

Abstract
Complete knowledge of all direct and indirect interactions between proteins in a given cell would represent an important milestone towards a comprehensive description of cellular mechanisms and functions. Although this goal is still elusive, considerable progress has been made—particularly for certain model organisms and functional systems. Currently, protein interactions and associations are annotated at various levels of detail in online resources, ranging from raw data repositories to highly formalized pathway databases. For many applications, a global view of all the available interaction data is desirable, including lower-quality data and/or computational predictions. The STRING database (http://string-db.org/) aims to provide such a global perspective for as many organisms as feasible. Known and predicted associations are scored and integrated, resulting in comprehensive protein networks covering >1100 organisms. Here, we describe the update to version 9.1 of STRING, introducing several improvements: (i) we extend the automated mining of scientific texts for interaction information, to now also include full-text articles; (ii) we entirely re-designed the algorithm for transferring interactions from one model organism to the other; and (iii) we provide users with statistical information on any functional enrichment observed in their networks.
INTRODUCTION
Highly complex organisms and behaviors can arise from a surprisingly restricted set of existing gene families (1,2), by a tightly regulated network of interactions among the proteins encoded by the genes. This functional web of protein–protein links extends well beyond direct physical interactions only; indeed, physical interactions might also be rather limited, covering perhaps <1% of the theoretically possible interaction space (3). Proteins do not necessarily need to undergo a stable physical interaction to have a specific, functional interplay: they can catalyze subsequent reactions in a metabolic pathway, regulate each other transcriptionally or post-transcriptionally, or jointly contribute to larger, structural assemblies without ever making direct contact. Together with direct, physical interactions, such indirect interactions constitute the larger superset of ‘functional protein–protein associations’ or ‘functional protein linkages’ (4,5).
Protein–protein associations have proven to be a useful concept, by which to group and organize all protein-coding genes in a genome. The complete set of associations can be assembled into a large network, which captures the current knowledge on the functional modularity and interconnectivity in the cell. Apart from ad hoc use—i.e. by browsing networks for genes of interest, inspecting interaction evidence or performing interactive clustering—a variety of systematic and large-scale usage scenarios for functional association networks have emerged. For example, (i) association networks have been frequently used to interpret the results of genome-wide genetic screens, in particular RNAi perturbation screens (6–9). Because such screens can be noisy and difficult to interpret, any protein-network information that may help to connect potential hits can serve to provide additional confidence, particularly if a number of hits can be observed in a densely connected functional module in the network. (ii) Protein network information can aid in the interpretation of functional genomics data, e.g. in systematic proteomics surveys (10–12). This is particularly useful when the proteomics data themselves contain a protein–protein association component, such as in MS-based interaction discovery or in large-scale enzyme/substrate analysis. (iii) Protein association networks have also proven surprisingly useful for the elucidation of disease genes, both for Mendelian and for complex diseases (13–15). For the latter application, the networks can help to constrain the search space—genomic regions encompassing more than one candidate gene, or lists of genes observed to be mutated in sequencing studies, can be filtered for those genes that have connections to known disease genes (or for genes having above-random connectivity among themselves).
The STRING database has been designed with the goal to assemble, evaluate and disseminate protein–protein association information, in a user-friendly and comprehensive manner. As interactions between proteins represent such a crucial component for modern biology, STRING is by far not the only online resource dedicated to this topic. Apart from the primary databases that hold the experimental data in this field (16–20) and hand-curated databases serving expert annotations (21,22), a number of resources take a meta-analysis approach, similar to STRING. These include GeneMANIA (23), ConsensusPathDB (24), I2D (25), VisANT (26) and, more recently, hPRINT (27), HitPredict (28), IMID (29) and IMP (30). Within this wide variety of online resources and databases dedicated to interactions, STRING specializes in three ways: (i) it provides uniquely comprehensive coverage, with >1000 organisms, 5 million proteins and >200 million interactions stored; (ii) it is one of very few sites to hold experimental, predicted and transferred interactions, together with interactions obtained through text mining; and (iii) it includes a wealth of accessory information, such as protein domains and protein structures, improving its day-to-day value for users.
We have already discussed many aspects of the STRING resource previously, e.g. (31,32), including its data-sources, prediction algorithms and user-interface. Here, we describe the current update to version 9.1 of the resource, focusing on new features and updated algorithms. In particular, we will describe how STRING increasingly makes use of externally provided orthology information [from the eggNOG database (33)] to better integrate evidence across distinct organisms.
