

Abstract
EcoGene (http://ecogene.org) is a database and website devoted to continuously improving the structural and functional annotation of Escherichia coli K-12, one of the most well understood model organisms, represented by the MG1655(Seq) genome sequence and annotations. Major improvements to EcoGene in the past decade include (i) graphic presentations of genome map features; (ii) ability to design Boolean queries and Venn diagrams from EcoArray, EcoTopics or user-provided GeneSets; (iii) the genome-wide clone and deletion primer design tool, PrimerPairs; (iv) sequence searches using a customized EcoBLAST; (v) a Cross Reference table of synonymous gene and protein identifiers; (vi) proteome-wide indexing with GO terms; (vii) EcoTools access to >2000 complete bacterial genomes in EcoGene-RefSeq; (viii) establishment of a MySql relational database; and (ix) use of web content management systems. The biomedical literature is surveyed daily to provide citation and gene function updates. As of September 2012, the review of 37 397 abstracts and articles led to creation of 98 425 PubMed-Gene links and 5415 PubMed-Topic links. Annotation updates to Genbank U00096 are transmitted from EcoGene to NCBI. Experimental verifications include confirmation of a CTG start codon, pseudogene restoration and quality assurance of the Keio strain collection.
INTRODUCTION
The EcoGene database evolved from pre-genome era studies anticipating the genome era (1). EcoSeq was a growing collection of Escherichia coli K-12 genomic sequence contigs that were shotgun-assembled from the numerous gene-length DNA sequences in Genbank. EcoSeq was aligned, using the novel restriction map alignment software MapSearch (2), to EcoMap, a digitized version of the Kohara map, a complete genome restriction map (3). The gel-derived Kohara restriction map was replaced by DNA sequence restriction maps derived from sequenced regions, creating a hybrid map. Sequenced genes were aligned and oriented: EcoMap became the model for transitional genetic maps to localize and orient genes on the chromosome based on DNA sequences, restriction maps and genetic crosses (4). EcoSeq was one of the first large sets of DNA sequence data for which the genome orientation was known (>1.6 megabases) and was used to localize both known and novel DNA repeats, including the extreme DNA strand bias (skew) of the recombination hotspot Chi (5).
EcoGene was created by the development and application of new methods for accurately re-annotating the gene content of EcoSeq. Most re-annotations were done one protein coding sequence at a time, including careful consideration of DNA and protein sequence alignments, signal peptide predictions, and ribosome binding site predictions. The gene finding software GeneMark was used in a collaborative endeavor that assisted GeneMark’s development (6,7). When the E. coli K-12 genome sequence was finished, these same methods were used to re-annotate the complete genome sequence for EcoGene.
The previous database publication described our basic annotation approaches including translation start site revisions, a compilation of the Verified Set of sequenced protein starts, as well as the identification and hypothetical reconstruction of deletion, frame-shifted and interrupted pseudogenes (8). The process of daily updating from the literature was described and currently 37 397 PubMed abstracts and full articles have been reviewed, creating 98 425 PubMed-to-Gene links. The EcoGene database and website previously described were rudimentary (see the EcoGene MySql section below). The map depictions at the original EcoGene.org website were static PDF maps produced by PrintMap (1).
The previous database article noted that the many updates in EcoGene differed substantially from the annotation in the E. coli K-12 MG1655(Seq) primary Genbank record U00096 (8,9). Genbank is the source database for gene names, product names and sequences used in many other databases, as well as source data for many bioinformatics studies. An annotation workshop held in 2005 included participants from several E. coli databases, and was a conduit for the EcoGene revisions, particularly the extensive start site revisions, to migrate into Genbank U00096 (10). This was a follow-up to a previous annotation workshop in 2003 that established the multi-database annotation collaboration. The workshop report included a description of many of the annotation methods used at EcoGene during the previous decade. Although the report states that the start site revisions were performed at the workshop, all revisions had been reviewed and corrected during the previous decade at EcoGene. One of us (K.E.R.) demonstrated and explained the unpublished methodology used to correct start sites and frame-shifted pseudogenes, using over 200 examples shown to other curators at the workshop. Later, in April 2007, EcoGene.org became the source database for the Genbank U00096 and companion RefSeq record NC_000913 annotations. Updated MySql tables are transferred to NCBI from EcoGene.org to facilitate periodic annotation updates. A file comparing the update to the previous annotations is produced at NCBI and reviewed for comments and corrections by senior curators at UniProtKB (11), EcoCyc (12), NCBI (13), ASAP (14) and EcoGene. Updates are performed at EcoGene using a suite of in-house curation tools, some of which were developed with funds from a 1-year EcoliHub NIH sub-award in 2008–09.
Although Genbank U00096 is titled as the genome sequence for E. coli K-12 MG1655, a semi-motile strain represented by Coli Genetic Stock Center strain CGSC#6300, the genome sequence actually corresponds to MG1655(Seq), a spontaneous hyper-motile variant (CGSC#7740) (15). The major difference between the two strains is the insertion of IS1H at flhDC, which causes hyper-motility. U00096.2 is a sequence-corrected version of U00096.1 (16). With the advent of next-generation sequencing, two groups have reported minor sequence errors in U00096.2 (17,18), and we have independently confirmed these few corrections (A. Richardson and K.E.R., unpublished). The corrections will appear in U00096.3.
