


Abstract
The SwissBioisostere database (http://www.swissbioisostere.ch) contains information on molecular replacements and their performance in biochemical assays. It is meant to provide researchers in drug discovery projects with ideas for bioisosteric modifications of their current lead molecule, as well as to give interested scientists access to the details on particular molecular replacements. As of August 2012, the database contains 21 293 355 datapoints corresponding to 5 586 462 unique replacements that have been measured in 35 039 assays against 1948 molecular targets representing 30 target classes. The accessible data were created through detection of matched molecular pairs and mining bioactivity data in the ChEMBL database. The SwissBioisostere database is hosted by the Swiss Institute of Bioinformatics and available via a web-based interface.
INTRODUCTION
During the course of drug discovery, a considerable amount of time is spent in lead optimization (1). In this phase, compounds that have been identified as active against a molecularly defined target of interest are modified to carefully balance potency and other relevant parameters, such as bioavailability, toxicity, metabolic stability or solubility. Issues with any of these parameters usually need to be addressed without losing the achieved bioactivity against the primary target. The knowledge of potential bioisosteres, i.e. pairs of replacements that share physical and chemical similarities, can be of great help to prioritize the synthesis of analogues with the purpose of modulating the biological and biophysical properties of the entire compound. Traditionally, these replacements have been identified by simple chemical considerations and trial-and-error and found their way into the common knowledge of medicinal chemists over time. Already in 1919, the concept of isosterism, i.e. molecules or ions that possess the same number of atoms and valence electrons, was termed by Langmuir (2) and developed further by Grimm (3), and later Erlenmeyer (4). Friedmann (5) and Thornber (6) described bioisosterism as a form of non-classical isosterism with relation to the modulation of a biological endpoint. Since then, interest and research on this concept have been gradually increasing, and efforts have been put into collecting anecdotes of historically successful applications. Several extensive reviews on examples for bioisosteric relations are available (7–9), and a commercial solution captures annotated data retrieved from primary literature (10). Although these examples certainly contain valuable information and are a great source of inspiration, from a practical point of view it is nevertheless questionable whether success stories are truly generalizable, and what the exhaustiveness of such lists is. We aim to improve on both aspects in SwissBioisostere.
As modern drug discovery projects produce a wealth of biochemical assay data for different chemical structures, it is possible to mine these data computationally by fragmenting the chemical structures and identifying pairs of molecules that differ from each other only by small substructural exchanges, i.e. the so-called matched molecular pairs (MMPs) (11). The additional information of the difference in bioactivity in the biochemical assay allows inferring the impact of this structural exchange.
The presented SwissBioisostere database contains such molecular replacements annotated with their differences in bioactivity against the primary target they were measured against. This development has been made possible as (i) extensive drug discovery data have been put in the public domain in a useable format by efforts such as ChEMBL (12) and (ii) fast algorithms to mine these data for MMPs have been developed (13,14). Due to these recent developments, MMP analysis has gained increased interest in the past years (15). Although historically, such analyses were almost exclusively possible within the proprietary data of large pharmaceutical companies and constrained to small data sets owing to the algorithmic complexity, nowadays even very large data sets can be analysed, and the increasing availability of public domain databases (16) offers the potential to perform these analyses also in academic settings.
With the SwissBioisostere database, we provide access to a large knowledgebase of molecular replacements and information on their observed impact on biological activity, to aid medicinal chemists in their quest to identify clinical candidates and to facilitate drug design research via an interface that requires no specific training.
DATABASE CONSTRUCTION AND FEATURES
Experimental data retrieval and preparation
The ChEMBL database was used as primary data source for experimental assay records. Version 13 (2012) was downloaded from the website of the European Bioinformatics Institute and held in a local MySQL database. Then, data points of interest were selected according to the following criteria: all compounds with annotated assay data in IC50, EC50, Ki or Kd, which underwent a standardization process by ChEMBL curators, were selected. Bioactivity values were transformed into their cologarithmic forms. Data points annotated with values >10 µM, were kept, but flagged as inactive. In case a limiting operator (e.g. < or >) was attributed to an activity, the corresponding data point was removed to keep only clearly annotated bioactivity values. Compounds with a molecular weight ≥1000 Da were filtered to avoid long runtimes during the fragmentation process. Assays were kept, if a curator of the ChEMBL database annotated the observed effect as directly related to an interaction with a particular molecularly defined target (confidences level 8 and 9). Some assays were manually removed owing to obvious errors in their annotated bioactivity data. If more than one target was attributed to a particular assay, all mentioned targets were stored as target ensemble for this particular assay. Target class tags were created by merging the tags of levels 1 and 2 in the target classification of the ChEMBL database.