UPDATED TEXT MINING
The new version of STRING features a redesigned text-mining pipeline. We have improved the named entity recognition engine to use custom-made hashing and string-compare functions to comprehensively and efficiently handle orthographic variation related to whether a name is written as one word, two words or with a hyphen. As in the previous versions of STRING, associations between proteins are derived from statistical analysis of co-occurrence in documents and from natural language processing. The latter combines part-of-speech tagging, semantic tagging and a chunking grammar to achieve rule-based extraction of physical and regulatory interactions, as described previously (34).
To improve the quality and number of links derived from co-occurrence, we have developed an entirely new scoring scheme, which takes into account co-occurrences within sentences, within paragraphs and within whole documents and combines them through an optimized weighting scheme.
The scoring scheme first calculates a weighted count (Cij) for each pair of entities i and j:
 |
where wd = 1, wp = 2 and ws = 0.2 are the weights for co-occurrence within the same document, same paragraph and same sentence, respectively. The delta functions δdijk, δpijk and δsijk are 1, if the entities i and j are co-mentioned in the document k, a paragraph of k or a sentence of k. Based on the weighted counts, the co-occurrence score (Sij) is defined as:
 |
where Ci• and C•j are the sums over all pairs involving i or j and an entity from the same taxon, C•• is the sum over all pairs of entities from the taxon, and α = 0.6. The parameters were optimized on the KEGG benchmark set.
This has substantially improved the quality and number of associations extracted (Table 1). The more efficient named entity recognition engine and the new scoring scheme also enabled us to move beyond the parsing of MEDLINE abstracts, and to now include text mining of 1 821 983 full-text articles, which were freely available from publishers web sites. This has further improved the comprehensiveness of the text mining in the new version of STRING (Table 1). The natural language processing part of the pipeline has also been standardized, to make use of an ontology that describes possible molecular modes of action by which proteins can influence each other (35). Finally, the new text-mining pipeline explicitly takes into account orthology information by treating each orthologous group as an entity that is considered whenever one of its member proteins is mentioned (33), thereby directly detecting associations between orthologous groups as well as between proteins.
Table 1.
Protein–protein associations based on automated text mining
| STRING v9.0 | STRING v9.1 | Fold increase | |
|---|---|---|---|
| Natural language processing | 38 859 | 63 331 | 1.629 |
| Cooccurrence, high confidence | 286 880 | 792 730 | 2.763 |
| Cooccurrence, medium confidence | 1 100 756 | 1 672 222 | 1.519 |
| Cooccurrence, low confidence | 3 214 754 | 4 270 322 | 1.328 |
This table quantifies non-redundant associations extracted by text mining in STRING, at various confidence levels; note that both STRING versions shown here are based on the same set of organisms and proteins. The increase in text-mining interactions is largest in the high confidence bracket, reflecting the increased performance enabled by the extension to full text articles, and by the improved entity recognition engine.
TRANSFER OF INTERACTIONS BETWEEN ORGANISMS
Evolutionarily related proteins are known to usually maintain their three-dimensional structure, even when they have become so diverged over time that there is hardly any detectable sequence similarity left between them (36,37). Similarly, most protein–protein interaction interfaces remain well-conserved over time, at least for the case of stably bound protein partners located next to each other in protein complexes (38,39). This means that a pair of proteins observed to be stably binding in one organism can be expected to be binding in another organism as well, provided both genes have been retained in both genomes. The term ‘interologs’ was coined for such pairs, a combination of the words ‘interaction’ and ‘ortholog’ (40). Whether this high degree of interaction conservation is true also for other, more indirect or transient types of protein–protein associations is less clear—although at least one such type, namely joint metabolic pathway membership, has also been shown to be generally well-conserved (41,42). Based on the principle of interaction conservation, evidence transfer from one model organism to the other seems feasible, and it has been implemented in several frameworks already.
