


Abstract
The concept of orthology provides a foundation for formulating hypotheses on gene and genome evolution, and thus forms the cornerstone of comparative genomics, phylogenomics and metagenomics. We present the update of OrthoDB—the hierarchical catalog of orthologs (http://www.orthodb.org). From its conception, OrthoDB promoted delineation of orthologs at varying resolution by explicitly referring to the hierarchy of species radiations, now also adopted by other resources. The current release provides comprehensive coverage of animals and fungi representing 252 eukaryotic species, and is now extended to prokaryotes with the inclusion of 1115 bacteria. Functional annotations of orthologous groups are provided through mapping to InterPro, GO, OMIM and model organism phenotypes, with cross-references to major resources including UniProt, NCBI and FlyBase. Uniquely, OrthoDB provides computed evolutionary traits of orthologs, such as gene duplicability and loss profiles, divergence rates, sibling groups, and now extended with exon–intron architectures, syntenic orthologs and parent–child trees. The interactive web interface allows navigation along the species phylogenies, complex queries with various identifiers, annotation keywords and phrases, as well as with gene copy-number profiles and sequence homology searches. With the explosive growth of available data, OrthoDB also provides mapping of newly sequenced genomes and transcriptomes to the current orthologous groups.
INTRODUCTION
Homology in molecular biology refers to a common ancestry. In practice, homologous genes are recognized through the assessment of the statistical significance of sequence similarities of aligned nucleotides or amino acids. With reference to a specific species radiation, homologous relations define orthologs—‘equivalent’ genes in different species descended from a single ancestral gene (1–3). Speciation events, gene duplications, losses and sequence mutations lead to the diversity of genes encoded in the genomes of modern species. For any given set of species, all the descendants of a single gene from their last common ancestor constitute an orthologous group of genes. Orthology is therefore inherently hierarchical, referring explicitly to the last common ancestor, such that mostly one-to-one orthologs are identified among closely related species, whereas among more distantly related species orthologous groups comprise all surviving descendants of the ancestral gene.
There are two main approaches for orthology delineation: (i) algorithms that cluster all-against-all pairwise sequence comparisons, usually first identifying best-reciprocal matches between genomes that correspond to the shortest path over the speciation node of a distance-based tree, e.g. (4–12); and (ii) phylogeny-based methods that first define homologous gene families, build gene trees for each family, and then explicitly or implicitly reconcile them with the species tree often employing assumptions on rates of gene losses and duplications, e.g. (13–18). Phylogeny-based approaches have more parameters and may therefore yield better accuracy given sufficient data, but are often limited by the quality of multiple sequence alignments. This approach also considerably increases computational demands and becomes impractical for hundreds of species.
Recent benchmarking of prominent orthology resources (19,20) show that in the trade-off between specificity and sensitivity, OrthoDB assignments favor greater specificity with reasonable sensitivity, a balance that is well-suited to the goal of inferring gene functions. Although orthology is strictly an evolutionary concept, it can support the tentative transfer of functional annotations from well-studied organisms to orthologs in newly sequenced species. The confidence of such hypotheses on gene function may be qualitatively gauged by the genes’ evolutionary histories, e.g. more confident inferences may be made for orthologs that are preserved across many species mostly as single-copy genes, with relatively low levels of sequence divergence, and consistent protein domain architectures. Gene duplicates in multi-copy orthologous groups often exhibit greater sequence divergence than single-copy orthologs (21), and as this may reflect biological innovation, any inferences on gene function should be made cautiously. OrthoDB classifications have proved to be accurate and biologically relevant as assessed within the framework of several recent genome projects, e.g. (22–26). Thus, the evolutionary characterization of orthologous groups in OrthoDB, collated with available gene functional annotations, provide a strong basis for making informed hypotheses that can drive evolutionary and molecular biology research.
SPECIES SAMPLING
The current OrthoDB release includes more than 250 eukaryotes and now also extends to cover prokaryotes with a total of 1115 bacterial species (Table 1, Supplementary Table S1). The predicted protein-coding gene sets and their corresponding General Feature Format (GFF) annotations for 52 vertebrate species were retrieved from Ensembl (27) (Release 67, May 2012). Data for the 45 arthropods were sourced from AphidBase (28), BeetleBase (29), FlyBase (30), Hymenoptera Genome Database (31), SilkDB (32), VectorBase (33), wFleaBase (34) and several genome consortia (as of July 2012). Gene sets for an additional 13 basal animal species were retrieved from Ensembl Genomes (35) and the Joint Genome Institute (36) (as of July 2012). The 142 fungal gene sets were retrieved from UniProt (37) (July 2012 release) and the bacteria were retrieved from NCBI (38) (Supplementary Table S1).
Table 1.