Description
GenePages with graphic map depictions
EcoGene (http://ecogene.org) displays dynamically generated maps of genes, proteins and other features on GenePages. The tab-oriented approach taken for the EcoGene 3.0 GenePage design is depicted in Figure 1. Every protein and RNA-encoding gene has a GenePage with additional tabs displaying the DNA sequence, protein sequence, microarray results, PubMed-linked bibliographies, and PDB-linked structures. The GenePage tab also includes links to TopicPages related to the specific gene of interest. A resources menu to gene-oriented pages at external database resources and a navigation menu for easy access to other EcoGene functions are provided. Many GenePages also have comments noting recent discoveries, conflicts in the literature or curator interpretations. Comprehensive mini-reviews are not presented; however, links to EcoCyc provide the user with immediate access to their many well-written gene and protein function summaries. The GenePage bibliographies provide access to a comprehensive collection of daily-annotated PubMed abstracts, research articles and review articles that also contain summary material. Navigation arrows move to adjacent genes or move to genes in alphabetical order. Additional GenePage features display the length of the adjacent intergenic regions, link to pre-run BLAST output files, provide details on N-terminal protein sequencing (the Verified Set), display any number of upstream and downstream basepairs, explain the gene name mnemonic and provide alternate gene symbols (synonyms). A primary gene name is assigned considering historical precedence, accuracy, uniqueness and current usage. In addition to the EcoGene EG accession number for the gene, which refers to both the K-12 gene and alleles of the same gene in other E. coli strains, the ECK accession number, also maintained at EcoGene, refers specifically to the E. coli K-12 MG1655 allele.
Figure 1.

The main GenePage tab of the EcoGene 3.0 GenePage for lacZ. The main areas of interest are circled and described in the text.
Three regional maps, in Portable Network Graphic (PNG) format that allows for the images to be saved and used in publications and presentations, are found at the bottom of each GenePage. The first map is the Gene Map, containing gene depictions along with kb and centisome (%) scales. The Gene Map depicts a default 10-kb region of genomic DNA and can be zoomed in by users to define shorter regions in more detail. The Gene Map also illustrates the position of transcription factor binding sites (TFBSs) and intergenic repeats. Graphic tracks, found at the bottom of the Gene Map, show whether the gene is a Core gene, present in most E. coli strains, or a non-Core gene labeled as ‘HT’ to indicate they are predicted to be inherited through horizontal transmission, although gene loss could also account for the non-Core status in some cases (19). Objects within the Gene Map contain hyperlinks to other GenePages and TopicPages. Another link found within the Gene Map is PDF EcoMap, which allows the user to download a regional PDF map created using the PrintMap program (1). Full genome PDF maps in PrintMap format are available on the EcoTools page. The Site Map, the second of the three maps, depicts all the restriction sites for three default restriction enzymes (BamHI, EcoRI and HindIII), and can be customized by the user to show up to seven restriction enzyme sites. The list of restriction enzymes and their DNA recognition sequences were obtained from REBASE (20). Clicking on the restriction enzyme name next to the restriction maps links to additional information for that enzyme at http://rebase.neb.com. An additional feature of the Site Map is that it magnifies in concert with the Gene Map. The Intergenic Region maps at the bottom of the GenePages offer a more detailed depiction of TFBSs and intergenes. The transcription factors binding to the region are listed with hyperlinks to their GenePages, and intergenes are listed with hyperlinks to their TopicPages. TFBS arrows are linked to the RegulonDB source database (21).
GeneSets: EcoSearch, EcoTopics, EcoArray, Boolean queries and interactive Venn diagrams
A graphic presentation of Boolean query comparisons using two or three genesets can be displayed in an interactive Venn diagram (Figure 2). Boolean queries can be executed using EcoGene genesets or user-specified genesets. EcoGene genesets are collections of genes clustered in EcoTopics or EcoArray. User-provided genesets are lists of EG accession numbers uploaded at a GeneSets Venn Diagram and Boolean Query page. EcoSearch can be used to obtain a list of user-specified EG numbers from a query or from an uploaded ID file containing gene names or any other unique gene identifiers; gene names that are synonyms are mapped to primary genes. The Cross Reference Mapping tool can also be used for this purpose (see EcoTools section). EcoSeach also allows searching of the extensive EcoRef bibliographies, including both GenePage and EcoTopic bibliographies. Both the EcoSearch results pages and the TopicPages display genesets on a small circular map linked to an interactive circular map design tool Circle Maps. For an example, see http://www.ecogene.org/genemap/map.php?search_topic=560. Circle Maps can also be accessed directly from the EcoTools menu.
Figure 2.

Interactive Venn diagrams and Boolean queries using genesets. Clicking on the number in any of the Venn diagram sectors produces a list of genes. The Venn diagrams can be saved as a PNG image for use in presentations or publications; a black-and-white version is available to save on printer and publishing costs.
EcoTopics cover a wide range of topics and are used to cluster genes for any reason; however, typical EcoTopics have a hierarchal nature with many topics having multiple sub-topics and super-topics. In all, 559 EcoTopics exist, and are linked to 21 154 genes and 5415 PubMed abstracts. EcoTopics can vary in the number of associated genes. An EcoTopic containing information from shotgun proteomics can contain hundreds of genes, whereas other EcoTopics link to more specific topics, such as the 17 IS elements, or intergenes like the 5 REP elements and the 10 CRISPR repeats families. Some EcoTopics have no associated genesets and were created to have a place to attach bibliographies that might not associate with a gene, such as the metabolic modeling literature. However, others only serve as placeholders to aggregate PubMed citations; many of these do not currently contain a text description. EcoTopics are not intended to contain comprehensive curator-authored reviews. Reading review articles, which are culled into separate bibliographies as well as being in the main bibliographies, and reading introductions to articles is a good way to get expert reviews of many EcoTopics. EcoArray was originally designed in 2007 to capture E. coli microarray results that were not deposited in pubic databases and to allow access to expression data for a particular gene from the GenePages. Although many public websites and databases populated with E. coli transcription array data now exist, some datasets in EcoArray are still missing from public repositories and other databases. We are currently importing and comparing data from numerous sources as we revamp EcoArray, as will be reported separately.