In practice, the search for potential interologs is not trivial, except for very closely related organisms. The reason for this lies in the high frequency of gene duplications, gene losses and gene re-arrangements, which makes it difficult to assign pairs of functionally equivalent genes across distant organisms. The best candidates for functionally equivalent genes in two organisms are ‘one-to-one’ orthologs, i.e. genes that track back to a single gene in the last common ancestor of both organisms, and have since undergone little or no duplication or loss events (43–45). In a large resource such as STRING, unequivocally identifying one-to-one orthologs for all pairs of organisms is not feasible: there are potentially more than a million pairs of organisms to study, each with thousands of genes, and the proper identification of orthologs would ideally entail exhaustive and time-consuming phylogenetic tree analysis. In the past, STRING has therefore used two distinct heuristic options: either to substitute homology for orthology (46) or to use pre-defined orthology relations described at high-level taxonomic groups, from the COG database (47). We found that both approaches were suboptimal; they both transferred evidence even when the presence of multiple paralogs indicated that the orthology situation was somewhat unclear—despite an explicit procedure to down-weigh the transferred scores in such cases, at least in the homology approach (46). We have, therefore, now devised a procedure that more explicitly considers the known phylogeny of organisms and which works on the basis of hierarchical orthologous groups maintained at the eggNOG database (33).
The taxonomy tree covering the 1133 species present in STRING consists of 495 branching nodes at different taxonomic positions (the tree is a down-sampled version of the taxonomy maintained at NCBI). Through experimentation and benchmarking, we have developed a new two-step procedure, which makes use of this tree for the transfer of functional associations. First, associations between proteins are transferred to the orthologous groups to which the proteins belong; this proceeds sequentially from lower to increasingly higher levels of taxonomic hierarchy. Second, associations are transferred in the opposite direction, i.e. from the orthologous groups back to their constituent proteins. Where available, the hierarchical orthology groups from eggNOG version 3 are used (33). As many of the taxonomic positions in the tree are not covered in eggNOG, we construct provisional groups for the missing positions by down-sampling the orthologous groups from the next higher taxonomy level present in eggNOG.
To compute a score of functional association (Sabk) between two orthologous groups a and b at the taxonomic level k, we sort the n associations (Pabi) between their member proteins from highest to lowest score, and then integrate them sequentially (Figure 1):
 |
where p′ is prior probability of two proteins being linked, which is 0.063 according to the KEGG benchmark set; fabi is a penalty dependent on the number of paralogs of a given protein pair and dij is a penalty dependent on the similarity of the species i and the other species j that have already been included in the score:
where cai and cbi are the number of proteins from a given species in the orthologous groups, and sij the median similarity between the given species, measured on a universal set of marker gene families (48) and expressed as the ‘self-normalized bit-score’ (i.e. the bit score of an alignment between two proteins, which is divided by the bit score of a self-alignment of the shorter of the two proteins; this measure always ranges from zero to one).
Figure 1.

Improved procedure for interaction transfer between organisms. Left: steps 1 and 2 of the functional association transfer pipeline. In the first step, the individual links between proteins are combined into a score between orthologous groups, sequentially, from the strongest link (thick line) to the weakest (thin). Each subsequent score is down-weighted, both based on the similarity of its organism to organisms that have already contributed to the combined scores, and on number of proteins from the same organism inside the orthologous group. In the second step of the transfer pipeline, the links between orthologous groups are transferred back to individual protein pairs belonging to these groups. This is done sequentially from the lowest to highest taxonomy level. In the above example, the two transferred links from the highest taxonomic level (orange links) are penalized for the increase in number of proteins from the target species in one of the orthologous groups. Right: ROC curves indicating the performance of predicted interolog scores, benchmarked against KEGG pathways; an inferred link between two proteins is considered to be a true positive when both proteins are annotated to be together in at least one shared KEGG pathway.
The process is repeated for all pairs of orthologous groups at every taxonomic level. Next, the scores between pairs of orthologous groups are transferred back to protein pairs; this finally results in the actual evidence transfer between organisms. To calculate the transferred score (Tim) from all taxonomic levels m to a protein pair from species i, we combine the scores (Sabk) from orthologous groups consecutively from the lowest to the highest taxonomy level, subtracting the contributions from all lower taxonomic levels (Figure 1):
 |
where at each taxonomic level, we subtract the part of the score that originates from the species itself (Pabi) while additionally penalizing it for the number of paralogs in the respective orthologous groups (fabi) and for the median self-normalized bit scores (sa and sb) of the proteins in the groups a and b.
