chen L, Maddula H, Akcan O, Oughtred R, Berman HM, Westbrook J. Ligand Depot: a data warehouse for ligands bound to macromolecules. Bioinformatics. 2004;20:2153–2155. doi: 10.1093/bioinformatics/bth214. [DOI] [PubMed] [Google Scholar]
- 33.Wang Y, Xiao J, Suzek TO, Zhang J, Wang J, Zhou Z, Han L, Karapetyan K, Dracheva S, Shoemaker BA, et al. PubChem's BioAssay Database. Nucleic Acids Res. 2012;40:D400–D412. doi: 10.1093/nar/gkr1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cardo-Vila M, Giordano RJ, Sidman RL, Bronk LF, Fan Z, Mendelsohn J, Arap W, Pasqualini R. From combinatorial peptide selection to drug prototype (II): targeting the epidermal growth factor receptor pathway. Proc. Natl Acad. Sci. USA. 2010;107:5118–5123. doi: 10.1073/pnas.0915146107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Funke SA, Willbold D. Mirror image phage display—a method to generate D-peptide ligands for use in diagnostic or therapeutical applications. Mol. Biosyst. 2009;5:783–786. doi: 10.1039/b904138a. [DOI] [PubMed] [Google Scholar]
- 36.Tonikian R, Xin X, Toret CP, Gfeller D, Landgraf C, Panni S, Paoluzi S, Castagnoli L, Currell B, Seshagiri S, et al. Bayesian modeling of the yeast SH3 domain interactome predicts spatiotemporal dynamics of endocytosis proteins. PLoS Biol. 2009;7:e1000218. doi: 10.1371/journal.pbio.1000218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gfeller D, Butty F, Wierzbicka M, Verschueren E, Vanhee P, Huang H, Ernst A, Dar N, Stagljar I, Serrano L, et al. The multiple-specificity landscape of modular peptide recognition domains. Mol. Syst. Biol. 2011;7:484. doi: 10.1038/msb.2011.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yongye AB, Li Y, Giulianotti MA, Yu Y, Houghten RA, Martinez-Mayorga K. Modeling of peptides containing D-amino acids: implications on cyclization. J. Comput. Aided. Mol. Des. 2009;23:677–689. doi: 10.1007/s10822-009-9295-y. [DOI] [PubMed] [Google Scholar]