OrthoDB species and gene content
| Lineage Representative species | Input genes |
Classified genes (%) | Percentage of classified genes |
||
|---|---|---|---|---|---|
| Total | Average | in groups with annotation(s)a | in groups with phenotype(s)b | ||
| 52 Vertebrates | 951 245 | 18 293 | 92.7 | 96.3 | 48.4 |
| Homo sapiens | 20 827 | na | 94.9 | 93.5 | 45.6 |
| Mus musculus | 23 075 | na | 87.0 | 96.5 | 47.9 |
| Danio rerio | 26 206 | na | 80.7 | 96.9 | 48.5 |
| 45 Arthropods | 746 324 | 16 585 | 71.1 | 87.1 | 25.1 |
| Drosophila melanogaster | 13 927 | na | 96.1 | 86.5 | 26.6 |
| 110c Metazoa | 1 974 947 | 17 954 | 81.9 | 93.5 | 60.8 |
| Caenorhabditis elegans | 20 517 | na | 71.5 | 84.7 | 61.4 |
| 142 Fungi | 1 223 848 | 8619 | 85.0 | 86.8 | 49.3 |
| Saccharomyces cerevisiae | 6652 | na | 96.2 | 91.9 | 94.8 |
| 1115 Bacteria | 3 532 434 | 3168 | 91.0 | 91.6 | 47.1 |
| Escherichia coli | 4149 | na | 97.8 | 97.7 | 98.8 |
| Haemophilus influenza | 1657 | na | 98.2 | 98.8 | 85.3 |
| Mycobacterium tuberculosis | 3977 | na | 95.5 | 93.3 | 35.9 |
Statistics describing OrthoDB species coverage of vertebrate, arthropod, basal metazoan, fungal and bacterial orthologs with rich functional annotations.
aGO terms or InterPro domains.
bFrom Online Mendelian Inheritance in Man, the Mouse Genome Database, the Zebrafish Model Organism Database, FlyBase, WormBase, Saccharomyces Genome Database, EcoGene or the Database of Essential Genes.
c13 basal metazoan species plus 52 vertebrates and 45 arthropods.
HIERARCHICAL ORTHOLOGOUS GROUPS
The OrthoDB orthology delineation procedure is based on clustering of best-reciprocal-hits (BRHs) between genes from each species pair, determined from all-against-all Smith–Waterman protein sequence comparisons now using SWIPE (39). The clustering procedure considers only the longest transcript per gene, and only the longest of all gene copies in a single genome with over 97% amino acid identity as determined by CD-HIT (40). Clusters are built progressively, with an e-value cutoff of 1e-3 for triangulating BRHs, and 1e-6 for pair-only BRHs, requiring an overall minimum sequence alignment overlap of 30 amino acids. The clusters of BRHs are subsequently further expanded to include all in-paralogs recognized as within-species homologs that are more closely related than the clustered BRHs.
Since its conception, OrthoDB (41) has promoted the concept of hierarchical orthology classifications by applying the clustering procedure at each radiation point of the considered species phylogeny and allowing users to explicitly select the most relevant level. It is rewarding to note that other resources e.g. (7,8) have embraced this concept and now provide orthology classifications at several major radiations across the tree of life. To determine the OrthoDB hierarchy, the species phylogenies in the current release were empirically computed using a maximum-likelihood approach as implemented in FastTree (42) over the super-alignment of mostly single-copy orthologs defined at the root node, multiply-aligned using MAFFT (43), and filtered using TrimAl (44), and corroborated with known taxonomies from the literature.
The hierarchical orthology delineation procedure of the sampled lineages of vertebrates, arthropods and fungi classified 84% of a total of 2 921 417 protein-coding genes into 25 371, 33 393 and 55 793 orthologous groups, respectively (Table 1). Root-level delineation across the 110 animal species defined 58 308 orthologous groups covering 82% of the 3 198 795 metazoan genes and clustering of the 1115 bacteria classified 91% of the 3 532 434 bacterial genes. In addition to the root-level orthologs, 11 subgroups of bacteria—corresponding to the NCBI taxonomy ‘class’ levels—were clustered to provide more fine-grained orthologous groups for Actinobacteria, Spirochetes, Tenericutes, Thermotage, two classes of Cyanobacteria and Firmicutes, and three classes of Proteobacteria.
MAPPED FUNCTIONAL ANNOTATIONS
As orthologous groups comprise genes descended from a common ancestor, functional attributes ascribed to one or more members can be tentatively extrapolated to the last common ancestor and describe the group as a whole. In this way, orthologous group summary annotations provide an overview of mapped functional attributes with links to respective source databases to allow further investigations of the putative biological roles of their member genes (Figure 1).
Figure 1.

Screenshot of a sample orthologous group results page, featuring functional and evolutionary annotations, the inferred parent–child gene tree and syntenic orthologs.