The parameters α, ε and γ are universal in the sense that they have the same values for all evidence channels in STRING, e.g. co-occurence, experiments and text mining, whereas β and δ are channel specific to take into account the different rate at which scores become independent from each other. The new transfer scheme was optimized and benchmarked on the set of known interactions in the KEGG database and achieves better performance than the previous method, both for orthologous groups and for individual proteins (Figure 1).
STATISTICAL ENRICHMENT ANALYSIS
STRING users that do not just query with a single protein of interest, but instead upload entire lists of proteins, are often interested in knowing whether their input shows evidence for a statistical enrichment of any known biological function or pathway. To address this question, a variety of dedicated online resources are already available (49,50), most notably the DAVID resource (51). However, entering gene lists at multiple websites can be cumbersome, and not all existing resources will make full use of the latest protein network information. Therefore, we have now included functionality to detect enrichment of functional systems in each currently displayed network in STRING, testing a number of functional annotation spaces including Gene Ontology, KEGG, Pfam and InterPro (see Figure 2). Any detected enrichments can be browsed interactively, visually highlighting the corresponding proteins in the network (Figure 2).
Figure 2.

Network visualization and statistical analysis of a user-supplied protein list. The STRING screenshot shows a user-supplied set of genes, here a selection of cancer genes as annotated at the COSMIC database (52). The set is restricted to those genes that are known to pre-dispose to cancer already when mutated in the germline, and that have at least one connection in STRING. The inset illustrates the website’s new functionality for automatically detecting statistically enriched functions or processes in a network. In this example, one of the detected processes (nucleotide excision repair) is of interest and has been selected; STRING automatically highlighted the corresponding nodes in the network, where they are seen to form a densely connected module.
In the Enrichment widget, STRING displays every functional pathway/term that can be associated to at least one protein in the network. The terms are sorted by their enrichment P-value, which we compute using a Hypergeometric test, as explained in (53). The P-values are corrected for multiple testing using the method of Benjamini and Hochberg (54), but we also provide options to either disable that correction or to select a more stringent statistical test (Bonferroni). In the case of testing for Gene Ontology enrichments, users have the additional options to exclude annotations inferred by automatic procedures only (Electronic Inferred Associations), to limit the testing to pre-defined higher level categories (GO Slim), or to prune away parent terms that are redundant with child terms (i.e. covering the exact same set of proteins).
Furthermore, we report to the user whether the protein list is enriched in STRING interactions per se, independent of known pathway annotations. The latter functionality is non-trivial and requires an explicit null model, owing to the non-uniform distribution of the connectivity degrees of proteins in networks (9,55–57). We chose a random background model that preserves the degree distribution of the proteins in a given list: the Random Graph with Given Degree Sequence (RGGDS), similar to references (55,57).
Given a list 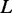 of proteins, let
of proteins, let 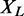 denote the number of edges connecting proteins in an RGGDS with similar size as
denote the number of edges connecting proteins in an RGGDS with similar size as 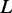 . For the given
. For the given 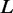 , a strong edge enrichment corresponds to a low probability of counting, in the RGGDS, at least the observed number
, a strong edge enrichment corresponds to a low probability of counting, in the RGGDS, at least the observed number 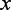 of edges connecting proteins in
of edges connecting proteins in 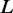 , i.e. a low value of:
, i.e. a low value of:
The random variable 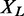 is a sum of Bernoulli variables with distinct parameters, and hence a Poisson–Binomial variable. If
is a sum of Bernoulli variables with distinct parameters, and hence a Poisson–Binomial variable. If 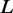 is large,
is large, 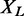 can thus be approximated by a Poisson random variable, whose cumulative probability function is:
can thus be approximated by a Poisson random variable, whose cumulative probability function is:
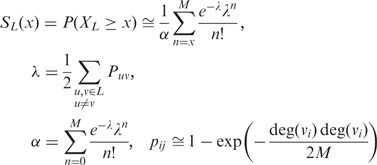 |
with M being the total number of interactions within L in STRING, and deg(v) denoting the degree of protein v, i.e. the number of interaction partners it has.
USER INTERFACE
The STRING website aims to provide easy and intuitive interfaces for searching and browsing the protein interaction data, as well as for inspecting the underlying evidence. Users can query for a single protein of interest, or for a set of proteins, using a variety of different identifier name spaces. The resulting network can then be inspected, rearranged interactively or clustered at variable stringency. Each protein node in the network shows a preview to 3D structural information, if available, and can be clicked to reveal a pop-up window with more information about the protein [including its annotation (58), SMART domain-structure (59), structure homology models from SWISS-MODEL Repository (60), etc.]. Each edge in the network denotes a known or predicted interaction, and leads to a pop-up window providing details on the underlying evidence and the interaction confidence scores.