The resulting entries were grouped by their assay ID. Members of such a group have been measured in the same assay under equivalent conditions. Duplicate structures were removed from each subset, and, in case their results were annotated with differing experimental values, replaced by the median. The compound structures were standardized using the ‘Standardize Molecule’ component in Pipeline Pilot (17) by de-salting, harmonizing the bonds and formal charges of particular substructures [e.g. nitro groups as − N + (=O)O−] and normalizing the chirality annotation (18). Furthermore, compounds were neutralized to the parent form by protonating acids and deprotonating bases. Molecules containing bad valences were filtered out. Finally, a canonical tautomer was defined for each compound, based on the algorithm by Sayle and Delaney (19).
For assays with more than one remaining compound, a canonical SMILES was calculated for all compounds and used together with information on assay ID, measured bioactivity, corresponding target and target class as input for the MMP identification process.
The number of entries, compounds and assays filtered at each step are listed in Table 1. Figure 1 gives an overview about the filtering cascade.
Table 1.
Overview of the number of entries filtered out during the different filter steps
| Steps | Entries | Compounds | Assays |
|---|---|---|---|
| ChEMBL v13 | 6 933 068 | 1 143 682 | 617 681 |
| Entry selection | −6 250 195 | −847 213 | −556 127 |
| Removal of unwanted entries | −60 840 | −14 219 | −20 070 |
| Compound treatment | −32 473 | −2462 | −632 |
| MMP identification | 589 560 | 279 788 | 40 852 |
Figure 1.

Overview of the different steps leading to the creation of the SwissBioisostere database.
MMP identification
To identify pairs of molecules differing from each other only by a small substructural exchange, the Hussain and Rea algorithm (14) was implemented in Java using libraries from the Chemistry Development Kit (20) and the Indigo library from GGA (http://ggasoftware.com/opensource/indigo). The algorithm fragments molecular structures by cutting up to three bonds simultaneously and stores one part of the molecule as key of a hash table while the other part becomes the corresponding object. For single cuts, in which only one bond is cut at a time, both fragments become key and object, respectively. For double and triple cuts, only the resulting fragments with single attachment points become the key, and scaffold (three attachment points) or linker (two attachment points) becomes the object. If the fragmentation of two different compounds results in equal keys, the two corresponding objects define the variable part of an identified MMP; i.e. a replacement has been found. Cuttable bonds were defined as single bonds that were neither part of a ring system nor part of a functional group such as, e.g. an amide group or a carboxylic acid (SMARTS: [#6+0;!$(*=,#[!#6&!R])]!@!#!=[*]). Furthermore, an additional rule was implemented, to be able to detect replacements of groups connected by exocyclic double bonds to a ring system (SMARTS: [#6+0;R]=[#7&!R,#8&!R,#16&!R,#6+0&!R]). Replacements are encoded as canonicalized SMIRKS. (21) Only replacements with a maximal number of 12 heavy atoms in the variable part were kept during the identification process. This threshold was set as a precautionary measure with respect to the resulting number of potential cuts and to ensure that replacements for larger scaffolds, e.g. bicyclic ring systems, could still be identified. Furthermore, the common part of a matched pair was required to have at least the same number of heavy atoms as the variable part. If compounds containing marked stereo centres in the constant part were matched, the match was verified for identical stereochemistry of all centres, and kept, if this was the case. Matches of defined stereo centres with non-annotated ones were allowed, but flagged in the database. Mismatched stereo centres led to rejection of a previously found replacement. Each performed fragmentation was validated by reconnection of the fragments and comparison with the original molecule. Hydrogen replacements were identified in a separate step. An a posteriori filter was implemented for replacements identified through triple cuts: Topological distances between all attachment points were calculated for both common parts of a replacement. In case the sum of the differences in topological distance exceeded the conservatively chosen threshold of 3, a triple cut replacement was not accepted. This filter prevents the occurrence of cases where the directionality of the sidechains (the common constant part) between the two fragments has significantly changed, whereas at the same time smaller changes (e.g. hexyl to pentyl or phenyl to benzyl) remain accepted. Studies by Papadatos and coworkers (22) showed the importance of the chemical context of an attachment point when comparing difference distributions of replacements observed in hERG, solubility and lipohilicity assay data. Contextual information was taken into account by annotating the corresponding attachment points with such information during the fragmentation process. Three classes of attachment points were defined: (i) connection to an aromatic ring; (ii) link to an aliphatic ring; and (iii) connection to an aliphatic linker. A finer differentiation like in the work of Papadatos has been considered, but not been implemented because it would have resulted in data subsets with only few members, preventing a subsequent statistical assessment of the impact of the chemical context. Furthermore, even if it would be differentiated in a more detailed fashion (e.g. between heteroaromatic rings containing atoms with potential H-bond acceptor and donor function), although keeping an adequate amount of data, there might still be an important difference in whether these atoms are located in ortho-, meta- or para-position to the attachment point for instance, which would prevent a relevant exploitation of the data [see a corresponding example in the review article of Griffen et al. (15)]. All calculated and accepted replacements were stored in a MySQL 5.5 database.