PrimerPairs
PrimerPairs is an embedded design tool for obtaining genome-wide polymerase chain reaction (PCR) primer sequences, which can be used to generate desired deletion–insertions or to create an ordered clone library, using the current EcoGene annotations of gene intervals. The user has the option to set primer lengths, offset positions inside or outside of genes, add-on sequences for adding restriction sites or sequences to amplify kanamycin (kan) or chloramphenicol (cat) antibiotic resistance cassettes. PrimerPairs has the ability to detect and correct primer sequences that delete part of adjacent overlapping genes. The offending primers are repositioned automatically to avoid the double-deletion problem found in the Keio mutant collection (see below). PrimerPairs has previously been described in detail (22).
EcoBlast
The NCBI BLAST program (13) runs locally on the EcoGene server and is used to prepare pre-run BLAST search results for all protein sequences on a weekly basis. Links to these pre-run results are provided on each GenePage. A local BLAST interface, called EcoBlast, provides users with the ability to query input sequences against a set of in-house databases. These specialized databases include EcoProt and EcoGene [sequence libraries derived from K-12 MG1655(Seq) gene-only sequences], the current (M56) and original (M54) versions of Genbank U00096, the Salmonella typhimurium LT2 genome and proteome, sequence libraries of all complete E. coli or Salmonella genomes and proteomes and in-house compiled libraries of complete genomes and proteomes from Enterobacteriales species (excluding E. coli). Automated downloads of the comprehensive NCBI NT and NR non-redundant nucleotide and protein sequence libraries are performed weekly. The UniProtKB UniRef100 and Swiss-Prot/TrEMBL proteomes are also included in the EcoBlast Database menu. Finally, a comprehensive NR+EnvNR protein sequence database combining genomic and environmental sequences from NCBI is compiled in-house and gives comprehensive EcoBlastP results populated with intermediate homologs from EnvNR.
EcoTools: downloads and cross reference tool
In addition to the Boolean Query tool and PrimerPairs tool described above, the EcoTools page of EcoGene 3.0 has links to other data download pages, which are also listed in the EcoTools sidebar sub-menu. The EcoGene Database Table Download page allows the user to select fields from the current daily-updated database for export to a tab-delimited text file. This table includes information present on the GenePage and Protein tabs, including gene name, protein name, gene synonyms, molecular weight and map position. The table also includes several important accession numbers, such as EcoGene (EG) number, E. coli K-12 ECK number, UniProtKB/Swiss-Prot ID, GenBank GI. Recently, the Cross Reference Mapping and Download page was created for user access to many additional accession numbers and other gene identifiers such as gene name and synonyms. The E. coli K-12 gene accession numbers from >30 other databases were collected to enable the construction of hyperlinks to gene pages at other websites and to easily update tables that lack EG or ECK ids to the most recent gene names and functions. A Genome Sequence Download page enables whole genome DNA sequence or user-specified sub-sequence extraction. Some basic EcoGene navigation and retrieval functions, including gene-start or gene-stop anchored sub-sequence extractions, have been presented as detailed protocols (22).
An Intergenic Region Download page enables the retrieval of the E. coli K-12 DNA sequences between genes. These intergenic sequences are named and oriented with respect to the flanking genes, and distances between genes are given. These intergenic sequences include many conserved features that lie between genes such as promoters, terminators, TFBSs and intergenic repeat families, e.g. CRISPR and REP elements (23). As these features are genetic determinants that lie outside of gene borders, they are termed intergenes. There are TopicPages for all the families of intergenic repeats. An option to exclude the intergenic repeats is provided causing the intergenic intervals to be broken up and named by the flanking genes and repeats. A link to the Intergenic Repeat Families super-TopicPage is provided on the EcoTools page. In addition to the DNA sequence files in FASTA format, a descriptive table of intergenic regions is provided for downloading. A download option to include adjacent gene overlaps as well as the intervals between genes is provided; the length of the overlap is provided as a negative interval length. Downloaded files have pseudogene gene names marked with apostrophes to indicate their pseudogene status. The pseudogene TopicPage explains the pseudogene annotation conventions used in EcoGene and can be accessed from the EcoTools page.
The EcoGene Tools page has files available for direct download, including EcoGene and EcoProt sequences files, with or without the partial and reconstructed pseudogene sequences, a PDF map of the entire chromosome in PrintMap format, and the Verified Set table (see below). Also provided is the EcoGene OpenSearch browser plug-in that enables EcoGene gene searches in browser toolbars.
GO term indexing
The Gene Ontology (GO) project is a collaborative system for gene products classification and description using three structured controlled vocabularies: cellular component, molecular function and biological process (http://www.geneontology.org/GO.doc.shtml) (24). All the GO terms currently associated with E. coli K-12 genes and the GO hierarchy of terms, names and definitions, including all ancestor and children relations, were obtained from the Amigo website (25). The E. coli GO term assignments at Amigo are compiled from several sources including EcoCyc (26), EcoliWiki (27), UniProtKB (11,28) and InterPro (29). We made two observations consistent with problems reported elsewhere (30). First, the GO annotations are incomplete and many proteins lack GO annotations consistent with their molecular function as currently understood. In some cases, a more specific child term can be applied to better describe the molecular function, whereas in other cases, the GO term is either missing or out-of-date. Second less specific ancestor terms are inconsistently and sporadically applied. Many new gene functions are first annotated in EcoGene and then propagated worldwide to other databases using the regularly updated Genbank U00096 record. We address the problem of incompleteness by monitoring and incorporating updates at Amigo and updating GO terms at EcoGene during the daily literature-based updating process. The EcoGene staff will periodically provide its updated GO term assignments to the GO consortium. To address the problem of sporadic ancestor term assignments, we remove all ancestors and retain only the most specific child terms for each function thread. We then derive all less-specific ancestor terms using the GO hierarchy.