An important new feature in version 9.1 of STRING is the possibility for users to identify themselves by logging in. Although this is not necessary for basic browsing and searching, it provides users with the option to browse their history of past searches, save visited pages for later return and upload lists of proteins that are of interest to them. In addition, logging in is useful for storing and retrieving ‘payload’ information to be shown and browsed alongside the network. As described previously (31), ‘payload’ information is user-provided extra data that can be projected onto the STRING network; it can consist of information regarding both nodes (proteins) and edges (interactions). Previously, any payload information had to be communicated to STRING via a set of files following a specific format—now, they can be uploaded and managed interactively.
FUNDING
The Swiss Institute of Bioinformatics (SIB) provides sustained funding for this project. Work on the project has also been supported in part by the Novo Nordisk Foundation Center for Protein Research and the European Molecular Biology Laboratory (EMBL). Funding for open access charge: University of Zurich.
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
The authors wish to thank Yan P. Yuan (EMBL) for excellent administrative support with the STRING backend servers, and Carlos García Girón (Sanger Institute) for help in implementing the user-payload-data mechanism.
REFERENCES
- 1.Chothia C. Proteins. One thousand families for the molecular biologist. Nature. 1992;357:543–544. doi: 10.1038/357543a0. [DOI] [PubMed] [Google Scholar]
- 2.Wolf YI, Grishin NV, Koonin EV. Estimating the number of protein folds and families from complete genome data. J.Mol. Biol. 2000;299:897–905. doi: 10.1006/jmbi.2000.3786. [DOI] [PubMed] [Google Scholar]
- 3.Aloy P, Russell RB. Ten thousand interactions for the molecular biologist. Nature Biotechnol. 2004;22:1317–1321. doi: 10.1038/nbt1018. [DOI] [PubMed] [Google Scholar]
- 4.Huynen M, Snel B, Lathe W, 3rd, Bork P. Predicting protein function by genomic context: quantitative evaluation and qualitative inferences. Genome Res. 2000;10:1204–1210. doi: 10.1101/gr.10.8.1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Eisenberg D, Marcotte EM, Xenarios I, Yeates TO. Protein function in the post-genomic era. Nature. 2000;405:823–826. doi: 10.1038/35015694. [DOI] [PubMed] [Google Scholar]
- 6.Gonzalez O, Zimmer R. Contextual analysis of RNAi-based functional screens using interaction networks. Bioinformatics. 2011;27:2707–2713. doi: 10.1093/bioinformatics/btr469. [DOI] [PubMed] [Google Scholar]
- 7.Simpson JC, Joggerst B, Laketa V, Verissimo F, Cetin C, Erfle H, Bexiga MG, Singan VR, Heriche JK, Neumann B, et al. Genome-wide RNAi screening identifies human proteins with a regulatory function in the early secretory pathway. Nature Cell Biol. 2012;14:764–774. doi: 10.1038/ncb2510. [DOI] [PubMed] [Google Scholar]
- 8.Moreau D, Kumar P, Wang SC, Chaumet A, Chew SY, Chevalley H, Bard F. Genome-wide RNAi screens identify genes required for Ricin and PE intoxications. Dev. Cell. 2011;21:231–244. doi: 10.1016/j.devcel.2011.06.014. [DOI] [PubMed] [Google Scholar]
- 9.Kaplow IM, Singh R, Friedman A, Bakal C, Perrimon N, Berger B. RNAiCut: automated detection of significant genes from functional genomic screens. Nat. Methods. 2009;6:476–477. doi: 10.1038/nmeth0709-476. [DOI] [PubMed] [Google Scholar]
- 10.Goh WW, Lee YH, Chung M, Wong L. How advancement in biological network analysis methods empowers proteomics. Proteomics. 2012;12:550–563. doi: 10.1002/pmic.201100321. [DOI] [PubMed] [Google Scholar]