Concise descriptors
Gene functional descriptions sourced from UniProt (37) and NCBI (38) provide succinct indications of known or inferred biological functions with coherent nomenclatures based on data from the literature as well as biocurator-evaluated and automatic computational classifications and annotations. In this OrthoDB release, frequently occurring phrases from member-gene descriptions label the group with a meaningful descriptor for each orthologous group.
Gene ontologies and InterPro domains
Molecular function, biological process and cellular component Gene Ontology (GO) (45) terms were retrieved from UniProt (37) and InterPro (46) protein domain signatures were sourced from the UniProt Archive of sequences. The available functional evidence for each orthologous group is summarized by listing the frequencies of associated GO terms and InterPro domains with concise attribute descriptions. Additionally, InterPro matches are displayed with domains ordered sequentially from the N- to C-terminus, describing the complete domain architecture of multi-domain genes, thereby allowing database queries with specific domain combinations. More than 85% of orthologs from each of the lineages are classified in groups that can be described by either GO terms or InterPro domains (Table 1).
Model organism phenotypes
OrthoDB gene annotations are enhanced with detailed functional data from well-studied model organisms in each lineage to highlight phenotypes associated with genes from Mus musculus, Drosophila melanogaster and Saccharomyces cerevisiae, sourced from the Mouse Genome Database (47), FlyBase (30) and Saccharomyces Genome Database (48), respectively. Eukaryotic model organism phenotypes now also include Danio rerio from the Zebrafish Model Organism Database (49) and Caenorhabditis elegans from WormBase (50). For bacteria, gene annotations are extended with phenotype data from EcoGene (51) for Escherichia coli genes and from the Database of Essential Genes (52) which covers 16 bacteria including E. coli, Haemophilus influenza and Mycobacterium tuberculosis (Table 1).
Online Mendelian inheritance in man
Human gene annotations are now enhanced with links to online Mendelian inheritance in man (OMIM®) (53), the catalog of associations between causative genes and human disease phenotypes, which describes thousands of allelic variants linked to numerous different disorders or susceptibilities. Mapping of human genes in OrthoDB to OMIM® records highlights known disease associations for almost 3000 genes (Table 1).
COMPUTED EVOLUTIONARY ANNOTATIONS
OrthoDB presents quantified orthologous group characteristics that describe evolutionary properties such as gene duplications or losses and rates of sequence divergence, these detail their evolutionary histories and provide a basis for the assessment of the confidence with which inferences on gene function may be made (Figure 1).
Phyletic profiles
Orthologous group phyletic profiles contrast the number of species with single-copy versus multi-copy orthologs and indicate the species coverage at the selected radiation point. The profiles thus highlight how descendant genes have been preserved across the phylogeny and whether gene duplications are widespread (‘multi-copy license’) or restricted (‘single-copy control’) as discussed in (21).
Evolutionary rates
The relative divergence among orthologous group member genes is quantified as the average of inter-species protein sequence identities normalized to the average identity of all inter-species BRHs. Appreciably higher or lower rates of divergence distinguish groups of orthologs with restrained or relaxed rates of protein sequence evolution, e.g. essential-gene-containing groups usually exhibit greater sequence conservation than those without.
Sibling groups
Homologous relations among genes from different orthologous groups at a given species radiation identify homologous or ‘sibling’ orthologous groups. These relations are quantified using data from all-against-all sequence comparisons by averaging over all pairs of homologs that link two orthologous groups with an e-value cutoff of 1e-3. This allows the user to retrieve sets of sibling orthologous groups that share significant sequence homology—which may therefore have some functional similarities—in an unbiased way that does not rely on protein domain or gene functional annotations.
Parent–child trees
Orthology delineation at each radiation along a given phylogeny hierarchically defines groups of orthologs with increasing resolution from the root level with the complete set of species to the most closely related species pairs. Parent–child relationships among orthologous groups delineated at each descendant radiation may therefore be defined by stepping along the phylogeny to identify orthologous groups with common subsets of genes (Figure 2). This new feature of OrthoDB represents these relationships as parent–child trees that illustrate the hierarchy of orthologous groups and their member genes, thereby building an inferred gene tree for a parent group by taking advantage of the greater resolution of its child groups. Users may view and edit the parent–child trees, as well as retrieve tree data formatted using Newick Utilities (54), from the ‘Display Tree’ window (Figure 1) that integrates the PhyloWidget (55) tool for the visualization and manipulation of phylogenetic tree data.
Figure 2.