SwissBioisostere database content
Currently (August 2012), SwissBioisostere contains information on 5 586 462 unique replacements that can be sub-divided into 33.03% Single Cuts (substituent replacements), 45.89% Double Cuts (linker replacements) and 21.08% Triple Cuts (scaffold replacements). In total, 21 293 355 data points can be accessed via the interface. Figure 2 highlights how often specific replacements have been observed in the database. Indeed, about half of the accessible replacements (46.61%) are singletons, i.e. have been only observed once, 13.23% have been observed ≥five times. A list of the 1948 targets from 30 target classes can be found in the Supplemental Material (Supplementary Tables S1 and S2).
Figure 2.
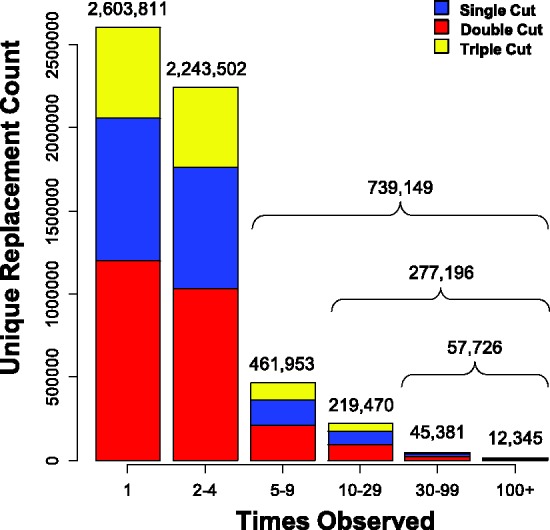
Information on counts of unique replacements stratified by the type of cut that has been performed.
Table 2 gives an overview about the parameters stored in the SwissBioisostere database and their origin. Differences in logP, tPSA and Molecular Weight of the fragments corresponding to a replacement are based on values that have been provided by ChEMBL for the corresponding compounds. In case a replacement was observed more than once, the mean of the differences over all observed MMPs is reported.
Table 2.
Properties available in SwissBioisostere
| Assays | Compounds | MMP | Replacement |
|---|---|---|---|
| Assay ID a | ChEMBL ID a | Compound IDs a | SMIRKS b |
| Target a | SMILES b | Bioactivities a | R group distance b |
| Target class a | ACD logP a | Bioactivity difference b | Score b |
| Assay type a | tPSA a | Attachment point context b | |
| Confidence a | Molecular weight a | Constant part b | |
| PubMed ID a | Murcko fragment b | Stereo tag b |
aEntries were taken directly from ChEMBL.
bEntries are calculated properties for SwissBioisostere.
To aid ranking of replacements, a scoring scheme has been implemented. It is based on the observed bioactivity differences of the replacement, its total count, as well as the number of different targets, target classes and Murcko scaffold families (23) in which it has been observed. For more details on the implemented score, the reader is referred to section 1 of the Supplemental Material.
We implemented an adapted version of the R group descriptor published by Holliday et al. (24) as a chemical similarity measure between fragments corresponding to a replacement. Details on the calculation can be found in section 2 of the Supplemental Material.