The GO terms are displayed on the Protein tabs associated with GenePages (Figure 3). In addition to the GO term accession numbers, the number of genes associated with specific GO terms are indicated in parentheses. Clicking the number of genes retrieves all those gene records. The link associated with the GO accession leads to the descriptive Amigo page for that GO term including a listing of all the ancestors and children for that GO term. Mousing over the GO term accession reveals its name and definition. All of the less-specific ancestors are hidden but can be revealed with a click. EcoGene is fully indexed with the expanded ancestor GO terms, and EcoSearch now has a dedicated search box for GO terms as an alternate way to retrieve genes without using the links on the Protein page. GO terms are being updated in-house by adding missing GO child terms, and by replacing child terms with more specific child terms if they exist.
Figure 3.

A GenePage Protein tab showing the GO terms with automatic expansion to a full list of less specific ancestor GO terms.
EcoGene-RefSeq
To make the maps and tools developed for EcoGene available for other genomes, EcoGene-RefSeq was developed. Currently, 2074 complete bacterial genomes from the NCBI RefSeq project (31) are accessible through EcoGene-RefSeq. EcoGene-RefSeq is organized independently from EcoGene, with its own MySql database and web interface accessible from the EcoGene home page. Genome data is imported from NCBI in Generic Feature Format Version 3.0 format and stored in MySql tables. The EcoTools currently ported to EcoGene-RefSeq include PrimerPairs, Search and Download, GenePage navigation and Cross Reference mapping. An alphabetical gene index is also provided. A detailed bioinformatic description of EcoGene-RefSeq will be published separately (J.Z. and K.E.R., manuscript in preparation).
The verified set
The E. coli K-12 MG1655(Seq) genome sequence annotation is kept current with the biomedical literature by extensive manual curation at EcoGene.org. This includes updating the Verified Set of proteins whose starts have been determined by protein sequencing using Edman degradation. In 2000, the Verified Set contained 717 protein starts and citations to their published protein sequencing results (8). Currently, the Verified Set is composed of 923 verified protein starts identified in 818 publications. The verified proteins include 419 unprocessed proteins, 356 proteins whose N-terminal methionine is removed by methionine aminopeptidase, 142 exported proteins cleaved by signal peptidase I and six starts generated by various proteolytic cleavages. In addition to compiling published verifications, the EcoGene staff performs selected experimental verifications of predicted structural and functional annotations. The signal peptidase I cleavage sites and periplasmic locations were experimentally determined for HiuH and YhcN, as noted on their GenePages (N. Hus, R. Mitchell, M. Del Campo, and K.E.R., unpublished results). In most cases, the Verified Set also confirms a predicted translation start codon. The EcoTools page has a full table of the Verified Set, including PubMed identifiers as documentation. In addition, 82 proteins have been verified as lipoproteins in prior publications, and 38 proteins have been predicted to be lipoproteins in a comparative analysis of lipoprotein predictions in an EcoGene collaborative publication (32). These lipoproteins are clustered in EcoTopics.
CTG start codon verification
Start codon mutagenesis was used to confirm the second instance of CTG being used as a start codon in the E. coli K-12 genome. The hda gene was the first E. coli chromosomal gene shown to initiate translation with CTG (33). As part of an ongoing bioinformatics analysis of predicted initiation codons (8), two additional CTG starts were predicted based on sequence conservation and inspection of potential ribosome binding sites. Using start codon mutagenesis and protein fusions, the CTG start codon predicted for yfjD was confirmed as the start codon, while mutating the previously annotated TTG start at codon 9 had no effect on gene expression (D. Dague and K.E.R., unpublished results). The pseudogene yghF’ encodes an N-terminal gene fragment that is also predicted to start with CTG at EcoGene.org, but this could not be confirmed because expression from YghF’-LacZ protein fusions was negligible even when additional yghF’ upstream DNA was included in the plasmid construct.
Small protein and sRNA verifications
The prediction, annotation and functional characterization of small proteins and sRNAs remain a challenge (34,35). Two EcoGene collaborative projects combined bioinformatic and experimental approaches to identify novel small proteins and sRNAs in E. coli. Protein translations of 20 previously annotated and 18 newly predicted small proteins (16–50 amino acids), including nine new membrane proteins, were experimentally verified (36). Additionally, the novel IbsA-E toxins, encoded opposite the SibA-E sRNA antitoxins, are composed of only 18 or 19 amino acids and were experimentally characterized (37). The Sib sRNAs were originally predicted to be sRNAs encoded in the novel QUAD repeats, as reported in an earlier EcoGene bioinformatics study (23). The Sib sRNAs were experimentally verified independently in both the EcoGene and Storz laboratories and the QUAD repeats were renamed as SIB repeats (37).
The COMBREX-EcoGene collaboration
The COMBREX project aims to identify the functions of uncharacterized genes in E. coli and other bacteria (38,39). A 1-year sub-award from the COMBREX project funded a COMBREX–EcoGene collaboration to functionally characterize E. coli genes. The identification of YhiQ as RsmJ, the only unidentified 16S rRNA methyltransferase in E. coli, responsible for the m(2)G1516 modification, was a result of that collaboration (40). In addition, the yaiX’ pseudogene was repaired and shown to facilitate clumping during vegetative growth. The previously uncharacterized YaiP, a predicted glucosyltransferase encoded in the yaiX operon was also demonstrated to be required for vegetative clumping, a result that could not be obtained until the yaiX’ pseudogene was repaired (Dague et al., abstract presented at The 2011 Madison Molecular Genetics of Bacteria and Phages Meeting, August 2–7, Madison, WI).
The E. coli K-12 ignome
The functional characterization of products from the y-genes (see below) yihA, yeeX, yhgF, ybcJ, yhbY, yjgA, yhdE, yceF and yhcN was initiated with the COMBREX funding. These are now ongoing EcoGene laboratory projects. These uncharacterized gene products are part of the E. coli K-12 ignome. We define an ignome as all the knowledge we lack about an organism, including structural, functional, regulatory, spatial and organization information about all cellular constituents. A practical ignome is all the knowable information about an organism, acknowledging that the acquisition of knowledge may be limited by costs (time, money, means) or by insolvable technical hurdles (Figure 4).
Figure 4.