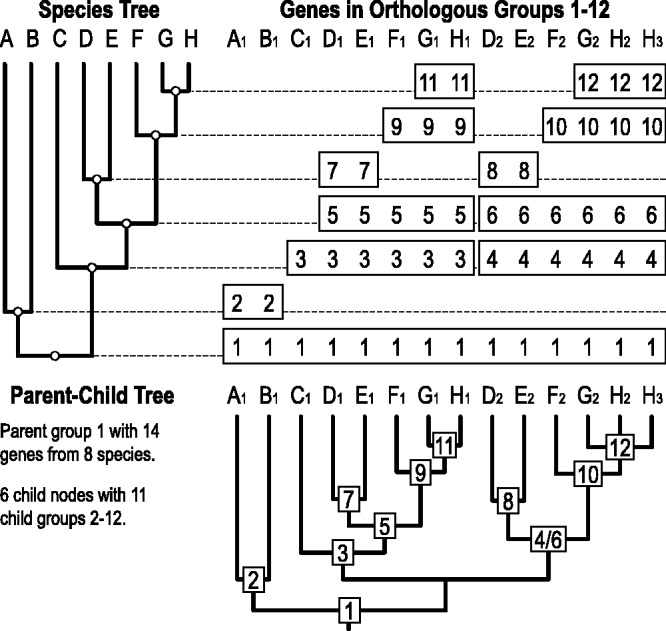
Hierarchical parent–child trees.
Gene architectures
Evolutionary annotations now also feature summary tables of protein lengths (all lineages) and exon counts (meatazoan lineages) that detail quantified mean, median and standard deviation values for each orthologous group, effectively describing a ‘consensus’ gene architecture. Amino acid and exon counts are also listed for each member gene, flagging those that are significantly shorter or longer than the consensus as potentially inaccurate gene model predictions.
Syntenic orthologs
Comparing the chromosomal arrangements of orthologous genes among sets of species from the OrthoDB arthropod lineage identifies conserved blocks of syntenic orthologs. Such genes have maintained their local gene neighborhoods in the face of continual genomic evolution through sequence deletions, insertions and inversions, which may suggest selective advantages associated with their genomic arrangements, e.g. the TipE gene cluster of insect Para sodium channel auxiliary subunits (56). Ortholog-anchored synteny delineation (57) first identifies pairwise blocks with a minimum of two orthologs, allowing at most two intervening orthologs for each pair of genomes, and then successively projects these blocks through each pair of species across the phylogeny. The ‘OrthoBlock’ viewer (Figure 1) displays the best block—weighted according to the evolutionary span of the species and the number of orthologous groups in the block—selected from all the resulting blocks with at least five species for each orthologous group.
ORTHODB ONLINE
Selecting any species radiation point of interest from the interactive species trees, users can navigate through the hierarchy of orthologous groups defined at each radiation of the eukaryotic species phylogenies and for 11 major bacterial clades. At each orthology level, text searches return results from matches to various database identifiers and annotation keywords or phrases that can be combined through logical operator syntax to build more complex queries (e.g. [‘cytochrome c'-mitochondrial]) using Sphinx indexing technology (http://sphinxsearch.com/). In addition, database cross-referencing of gene identifiers enhances search term matches through available gene names and synonyms, InterPro, or GO identifiers, as well as secondary identifiers from UniProt, Entrez GeneID, RefSeq, Protein Data Bank, OMIM, PubMed and model organism databases. Copy-number profile searches retrieve groups matching specific user-defined or general pre-defined phyletic profiles by combining the criteria of absent, present, single-copy, multi-copy or no restriction, for each species within any selected clade. BLAST (58) sequence similarity searches identify the best matches to genes from different species classified in OrthoDB, thereby allowing database querying with protein sequence data from any species. Importantly, although such sequence similarity searches with a single gene can recognize its homologs, accurate mapping to the defined orthologous groups requires assessment of the organism’s complete gene set (see ortholog mapping section below). Searches stored during each user’s web browser session provide a query history facility to allow recently executed queries to be reviewed, re-run or combined, e.g. a profile search for ‘single-copy in >90% of species’ could be combined with a text search with the GO identifier for ‘receptor activity’ to retrieve groups of mostly single-copy receptors. All search results may be easily exported as either Fasta-formatted files of protein sequences or tab-delimited text files of gene annotations, and the complete datasets are provided for download. All OrthoDB features are described in a comprehensive online help page and users may contact support@orthodb.org for additional information or specific requests, they may also subscribe to the low-traffic ‘orthodb-news’ mailing list (https://list.unige.ch/mailman/listinfo/orthodb-news) to keep abreast of the latest developments.
OrthoDB links
Search results present annotations for each orthologous group and tabulate all member genes with links to their respective sources e.g. Ensembl, UniProt, NCBI and FlyBase. Concise descriptors displayed for GO terms and InterPro domains are hyperlinked to their source records, and hyperlinks to OMIM and model organism databases provide direct access to all supporting data for genes with mapped phenotypes and synonyms. OrthoDB now provides FlyBase with orthology calls for the 12 Drosophila species as well as to selected arthropods and other animals. In addition, classified genes in OrthoDB are referenced with link-outs from UniProt records and NCBI gene link-outs.