- 11.Oppermann FS, Grundner-Culemann K, Kumar C, Gruss OJ, Jallepalli PV, Daub H. Combination of chemical genetics and phosphoproteomics for kinase signaling analysis enables confident identification of cellular downstream targets. Mol. Cell. Proteomics. 2012;11 doi: 10.1074/mcp.O111.012351. O111 012351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Olsson N, James P, Borrebaeck CA, Wingren C. Quantitative proteomics targeting classes of motif-containing peptides using immunoaffinity-based mass spectrometry. Mol. Cell. Proteomics. 2012;11:342–354. doi: 10.1074/mcp.M111.016238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee I, Blom UM, Wang PI, Shim JE, Marcotte M. Prioritizing candidate disease genes by network-based boosting of genome-wide association data. Genome Res. 2011;21:1109–1121. doi: 10.1101/gr.118992.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moreau Y, Tranchevent LC. Computational tools for prioritizing candidate genes: boosting disease gene discovery. Nat. Rev. Genet. 2012;13:523–536. doi: 10.1038/nrg3253. [DOI] [PubMed] [Google Scholar]
- 15.Piro RM, Di Cunto F. Computational approaches to disease-gene prediction: rationale, classification and successes. FEBS J. 2012;279:678–696. doi: 10.1111/j.1742-4658.2012.08471.x. [DOI] [PubMed] [Google Scholar]
- 16.Stark C, Breitkreutz BJ, Chatr-Aryamontri A, Boucher L, Oughtred R, Livstone MS, Nixon J, Van Auken K, Wang X, Shi X, et al. The BioGRID interaction database: 2011 update. Nucleic Acids Res. 2011;39:D698–D704. doi: 10.1093/nar/gkq1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kerrien S, Aranda B, Breuza L, Bridge A, Broackes-Carter F, Chen C, Duesbury M, Dumousseau M, Feuermann M, Hinz U, et al. The IntAct molecular interaction database in 2012. Nucleic Acids Res. 2012;40:D841–D846. doi: 10.1093/nar/gkr1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Salwinski L, Miller CS, Smith AJ, Pettit FK, Bowie JU, Eisenberg D. The database of interacting proteins: 2004 update. Nucleic Acids Res. 2004;32:D449–D451. doi: 10.1093/nar/gkh086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Licata L, Briganti L, Peluso D, Perfetto L, Iannuccelli M, Galeota E, Sacco F, Palma A, Nardozza AP, Santonico E, et al. MINT, the molecular interaction database: 2012 update. Nucleic Acids Res. 2012;40:D857–D861. doi: 10.1093/nar/gkr930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Goll J, Rajagopala SV, Shiau SC, Wu H, Lamb BT, Uetz P. MPIDB: the microbial protein interaction database. Bioinformatics. 2008;24:1743–1744. doi: 10.1093/bioinformatics/btn285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goel R, Harsha HC, Pandey A, Prasad TS. Human protein reference database and human proteinpedia as resources for phosphoproteome analysis. Mol. Biosyst. 2012;8:453–463. doi: 10.1039/c1mb05340j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Croft D, O’Kelly G, Wu G, Haw R, Gillespie M, Matthews L, Caudy M, Garapati P, Gopinath G, Jassal B, et al. Reactome: a database of reactions, pathways and biological processes. Nucleic Acids Res. 2011;39:D691–D697. doi: 10.1093/nar/gkq1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Warde-Farley D, Donaldson SL, Comes O, Zuberi K, Badrawi R, Chao P, Franz M, Grouios C, Kazi F, Lopes CT, et al. The GeneMANIA prediction server: biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 2010;38:W214–W220. doi: 10.1093/nar/gkq537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kamburov A, Pentchev K, Galicka H, Wierling C, Lehrach H, Herwig R. ConsensusPathDB: toward a more complete picture of cell biology. Nucleic Acids Res. 2011;39:D712–D717. doi: 10.1093/nar/gkq1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Niu Y, Otasek D, Jurisica I. Evaluation of linguistic features useful in extraction of interactions from PubMed; application to annotating known, high-throughput and predicted interactions in I2D. Bioinformatics. 2010;26:111–119. doi: 10.1093/bioinformatics/btp602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hu Z, Hung JH, Wang Y, Chang YC, Huang CL, Huyck M, DeLisi C. VisANT 3.5: multi-scale network visualization, analysis and inference based on the gene ontology. Nucleic Acids Res. 2009;37:W115–W121. doi: 10.1093/nar/gkp406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Elefsinioti A, Sarac OS, Hegele A, Plake C, Hubner NC, Poser I, Sarov M, Hyman A, Mann M, Schroeder M, et al. Large-scale de novo prediction of physical protein-protein association. Mol. Cell. Proteomics. 