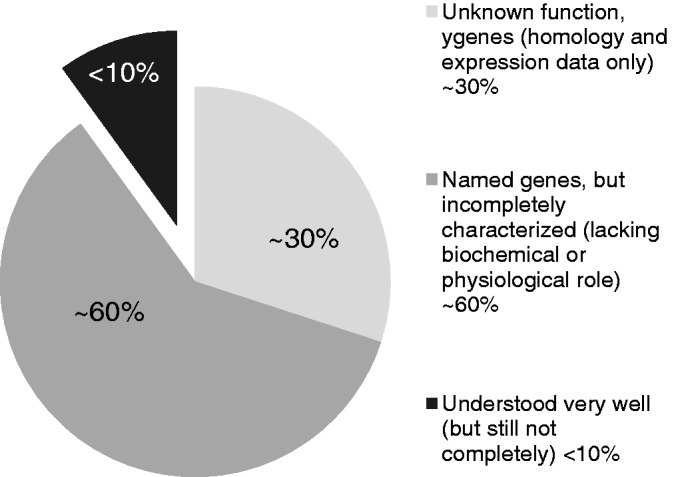
The E. coli K-12 ignome.
Keio collection and ASKA set verifications
The Keio collection contains deletions tagged with kanamycin resistance cassettes in the non-essential genes of E. coli K-12, created using high-throughput recombineering methodology (41,42). The Keio collection is a widely distributed and extensively used genetic resource that has a number of problems discovered during our extensive quality control assessment. Although the verifications are not completed, a preliminary summary of the problems we found has been published (22). One of the major problems relates to the lack of isogenicity for ∼65% of the frozen cultures in the collection. These cultures contain variants that have acquired hyper-motility mutations mostly due to IS1 or IS5 insertions upregulating the flhDC locus. We have also observed that the semi-motile parent strain BW25113 is genetically unstable in the laboratory and can acquire hyper-motility mutations, as has been demonstrated for other semi-motile E. coli K-12 strains (15,43). This instability at flhDC is responsible for the difference between MG1655 and MG1655(Seq), as noted in the Introduction. To stabilize the Keio collection, we crossed the entire collection into the hyper-motile strain KRE10000 [MG1655(Seq) rph+] by P1 transduction and re-recombineering, as noted previously (22). This whole collection transduction outcross revealed that ∼100 slow-growing strains had picked up unlinked growth defect suppressor mutations in the Keio collection. We also identified the same tandem duplications that were also found by the Keio group using their PCR primers (42).
As part of our quality assessment of the Keio collection, we confirmed the auxotrophic phenotypes by feeding with specific nutrients (D. Dague and K.E.R., unpublished). A companion genome-wide set of clones, the ASKA library, has also been constructed and widely distributed (44). We tested the ASKA collection for the ability to complement Keio mutants and found that all the auxotrophs we tested were able to grow without supplements when they harbor un-induced cognate ASKA clones (D. Dague and K.E.R., unpublished). Bioinformatic validation of the deletion primers used to make the Keio collection found numerous adjacent-gene double deletions. Identification of these adjacent-gene double deletions inspired development of the PrimerPairs genome-wide primer design tool used to eliminate double deletions when designing PCR primers (22). We used ASKA clone complementation to show that the adjacent-gene 3′ double deletions in amino acid biosynthetic operons are innocuous, that is, complementing only the targeted gene restored prototrophy (D. Dague and K.E.R., unpublished). This demonstrates that most, if not all, of the small 3′ deletions in the genes adjacent to the genes targeted in 468 strains do not cause a mutant phenotype, and this is not unexpected, as proteins generally tolerate modifications at their C-termini, such as hexa-histidine tags. In contrast, the small adjacent-gene 5′ deletions completely eliminate adjacent-gene function in the few mutants that could be easily tested (D. Dague and K.E.R., unpublished). The 32 adjacent-gene 5′ deletion mutants have been reconstructed by de novo recombineering in KRE10000 (D. Dague and K.E.R., unpublished).
Additional information regarding the Keio strains and ASKA clones can be found through links on GenePages to the Genobase website (http://ecoli.naist.jp/GB8-dev/). GenePages also contain a Keio/ASKA tab with specific information on strains and clones, including their locations in microtiter trays, along with pictures of PCR-amplified fragments from the flhDC locus of each mutant in the Keio collection, indicating the presence or absence of IS1 or IS5 insertion sequences responsible for hyper-motility phenotypes. The PCR reactions were performed on the original frozen cells and identified many mixed wells with both semi-motile (IS−) and hyper-motile (IS+) cells (K.E.R. unpublished). When using the Keio mutants, care should be taken to follow standard bacterial genetics methodology of single colony purification, phenotypic (motility) testing and P1 transduction as necessary to restore isogenicity. Published high-throughput results that used Keio mutants directly from trays without single colony purification need to be carefully re-examined.
Y-gene nomenclature
Genes without names or known functions were previously assigned y-gene names using a systematic nomenclature based on map location (4). Although initially assigned based on map position, e.g. yaaA-Z are genes in the first centisome (map minute, %) interval, the y-gene names are not intended to denote map position and have been used to denote the orthologous gene in S. typhimurium despite having a different map location. If any particular centisome interval has more than 26 unnamed genes, a second set of y-names can be used, e.g. ykaA-Z for the first centisome interval (Figure 5). Many y-genes have now been assigned functions, but authors often choose not to assign new meaningful mnemonic gene names as previously suggested (4) because they may not fully understand the biochemical activity or physiological role of the newly characterized gene. However, E. coli genes have been traditionally assigned names at the first, not final, characterization effort; as long as the initial mnemonic does not turn out to be completely incorrect, which rarely happens, it should be retained. This means that mnemonic names describing the functions or phenotypes can and should be assigned after their initial characterization. On the other hand, some authors assign new genes names to y-genes without first checking at EcoGene.org to see if the gene name has been previously used. Mnemonic gene names that do not refer to gene function or mnemonics that are common words, which are poor terms for computer searches, are discouraged. In addition, some authors assign new genes even when the primary gene name mnemonic is still valid just because they have discovered more detailed functional information. This re-naming can go on indefinitely, engenders confusion and is to be discouraged. These superfluous, albeit informative, re-namings are entered in EcoGene as gene synonyms; the primary gene names and synonyms for E. coli K-12 are maintained at EcoGene.org. Authors can consult the EcoGene curator (K.E.R.) on the choice of new gene names. A TopicPage devoted to the y-genes with additional information is available at EcoGene.org (http://www.ecogene.org/topic/364).