Mapping of new species
Through a recently developed ortholog mapping procedure and corresponding web interfaces, OrthoDB now provides orthology classifications for genes from species with newly sequenced genomes mapped to existing orthologous groups. The mapping procedure first compares all genes from the new organism to all genes in OrthoDB groups, and then performs the BRH clustering procedure only allowing new genes to be added to existing clusters. The web interfaces list mapped genes and mirror OrthoDB data from the lineage(s) to which the new species is mapped. Thus, OrthoDB now provides online browsing of mapped orthologs for new species with publically available gene sets such as the Chinese softshell turtle, Pelodiscus sinensis, (from Ensembl Release 68) (Supplementary Figure S1). Portals with restricted access provide the same functionality for private gene sets from organisms with recently sequenced genomes. For example, mapping the initial gene annotations of the genome of the alfalfa leafcutting bee, Megachile rotundata, helped to assess their quality and completeness, as well as providing a user-friendly portal to identify orthologs from other insects (G. Robinson, personal communication).
BENCHMARKING SETS OF UNIVERSAL SINGLE-COPY ORTHOLOGS
The fast-growing number of sequenced genomes and transcriptomes vary substantially in their completeness of sequencing, quality of read assembly and accuracy of gene annotation. A complementary approach to technical statistics such as the widely used N50 measure of genome assemblies, is to gauge the quality by examining the coverage of an expected gene set. This approach can assess not only completeness of genome coverage and fragmentation of the assembly, but also misassembly of haplotypes when the marker genes are known to exist only in single-copy, as well as the accuracy of annotation of such genes. For this purpose—of quality assessment of genomic data—we compiled benchmarking sets of universal single-copy orthologs (abbreviated BUSCOs) identified using OrthoDB for the Metazoan, Vertebrate, Arthropod and Fungal lineages (respectively, named BUSCO-Me, -Ve, -Ar, -Fu). Although these sets are intentionally conservative, they comprehensively sample each lineage and select representative genes from orthologous groups with single-copy orthologs in at least 90% of the species. The BUSCOs are available for download as Fasta-formatted protein sequences with corresponding gene, species and orthologous group identifiers.
PERSPECTIVES
The current OrthoDB release demonstrates the scalability of our computational procedures for the ab initio analysis of several millions of genes within a reasonable timeframe, e.g. with a 150 CPU-core computer cluster the total all-against-all sequence comparisons took about 1 month and the subsequent clustering procedures required from 1 day for the arthropod set to 4 weeks for the largest bacteria dataset on a single machine using a multi-threaded algorithm. Nevertheless, its comprehensive application to all emerging data will become prohibitive in a few years due to the exponential scaling of genome sequencing as well as to the variable completeness and quality of new genome annotations. Thus, our approach will be to focus the complete clustering analyses on only a representative selection of the best annotated species and those that maximize phylogenetic coverage, corroborating the results with curated classifications. These will form a comprehensive set of well-annotated and trusted orthologies to which genes from the other genomes, e.g. the thousands of insects to be sequenced through the i5K initiative (59), and new transcriptomes, e.g. from the 1KITE project (http://www.1kite.org), can be mapped.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Table 1 and Supplementary Figure 1.
FUNDING
Swiss National Science Foundation [31003A-125350]; ‘Commission Informatique’ of the University of Geneva; and Schmidheiny Foundation. Funding for open access charge: Swiss Institute of Bioinformatics.
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
The authors would like to thank all members of the Computational Evolutionary Genomics Group and Dr Ivo Pedruzzi for useful discussions and suggestions, the Swiss Institute of Bioinformatics for pledging funds to support the maintenance and future development of OrthoDB, and the anonymous reviewers for their valuable comments and suggestions.