2011;10 doi: 10.1074/mcp.M111.010629. M111 010629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Patil A, Nakai K, Nakamura H. HitPredict: a database of quality assessed protein-protein interactions in nine species. Nucleic Acids Res. 2011;39:D744–D749. doi: 10.1093/nar/gkq897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Balaji S, McClendon C, Chowdhary R, Liu JS, Zhang J. IMID: integrated molecular interaction database. Bioinformatics. 2012;28:747–749. doi: 10.1093/bioinformatics/bts010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wong AK, Park CY, Greene CS, Bongo LA, Guan Y, Troyanskaya OG. IMP: a multi-species functional genomics portal for integration, visualization and prediction of protein functions and networks. Nucleic Acids Res. 2012;40:W484–W490. doi: 10.1093/nar/gks458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Szklarczyk D, Franceschini A, Kuhn M, Simonovic M, Roth A, Minguez P, Doerks T, Stark M, Muller J, Bork P, et al. The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 2011;39:D561–D568. doi: 10.1093/nar/gkq973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jensen LJ, Kuhn M, Stark M, Chaffron S, Creevey C, Muller J, Doerks T, Julien P, Roth A, Simonovic M, et al. STRING 8–a global view on proteins and their functional interactions in 630 organisms. Nucleic Acids Res. 2009;37:D412–D416. doi: 10.1093/nar/gkn760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Powell S, Szklarczyk D, Trachana K, Roth A, Kuhn M, Muller J, Arnold R, Rattei T, Letunic I, Doerks T, et al. eggNOG v3.0: orthologous groups covering 1133 organisms at 41 different taxonomic ranges. Nucleic Acids Res. 2012;40:D284–D289. doi: 10.1093/nar/gkr1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Saric J, Jensen LJ, Ouzounova R, Rojas I, Bork P. Extraction of regulatory gene/protein networks from Medline. Bioinformatics. 2006;22:645–650. doi: 10.1093/bioinformatics/bti597. [DOI] [PubMed] [Google Scholar]
- 35.Minguez P, Parca L, Diella F, Mende DR, Kumar R, Helmer-Citterich M, Gavin AC, van Noort V, Bork P. Deciphering a global network of functionally associated post-translational modifications. Mol. Syst. Biol. 2012;8:599. doi: 10.1038/msb.2012.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thornton JM, Orengo CA, Todd AE, Pearl FM. Protein folds, functions and evolution. J. Mol. Biol. 1999;293:333–342. doi: 10.1006/jmbi.1999.3054. [DOI] [PubMed] [Google Scholar]
- 37.Koonin EV, Wolf YI, Karev GP. The structure of the protein universe and genome evolution. Nature. 2002;420:218–223. doi: 10.1038/nature01256. [DOI] [PubMed] [Google Scholar]
- 38.Zhang QC, Petrey D, Norel R, Honig BH. Protein interface conservation across structure space. Proc. Natl Acad. Sci. USA. 2010;107:10896–10901. doi: 10.1073/pnas.1005894107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Qian W, He X, Chan E, Xu H, Zhang J. Measuring the evolutionary rate of protein-protein interaction. Proc. Natl Acad. Sci. USA. 2011;108:8725–8730. doi: 10.1073/pnas.1104695108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Walhout AJ, Sordella R, Lu X, Hartley JL, Temple GF, Brasch MA, Thierry-Mieg N, Vidal M. Protein interaction mapping in C. elegans using proteins involved in vulval development. Science. 2000;287:116–122. doi: 10.1126/science.287.5450.116. [DOI] [PubMed] [Google Scholar]
- 41.Caspi R, Foerster H, Fulcher CA, Kaipa P, Krummenacker M, Latendresse M, Paley S, Rhee SY, Shearer AG, Tissier C, et al. The MetaCyc Database of metabolic pathways and enzymes and the BioCyc collection of Pathway/Genome Databases. Nucleic Acids Res. 2008;36:D623–D631. doi: 10.1093/nar/gkm900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Teichmann SA, Rison SC, Thornton JM, Riley M, Gough J, Chothia C. The evolution and structural anatomy of the small molecule metabolic pathways in Escherichia coli. J. Mol. Biol. 2001;311:693–708. doi: 10.1006/jmbi.2001.4912. [DOI] [PubMed] [Google Scholar]