Figure 5.

The y-gene code wheel depicting the systematic uncharacterized gene nomenclature rationale for E. coli K-12 is shown.
The EcoGene MySql relational database
The first edition of EcoGene used static HTML pages, essentially publishing rows from a spreadsheet into early GenePages as periodic updates (8). To provide continuously updated information to EcoGene.org, a live relational database is required. In 2005, a relational schema for EcoGene was created to build cross-references for the breadth of information being published describing the biology of the E. coli K-12 laboratory workhorse strain and to accommodate a detailed level of structural annotation, including intergenes and pseudogenes (Figure 6). The schema is modular, which assists the database to be robust, efficient and flexible in our experience. Five data-specific modules are displayed: Core EcoGene, Shared Sources, Protein & RNA, EcoTopic and EcoArray. Additional modules include the MySql tables for the Drupal content management system (http://drupal.org/) and recently added tables for GO term descriptions and an ancestor-expanded gene-GO linking table.
Figure 6.

EcoGene 3.0 MySql database schema. Some minor fields have been deleted for clarity. The five modules are color-coded.
The Core EcoGene module holds the genome sequence, the gene-pubmed cross-references and genomic address for genes and intergenes. It includes information for genes, synonyms, IS elements, repeat DNA, e.g. REPs and CRISPRs, and TFBSs (the latter imported from RegulonDB). Pseudogenes interrupted by IS insertion have split addresses analogous to exons. The Genbank format ‘join statements’ describing hypothetical reconstructions of the split genes that are displayed on their GenePages are derived from the t_split_gene_address table. IS elements present at multiple locations are represented in the t_gene_multi_address table, allowing for direct GenePage navigation among the various IS alleles and locations. Information related to the widely used Keio mutant and ASKA clone collections are also stored here.
The Shared Sources module holds tables for genomic gene and feature basepair positions, data derived from PubMed records and curated links to external databases. The Protein & RNA module contains product information derived from bacterial RefSeq records; for EcoGene-curated E. coli K-12 MG1655, this is where the GenBank record product descriptions are updated and stored between U00096 updates. The EcoTopic and EcoArray modules were added to support the ongoing development of two ambitious EcoGene projects.
EcoGene.org content management platform
EcoGene 2.0 was the default EcoGene.org website from 2006 until EcoGene 3.0 was officially launched in September 2012. During a 1-year beta deployment period, >80% of EcoGene.org usage switched to EcoGene 3.0. EcoGene 2.0 will remain available for users who are accustomed to its interface; it accesses the same daily updated MySql database as EcoGene 3.0, but all new development projects will be restricted to EcoGene 3.0. For example, the interactive Venn diagram tool is only available in EcoGene 3.0. EcoGene.org, launched in 2006, is powered by the open source content management software PHP-Nuke (http://phpnuke.org) and supported by a MySql database (http://www.mysql.com). However, in 2010 we desired improved capabilities and support, as well as a new look-and-feel for the EcoGene.org interface, so we evaluated popular open source content management platforms and chose Drupal to build EcoGene 3.0. Most of the EcoGene.org 2.0 PHP (http://www.php.net) software was rewritten to be compatible with Drupal (http://drupal.org). The EcoGene.org website operational algorithms, scripts and routines were also renovated and streamlined to facilitate future improvement and expansion.
FUNDING
National Institutes of Health [1-R01-GM58560, 3-U24-GM077905-3S1 subaward #4102-25541, RC2-GM92602 subaward]. Funding for open access charge: Waived by Oxford University Press.
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
We thank Fred Blattner and Guy Plunkett for facilitating the transfer of annotation update responsibility for Genbank U00096 to K.E.R. We acknowledge the participation of William Klimke (NCBI), Ingrid Keseler (EcoCyc), Guy Plunkett (ASAP), Eric Neeno-Eckwall (ASAP) and Andrea Auchincloss (UniProtKB) for reviewing the periodic annotation updates sent from EcoGene to NCBI. We acknowledge computer programming on EcoGene 2.0 by Dian Fan and Feng Liu. We would like to thank Julio Collado-Vides of the University Nationale de Mexico for permission to use data from RegulonDB in EcoGene. We thank Andrew Richardson for technical assistance, use of unpublished results, and a critical reading of the manuscript. We acknowledge Andrew Richardson, Yusuf Kaya, Kristi Jones, Darryl Dague, Joseph Reed, and Christopher Crowley for work on the Keio collection validation and various laboratory projects associated with EcoGene and COMBREX-EcoGene. We also acknowledge technical assistance from Nir Hus, Mark Del Campo and Rick Mitchell for unpublished results from verification studies. We acknowledge Kristi Jones for her help in designing and curating EcoArray.