REFERENCES
- 1.Fitch W. Distinguishing homologous from analogous proteins. Syst. Zool. 1970;19:99–113. [PubMed] [Google Scholar]
- 2.Koonin E. Orthologs, paralogs, and evolutionary genomics. Annu. Rev. Genet. 2005;39:309–338. doi: 10.1146/annurev.genet.39.073003.114725. [DOI] [PubMed] [Google Scholar]
- 3.Sonnhammer E, Koonin E. Orthology, paralogy and proposed classification for paralog subtypes. Trends Genet. 2002;18:619–620. doi: 10.1016/s0168-9525(02)02793-2. [DOI] [PubMed] [Google Scholar]
- 4.Tatusov R, Fedorova N, Jackson J, Jacobs A, Kiryutin B, Koonin E, Krylov D, Mazumder R, Mekhedov S, Nikolskaya A, et al. The COG database: an updated version includes eukaryotes. BMC Bioinform. 2003;4:41. doi: 10.1186/1471-2105-4-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen F, Mackey A, Stoeckert CJ, Roos D. OrthoMCL-DB: querying a comprehensive multi-species collection of ortholog groups. Nucleic Acids Res. 2006;34:D363–D368. doi: 10.1093/nar/gkj123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.DeLuca TF, Cui J, Jung JY, St Gabriel KC, Wall DP. Roundup 2.0: enabling comparative genomics for over 1800 genomes. Bioinformatics. 2012;28:715–716. doi: 10.1093/bioinformatics/bts006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Altenhoff AM, Schneider A, Gonnet GH, Dessimoz C. OMA 2011: orthology inference among 1000 complete genomes. Nucleic Acids Res. 2011;39:D289–D294. doi: 10.1093/nar/gkq1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Powell S, Szklarczyk D, Trachana K, Roth A, Kuhn M, Muller J, Arnold R, Rattei T, Letunic I, Doerks T, et al. eggNOG v3.0: orthologous groups covering 1133 organisms at 41 different taxonomic ranges. Nucleic Acids Res. 2012;40:D284–D289. doi: 10.1093/nar/gkr1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ostlund G, Schmitt T, Forslund K, Köstler T, Messina D, Roopra S, Frings O, Sonnhammer E. InParanoid 7: new algorithms and tools for eukaryotic orthology analysis. Nucleic Acids Res. 2010;38:D196–D203. doi: 10.1093/nar/gkp931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yu C, Desai V, Cheng L, Reifman J. QuartetS-DB: a large-scale orthology database for prokaryotes and eukaryotes inferred by evolutionary evidence. BMC Bioinform. 2012;13:143. doi: 10.1186/1471-2105-13-143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Waterhouse RM, Zdobnov EM, Tegenfeldt F, Li J, Kriventseva EV. OrthoDB: the hierarchical catalog of eukaryotic orthologs in 2011. Nucleic Acids Res. 2011;39:D283–D288. doi: 10.1093/nar/gkq930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Linard B, Thompson JD, Poch O, Lecompte O. OrthoInspector: comprehensive orthology analysis and visual exploration. BMC Bioinform. 2011;12 doi: 10.1186/1471-2105-12-11. Article 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Penel S, Arigon AM, Dufayard JF, Sertier AS, Daubin V, Duret L, Gouy M, Perrière G. Databases of homologous gene families for comparative genomics. BMC Bioinform. 2009;10(Suppl. 6):S3. doi: 10.1186/1471-2105-10-S6-S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huerta-Cepas J, Capella-Gutierrez S, Pryszcz LP, Denisov I, Kormes D, Marcet-Houben M, Gabaldón T. PhylomeDB v3.0: an expanding repository of genome-wide collections of trees, alignments and phylogeny-based orthology and paralogy predictions. Nucleic Acids Res. 2011;39:D556–D560. doi: 10.1093/nar/gkq1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ruan J, Li H, Chen Z, Coghlan A, Coin L, Guo Y, Hériché J, Hu Y, Kristiansen K, Li R, et al. TreeFam: 2008 Update. Nucleic Acids Res. 2008;36:D735–D740. doi: 10.1093/nar/gkm1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Datta R, Meacham C, Samad B, Neyer C, Sjölander K. Berkeley PHOG: PhyloFacts orthology group prediction web server. Nucleic Acids Res. 2009;37:W84–W89. doi: 10.1093/nar/gkp373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vilella A, Severin J, Ureta-Vidal A, Heng L, Durbin R, Birney E. EnsemblCompara GeneTrees: complete, duplication-aware phylogenetic trees in vertebrates. Genome Res. 2009;19:327–335. doi: 10.1101/gr.073585.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mi H, Dong Q, Muruganujan A, Gaudet P, Lewis S, Thomas P. PANTHER version 7: improved phylogenetic trees, orthologs and collaboration with the Gene Ontology Consortium. Nucleic Acids Res. 2010;38:D204–D210. doi: 10.1093/nar/gkp1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Trachana K, Larsson TA, Powell S, Chen W-H, Doerks T, Muller J, Bork P. Orthology prediction methods: a quality assessment using curated protein families. Bioessays. 2011;33:769–780. doi: 10.1002/bies.201100062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Boeckmann B, Robinson-Rechavi M, Xenarios I, Dessimoz C. Conceptual framework and pilot study to benchmark phylogenomic databases based on reference gene trees. Brief. Bioinform. 