- 43.Conant GC, Wolfe KH. Turning a hobby into a job: how duplicated genes find new functions. Nat. Rev. Genet. 2008;9:938–950. doi: 10.1038/nrg2482. [DOI] [PubMed] [Google Scholar]
- 44.Koonin EV. Orthologs, paralogs, and evolutionary genomics. Ann. Rev. Genet. 2005;39:309–338. doi: 10.1146/annurev.genet.39.073003.114725. [DOI] [PubMed] [Google Scholar]
- 45.Altenhoff AM, Studer RA, Robinson-Rechavi M, Dessimoz C. Resolving the ortholog conjecture: orthologs tend to be weakly, but significantly, more similar in function than paralogs. PLoS Comput. Biol. 2012;8:e1002514. doi: 10.1371/journal.pcbi.1002514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.von Mering C, Jensen LJ, Snel B, Hooper SD, Krupp M, Foglierini M, Jouffre N, Huynen MA, Bork P. STRING: known and predicted protein-protein associations, integrated and transferred across organisms. Nucleic Acids Res. 2005;33:D433–D437. doi: 10.1093/nar/gki005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tatusov RL, Galperin MY, Natale DA, Koonin EV. The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000;28:33–36. doi: 10.1093/nar/28.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ciccarelli FD, Doerks T, von Mering C, Creevey CJ, Snel B, Bork P. Toward automatic reconstruction of a highly resolved tree of life. Science. 2006;311:1283–1287. doi: 10.1126/science.1123061. [DOI] [PubMed] [Google Scholar]
- 49.Huang DW, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Khatri P, Sirota M, Butte AJ. Ten years of pathway analysis: current approaches and outstanding challenges. PLoS Comput. Biol. 2012;8:e1002375. doi: 10.1371/journal.pcbi.1002375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 52.Forbes SA, Bindal N, Bamford S, Cole C, Kok CY, Beare D, Jia M, Shepherd R, Leung K, Menzies A, et al. COSMIC: mining complete cancer genomes in the Catalogue of somatic mutations in cancer. Nucleic Acids Res. 2011;39:D945–D950. doi: 10.1093/nar/gkq929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rivals I, Personnaz L, Taing L, Potier MC. Enrichment or depletion of a GO category within a class of genes: which test? Bioinformatics. 2007;23:401–407. doi: 10.1093/bioinformatics/btl633. [DOI] [PubMed] [Google Scholar]
- 54.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. Roy. Statist. Soc. B. 1995;57:289–300. [Google Scholar]
- 55.Maslov S, Sneppen K. Specificity and stability in topology of protein networks. Science. 2002;296:910–913. doi: 10.1126/science.1065103. [DOI] [PubMed] [Google Scholar]
- 56.Minguez P, Gotz S, Montaner D, Al-Shahrour F, Dopazo J. SNOW, a web-based tool for the statistical analysis of protein-protein interaction networks. Nucleic Acids Res. 2009;37:W109–W114. doi: 10.1093/nar/gkp402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pradines JR, Farutin V, Rowley S, Dancik V. Analyzing protein lists with large networks: edge-count probabilities in random graphs with given expected degrees. J. Comput. Biol. 2005;12:113–128. doi: 10.1089/cmb.2005.12.113. [DOI] [PubMed] [Google Scholar]
- 58.Apweiler R, Martin MJ, O’Donovan C, Magrane M, Alam-Faruque Y, Antunes R, Barrell D, Bely B, Bingley M, Binns D, et al. Ongoing and future developments at the Universal Protein Resource. Nucleic Acids Res. 2011;39:D214–D219. doi: 10.1093/nar/gkq1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Letunic I, Doerks T, Bork P. SMART 7: recent updates to the protein domain annotation resource. Nucleic Acids Res. 2012;40:D302–D305. doi: 10.1093/nar/gkr931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kiefer F, Arnold K, Kunzli M, Bordoli L, Schwede T. The SWISS-MODEL Repository and associated resources. Nucleic Acids Res. 2009;37:D387–D392. doi: 10.1093/nar/gkn750. [DOI] [PMC free article] [PubMed] [Google Scholar]