REFERENCES
- 1.Rudd KE, Miller W, Werner C, Ostell J, Tolstoshev C, Satterfield SG. Mapping sequenced E. coli genes by computer: software, strategies and examples. Nucleic Acids Res. 1991;19:637–647. doi: 10.1093/nar/19.3.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Miller W, Ostell J, Rudd KE. An algorithm for searching restriction maps. Comput. Appl. Biosci. 1990;6:247–252. doi: 10.1093/bioinformatics/6.3.247. [DOI] [PubMed] [Google Scholar]
- 3.Kohara Y, Akiyama K, Isono K. The physical map of the whole E. coli chromosome: application of a new strategy for rapid analysis and sorting of a large genomic library. Cell. 1987;50:495–508. doi: 10.1016/0092-8674(87)90503-4. [DOI] [PubMed] [Google Scholar]
- 4.Rudd KE. Linkage map of Escherichia coli K-12, edition 10: the physical map. Microbiol. Mol. Biol. Rev. 1998;62:985–1019. doi: 10.1128/mmbr.62.3.985-1019.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blaisdell BE, Rudd KE, Matin A, Karlin S. Significant dispersed recurrent DNA sequences in the Escherichia coli genome. Several new groups. J. Mol. Biol. 1993;229:833–848. doi: 10.1006/jmbi.1993.1090. [DOI] [PubMed] [Google Scholar]
- 6.Borodovsky M, Rudd KE, Koonin EV. Intrinsic and extrinsic approaches for detecting genes in a bacterial genome. Nucleic Acids Res. 1994;22:4756–4767. doi: 10.1093/nar/22.22.4756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Borodovsky M, McIninch JD, Koonin EV, Rudd KE, Medigue C, Danchin A. Detection of new genes in a bacterial genome using Markov models for three gene classes. Nucleic Acids Res. 1995;23:3554–3562. doi: 10.1093/nar/23.17.3554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rudd KE. EcoGene: a genome sequence database for Escherichia coli K-12. Nucleic Acids Res. 2000;28:60–64. doi: 10.1093/nar/28.1.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Blattner FR, Plunkett G, 3rd, Bloch CA, Perna NT, Burland V, Riley M, Collado-Vides J, Glasner JD, Rode CK, Mayhew GF, et al. The complete genome sequence of Escherichia coli K-12. Science. 1997;277:1453–1474. doi: 10.1126/science.277.5331.1453. [DOI] [PubMed] [Google Scholar]
- 10.Riley M, Abe T, Arnaud MB, Berlyn MK, Blattner FR, Chaudhuri RR, Glasner JD, Horiuchi T, Keseler IM, Kosuge T, et al. Escherichia coli K-12: a cooperatively developed annotation snapshot—2005. Nucleic Acids Res. 2006;34:1–9. doi: 10.1093/nar/gkj405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Apweiler R, Jesus Martin M, O'onovan C, Magrane M, Alam-Faruque Y, Antunes R, Barrera Casanova E, Bely B, Bingley M, Bower L, et al. Reorganizing the protein space at the Universal Protein Resource (UniProt) Nucleic Acids Res. 2012;40:D71–D75. doi: 10.1093/nar/gkr981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Keseler IM, Collado-Vides J, Santos-Zavaleta A, Peralta-Gil M, Gama-Castro S, Muniz-Rascado L, Bonavides-Martinez C, Paley S, Krummenacker M, Altman T, et al. EcoCyc: a comprehensive database of Escherichia coli biology. Nucleic Acids Res. 2011;39:D583–D590. doi: 10.1093/nar/gkq1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sayers EW, Barrett T, Benson DA, Bolton E, Bryant SH, Canese K, Chetvernin V, Church DM, Dicuccio M, Federhen S, et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2012;40:D13–D25. doi: 10.1093/nar/gkr1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Glasner JD, Rusch M, Liss P, Plunkett G, 3rd, Cabot EL, Darling A, Anderson BD, Infield-Harm P, Gilson MC, Perna NT. ASAP: a resource for annotating, curating, comparing, and disseminating genomic data. Nucleic Acids Res. 2006;34:D41–D45. doi: 10.1093/nar/gkj164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barker CS, Pruss BM, Matsumura P. Increased motility of Escherichia coli by insertion sequence element integration into the regulatory region of the flhD operon. J. Bacteriol. 2004;186:7529–7537. doi: 10.1128/JB.186.22.7529-7537.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hayashi K, Morooka N, Yamamoto Y, Fujita K, Isono K, Choi S, Ohtsubo E, Baba T, Wanner BL, Mori H, et al. Highly accurate genome sequences of Escherichia coli K-12 strains MG1655 and W3110. Mol. Syst. Biol. 2006;2 doi: 10.1038/msb4100049. 2006 0007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Freddolino PL, Amini S, Tavazoie S. Newly identified genetic variations in common Escherichia coli MG1655 stock cultures. J. Bacteriol. 2012;194:303–306. doi: 10.1128/jb.06087-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fabich AJ, Leatham MP, Grissom JE, Wiley G, Lai H, Najar F, Roe BA, Cohen PS, Conway T. Genotype and phenotypes of an intestine-adapted Escherichia coli K-12 mutant selected by animal passage for superior colonization. Infect. Immun. 2011;79:2430–2439. doi: 10.1128/IAI.01199-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Davids W, Zhang Z. The impact of horizontal gene transfer in shaping operons and protein interaction networks—direct evidence of preferential attachment. BMC Evol. Biol. 2008;8:23. doi: 10.1186/1471-2148-8-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Roberts RJ, Vincze T, Posfai J, Macelis D. REBASE—a database for DNA restriction and modification: enzymes, genes and genomes. Nucleic Acids Res. 2010;38:D234–D236. doi: 10.1093/nar/gkp874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gama-Castro S, Salgado H, Peralta-Gil M, Santos-Zavaleta A, Muniz-Rascado L, Solano-Lira H, Jimenez-Jacinto V, Weiss V, Garcia-Sotelo JS, Lopez-Fuentes A, et al. RegulonDB version 7.0: transcriptional regulation of Escherichia coli K-12 integrated within genetic sensory response units (Gensor Units) Nucleic Acids Res. 2011;39:D98–D105. doi: 10.1093/nar/gkq1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhou J, Rudd KE. Bacterial genome reengineering. Methods Mol. Biol. 2011;765:3–25. doi: 10.1007/978-1-61779-197-0_1. [DOI] [PubMed] [Google Scholar]