2011;12:423–435. doi: 10.1093/bib/bbr034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Waterhouse RM, Zdobnov EM, Kriventseva EV. Correlating traits of gene retention, sequence divergence, duplicability and essentiality in vertebrates, arthropods, and fungi. Genome Biol. Evol. 2011;3:75–86. doi: 10.1093/gbe/evq083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Colbourne JK, Pfrender ME, Gilbert D, Thomas WK, Tucker A, Oakley TH, Tokishita S, Aerts A, Arnold GJ, Basu MK, et al. The ecoresponsive genome of Daphnia pulex. Science. 2011;331:555–561. doi: 10.1126/science.1197761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Werren JH, Richards S, Desjardins CA, Niehuis O, Gadau J, Colbourne JK, Beukeboom LW, Desplan C, Elsik CG, Grimmelikhuijzen CJP, et al. Functional and evolutionary insights from the genomes of three parasitoid Nasonia species. Science. 2010;327:343–348. doi: 10.1126/science.1178028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kirkness EF, Haas BJ, Sun W, Braig HR, Perotti MA, Clark JM, Lee SH, Robertson HM, Kennedy RC, Elhaik E, et al. Genome sequences of the human body louse and its primary endosymbiont provide insights into the permanent parasitic lifestyle. Proc. Natl Acad. Sci. USA. 2010;107:12168–12173. doi: 10.1073/pnas.1003379107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Arensburger P, Megy K, Waterhouse RM, Abrudan J, Amedeo P, Antelo B, Bartholomay L, Bidwell S, Caler E, Camara F, et al. Sequencing of Culex quinquefasciatus establishes a platform for mosquito comparative genomics. Science. 2010;330:86–88. doi: 10.1126/science.1191864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bartholomay LC, Waterhouse RM, Mayhew GF, Campbell CL, Michel K, Zou Z, Ramirez JL, Das S, Alvarez K, Arensburger P, et al. Pathogenomics of Culex quinquefasciatus and meta-analysis of infection responses to diverse pathogens. Science. 2010;330:88–90. doi: 10.1126/science.1193162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Flicek P, Amode MR, Barrell D, Beal K, Brent S, Carvalho-Silva D, Clapham P, Coates G, Fairley S, Fitzgerald S, et al. Ensembl 2012. Nucleic Acids Res. 2012;40:D84–D90. doi: 10.1093/nar/gkr991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Legeai F, Shigenobu S, Gauthier J, Colbourne J, Rispe C, Collin O, Richards S, Wilson A, Murphy T, Tagu D. AphidBase: a centralized bioinformatic resource for annotation of the pea aphid genome. Insect Mol. Biol. 2010;19(Suppl. 2):5–12. doi: 10.1111/j.1365-2583.2009.00930.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim H, Murphy T, Xia J, Caragea D, Park Y, Beeman R, Lorenzen M, Butcher S, Manak J, Brown S. BeetleBase in 2010: revisions to provide comprehensive genomic information for Tribolium castaneum. Nucleic Acids Res. 2010;38:D437–D442. doi: 10.1093/nar/gkp807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McQuilton P, St Pierre SE, Thurmond J, Consortium F. FlyBase 101–the basics of navigating FlyBase. Nucleic Acids Res. 2012;40:D706–D714. doi: 10.1093/nar/gkr1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Munoz-Torres MC, Reese JT, Childers CP, Bennett AK, Sundaram JP, Childs KL, Anzola JM, Milshina N, Elsik CG. Hymenoptera Genome Database: integrated community resources for insect species of the order Hymenoptera. Nucleic Acids Res. 2011;39:D658–D662. doi: 10.1093/nar/gkq1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Duan J, Li R, Cheng D, Fan W, Zha X, Cheng T, Wu Y, Wang J, Mita K, Xiang Z, et al. SilkDB v2.0: a platform for silkworm (Bombyx mori) genome biology. Nucleic Acids Res. 2010;38:D453–D456. doi: 10.1093/nar/gkp801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Megy K, Emrich SJ, Lawson D, Campbell D, Dialynas E, Hughes DS, Koscielny G, Louis C, Maccallum RM, Redmond SN, et al. VectorBase: improvements to a bioinformatics resource for invertebrate vector genomics. Nucleic Acids Res. 2012;40:D729–D734. doi: 10.1093/nar/gkr1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Colbourne J, Singan V, Gilbert D. wFleaBase: the Daphnia genome database. BMC Bioinform. 2005;6:45. doi: 10.1186/1471-2105-6-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kersey PJ, Staines DM, Lawson D, Kulesha E, Derwent P, Humphrey JC, Hughes DS, Keenan S, Kerhornou A, Koscielny G, et al. Ensembl Genomes: an integrative resource for genome-scale data from non-vertebrate species. Nucleic Acids Res. 2012;40:D91–D97. doi: 10.1093/nar/gkr895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Grigoriev IV, Nordberg H, Shabalov I, Aerts A, Cantor M, Goodstein D, Kuo A, Minovitsky S, Nikitin R, Ohm RA, et al. The genome portal of the Department of Energy Joint Genome Institute. Nucleic Acids Res. 2012;40:D26–D32. doi: 10.1093/nar/gkr947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.UniProt-Consortium. Reorganizing the protein space at the Universal Protein Resource (UniProt) Nucleic Acids Res. 2012;40:D71–D75. doi: 10.1093/nar/gkr981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sayers EW, Barrett T, Benson DA, Bolton E, Bryant SH, Canese K, Chetvernin V, Church DM, Dicuccio M, Federhen S, et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2012;40:D13–D25. doi: 10.1093/nar/gkr1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rognes T. Faster Smith-Waterman database searches with inter-sequence SIMD parallelisation. BMC Bioinform. 2011;12:221. doi: 10.1186/1471-2105-12-221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li W, Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;22:1658–1659. doi: 10.1093/bioinformatics/btl158. [DOI] [PubMed] [Google Scholar]
- 41.Kriventseva E, Rahman N, Espinosa O, Zdobnov E. OrthoDB: the hierarchical catalog of eukaryotic orthologs. Nucleic Acids Res. 2008;36:D271–D275. doi: 10.1093/nar/gkm845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Price MN, Dehal PS, Arkin AP. FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS One. 2010;5:e9490. doi: 10.1371/journal.pone.0009490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Katoh K, Toh H. Recent developments in the MAFFT multiple sequence alignment program. Brief. Bioinform. 2008;9:286–298. doi: 10.1093/bib/bbn013. [DOI] [PubMed] [Google Scholar]
- 44.Capella-Gutiérrez S, Silla-Martínez JM, Gabaldón T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics. 2009;25:1972–1973. doi: 10.1093/bioinformatics/btp348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.GO-Consortium. The Gene Ontology: enhancements for 2011. Nucleic Acids Res. 2012;40:D559–D564. doi: 10.1093/nar/gkr1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hunter S, Jones P, Mitchell A, Apweiler R, Attwood TK, Bateman A, Bernard T, Binns D, Bork P, Burge S, et al. InterPro in 2011: new developments in the family and domain prediction database. Nucleic Acids Res. 2012;40:D306–D312. doi: 10.1093/nar/gkr948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Eppig JT, Blake JA, Bult CJ, Kadin JA, Richardson JE, Group MGD. The Mouse Genome Database (MGD): comprehensive resource for genetics and genomics of the laboratory mouse. Nucleic Acids Res. 2012;40:D881–D886. doi: 10.1093/nar/gkr974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cherry JM, Hong EL, Amundsen C, Balakrishnan R, Binkley G, Chan ET, Christie KR, Costanzo MC, Dwight SS, Engel SR, et al. Saccharomyces Genome Database: the genomics resource of budding yeast. Nucleic Acids Res. 2012;40:D700–D705. doi: 10.1093/nar/gkr1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bradford Y, Conlin T, Dunn N, Fashena D, Frazer K, Howe DG, Knight J, Mani P, Martin R, Moxon SA, et al. ZFIN: enhancements and updates to the Zebrafish Model Organism Database. Nucleic Acids Res. 2011;39:D822–D829. doi: 10.1093/nar/gkq1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yook K, Harris TW, Bieri T, Cabunoc A, Chan J, Chen WJ, Davis P, de la Cruz N, Duong A, Fang R, et al. WormBase 2012: more genomes, more data, new website. Nucleic Acids Res. 2012;40:D735–D741. doi: 10.1093/nar/gkr954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rudd KE. EcoGene: a genome sequence database for Escherichia coli K-12. Nucleic Acids Res. 2000;28:60–64. doi: 10.1093/nar/28.1.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang R, Lin Y. DEG 5.0, a database of essential genes in both prokaryotes and eukaryotes. Nucleic Acids Res. 2009;37:D455–D458. doi: 10.1093/nar/gkn858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Amberger J, Bocchini C, Hamosh A. A new face and new challenges for Online Mendelian Inheritance in Man (OMIM®) Hum. Mutat. 2011;32:564–567. doi: 10.1002/humu.21466. [DOI] [PubMed] [Google Scholar]
- 54.Junier T, Zdobnov EM. The Newick utilities: high-throughput phylogenetic tree processing in the Unix shell. Bioinformatics. 2010;26:1669–1670. doi: 10.1093/bioinformatics/btq243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jordan GE, Piel WH. PhyloWidget: web-based visualizations for the tree of life. Bioinformatics. 2008;24:1641–1642. doi: 10.1093/bioinformatics/btn235. [DOI] [PubMed] [Google Scholar]
- 56.Li J, Waterhouse RM, Zdobnov EM. A remarkably stable TipE gene cluster: evolution of insect Para sodium channel auxiliary subunits. BMC Evol. Biol. 2011;11:337. doi: 10.1186/1471-2148-11-337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zdobnov EM, Bork P. Quantification of insect genome divergence. Trends Genet. 2007;23:16–20. doi: 10.1016/j.tig.2006.10.004. [DOI] [PubMed] [Google Scholar]
- 58.Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Robinson GE, Hackett KJ, Purcell-Miramontes M, Brown SJ, Evans JD, Goldsmith MR, Lawson D, Okamuro J, Robertson HM, Schneider DJ. Creating a buzz about insect genomes. Science. 2011;331:1386. doi: 10.1126/science.331.6023.1386. [DOI] [PubMed] [Google Scholar]