- 23.Rudd KE. Novel intergenic repeats of Escherichia coli K-12. Res. Microbiol. 1999;150:653–664. doi: 10.1016/s0923-2508(99)00126-6. [DOI] [PubMed] [Google Scholar]
- 24.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Carbon S, Ireland A, Mungall CJ, Shu S, Marshall B, Lewis S. AmiGO: online access to ontology and annotation data. Bioinformatics. 2009;25:288–289. doi: 10.1093/bioinformatics/btn615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hu JC, Karp PD, Keseler IM, Krummenacker M, Siegele DA. What we can learn about Escherichia coli through application of Gene Ontology. Trends Microbiol. 2009;17:269–278. doi: 10.1016/j.tim.2009.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McIntosh BK, Renfro DP, Knapp GS, Lairikyengbam CR, Liles NM, Niu L, Supak AM, Venkatraman A, Zweifel AE, Siegele DA, et al. EcoliWiki: a wiki-based community resource for Escherichia coli. Nucleic Acids Res. 2012;40:D1270–D1277. doi: 10.1093/nar/gkr880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dimmer EC, Huntley RP, Alam-Faruque Y, Sawford T, O'Donovan C, Martin MJ, Bely B, Browne P, Mun Chan W, Eberhardt R, et al. The UniProt-GO Annotation database in 2011. Nucleic Acids Res. 2012;40:D565–D570. doi: 10.1093/nar/gkr1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Burge S, Kelly E, Lonsdale D, Mutowo-Muellenet P, McAnulla C, Mitchell A, Sangrador-Vegas A, Yong SY, Mulder N, Hunter S. Manual GO annotation of predictive protein signatures: the InterPro approach to GO curation. Database (Oxford) 2012;2012:bar068. doi: 10.1093/database/bar068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Faria D, Schlicker A, Pesquita C, Bastos H, Ferreira AE, Albrecht M, Falcao AO. Mining GO annotations for improving annotation consistency. PLoS One. 2012;7:e40519. doi: 10.1371/journal.pone.0040519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pruitt KD, Tatusova T, Brown GR, Maglott DR. NCBI Reference Sequences (RefSeq): current status, new features and genome annotation policy. Nucleic Acids Res. 2012;40:D130–D135. doi: 10.1093/nar/gkr1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gonnet P, Rudd KE, Lisacek F. Fine-tuning the prediction of sequences cleaved by signal peptidase II: a curated set of proven and predicted lipoproteins of Escherichia coli K-12. Proteomics. 2004;4:1597–1613. doi: 10.1002/pmic.200300749. [DOI] [PubMed] [Google Scholar]
- 33.Su'etsugu M, Nakamura K, Keyamura K, Kudo Y, Katayama T. Hda monomerization by ADP binding promotes replicase clamp-mediated DnaA-ATP hydrolysis. J. Biol. Chem. 2008;283:36118–36131. doi: 10.1074/jbc.M803158200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rudd KE, Humphery-Smith I, Wasinger VC, Bairoch A. Low molecular weight proteins: a challenge for post-genomic research. Electrophoresis. 1998;19:536–544. doi: 10.1002/elps.1150190413. [DOI] [PubMed] [Google Scholar]
- 35.Hemm MR, Paul BJ, Miranda-Rios J, Zhang A, Soltanzad N, Storz G. Small stress response proteins in Escherichia coli: proteins missed by classical proteomic studies. J. Bacteriol. 2010;192:46–58. doi: 10.1128/JB.00872-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hemm MR, Paul BJ, Schneider TD, Storz G, Rudd KE. Small membrane proteins found by comparative genomics and ribosome binding site models. Mol. Microbiol. 2008;70:1487–1501. doi: 10.1111/j.1365-2958.2008.06495.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fozo EM, Kawano M, Fontaine F, Kaya Y, Mendieta KS, Jones KL, Ocampo A, Rudd KE, Storz G. Repression of small toxic protein synthesis by the Sib and OhsC small RNAs. Mol. Microbiol. 2008;70:1076–1093. doi: 10.1111/j.1365-2958.2008.06394.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Roberts RJ. COMBREX: COMputational BRidge to EXperiments. Biochem. Soc. Trans. 2011;39:581–583. doi: 10.1042/BST0390581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Roberts RJ, Chang YC, Hu Z, Rachlin JN, Anton BP, Pokrzywa RM, Choi HP, Faller LL, Guleria J, Housman G, et al. COMBREX: a project to accelerate the functional annotation of prokaryotic genomes. Nucleic Acids Res. 2011;39:D11–D14. doi: 10.1093/nar/gkq1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Basturea GN, Dague DR, Deutscher MP, Rudd KE. YhiQ is RsmJ, the methyltransferase responsible for methylation of G1516 in 16S rRNA of E. coli. J. Mol. Biol. 2012;415:16–21. doi: 10.1016/j.jmb.2011.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol. 2006;2 doi: 10.1038/msb4100050. 2006.0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yamamoto N, Nakahigashi K, Nakamichi T, Yoshino M, Takai Y, Touda Y, Furubayashi A, Kinjyo S, Dose H, Hasegawa M, et al. Update on the Keio collection of Escherichia coli single-gene deletion mutants. Mol. Syst. Biol. 2009;5:335. doi: 10.1038/msb.2009.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang X, Wood TK. IS5 inserts upstream of the master motility operon flhDC in a quasi-Lamarckian way. ISME J. 2011;5:1517–1525. doi: 10.1038/ismej.2011.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kitagawa M, Ara T, Arifuzzaman M, Ioka-Nakamichi T, Inamoto E, Toyonaga H, Mori H. Complete set of ORF clones of Escherichia coli ASKA library (a complete set of E. coli K-12 ORF archive): unique resources for biological research. DNA Res. 2005;12:291–299. doi: 10.1093/dnares/dsi012. [DOI] [PubMed] [Google Scholar]