DATA ACCESS
Interface description
Access to the SwissBioisostere database is provided via a web-based interface accessible at http://www.swissbioisostere.ch. The starting page contains two chemical sketchers and allows the user to perform two different types of query: (i) query for observed replacements of a particular substructure or (ii) query for a particular replacement. Scenario 1 is typically a question that is raised if a New Chemical Entity drug discovery project is running into an issue with a particular property and aims to escape this situation by chemical modification of the lead compound. When entering a single substructure into sketcher 1 on the start page of the website, the database is searched for replacements that have been used in the past for this exact substructure. An exemplary screenshot (Figure 3) shows the results for the nitro function. This page gives a broad overview of observed replacements for this particular substructure. For more details for a particular replacement, the user is able to follow the links provided on each fragment. As medicinal chemists are often interested in modifying lipophilicity and/or polar surface area of a compound, these values are reported in a dynamic plot, which allows to select on the difference that a replacement would cause in these two properties. The performance of each replacement in biochemical assays as stored in the ChEMBL database is presented as a three-class distribution (improved, worsened or isoactive). Class membership has been attributed with regards to the observed change in bioactivity for a replacement: improved (Δ bioactivity < −0.5 log units or transformed inactive molecule to an active form), worsened (Δ bioactivity > 0.5 log units or transformed active molecule to an inactive form) or isoactive (else). The threshold of 0.5 log units, i.e. a factor of 3.2 in activity, has been chosen to account for the unknown experimental uncertainty in the underlying assays. It is possible to sort the given replacements by values of the different parameters—ΔtPSA, ΔlogP, ΔMW, total count, class counts, success-based score and chemical similarity measure—provided in the table of results. Filtered results can then be exported via a copy-to-clipboard function or to a CSV file.
Figure 3.

Overview about the fragment replacement proposal process. The user enters a chemical substructure in sketcher 1 (1). The query results in an overview page showing all replacements that have been performed for this particular substructure: (2) ΔlogP, ΔtPSA overview that can be used to select subsets, (3) results table, (4) image of performed replacement, which links to a detailed overview page, (5) bioactivity difference distribution, three class classification, (6) sorts columns, (7) data export.
Via the link of a proposed replacement, or in case the query was performed by adding two fragments in sketcher 1 and 2, respectively, the user obtains a detailed overview page presenting data concerning the effects this particular replacement showed experimentally with regards to the activity against molecularly defined targets. Figure 4 shows the results of the exchange of a pyridine with a morpholine. The distribution of bioactivity differences is visualized in two different manners: an overview of the full distribution and a split by the chemical context of the attachment points (in case of single and double cuts). The plots are sensitive to a performed selection via the available filters. The column filter within the table provides the user with options to filter for particular target classes, targets or assay IDs. An additional filter box provides the user with more possibilities to fine-grain the data. Here, it is possible to select the range of activity of compound A of a matched pair A–B. Thereby, it is possible to analyse, e.g. whether a replacement showed a similar or improved bioactivity over the whole activity range, or only in a particular region. Furthermore, the data can be split to focus on results from different assay types (functional or binding assays), according to the confidence the ChEMBL curators attributed to a particular assay/target annotation, or if only perfectly matched stereochemistry or also MMPs, in which stereochemistry was not completely defined for one or both of the two molecules, should be considered. The table of results contains additional information on each measured data point. Under each entry, representations of the corresponding molecules to a MMP can be retrieved, as well as their measured bioactivity values in the biochemical assay. Furthermore, a link to the respective website at the European Bioinformatics Institute provides access to the corresponding compound reports in the ChEMBL database. Again, all filtered results can be exported via a copy to the clipboard function or download of a CSV file.
Figure 4.

Retrieval of details for a particular molecular replacement. The user enters the replacement as chemical substructures in sketcher 1 and 2 (1), or follows a link from the result page presented in Figure 3. The query results in a detailed overview page that presents the underlying data for this particular replacement: (2) general statistics, (3) bioactivity difference distribution plot, (4) bioactivity difference distributions by attachment point context, (5) filter for assay and compound properties, (6) results table, (7) sorts columns, (8) column filter for assays, targets, target classes, (9) detail view for particular data record and (10) data export.
Applied technologies
In its current design, SwissBioisostere is built on Apache, MySQL, Java, Javascript and Flash technologies. The underlying RDBS is based on MySQL 5.5 (http://www.mysql.com). The host server runs an Apache Tomcat 7 server (http://tomcat.apache.org). In the current version, the molecular sketchers are based on MarvinSketch by Chemaxon (http://www.chemaxon.com). All presented plots are created using the flot package developed by Ole Laursen (http://code.google.com/p/flot), result tables and capabilities such as ranking, filtering and exporting data were implemented using the DataTables package by SpryMedia (http://datatables.net). JQuery (http://jquery.com) is the underlying javascript library that is accessed by those components.
CONCLUSIONS AND FUTURE DIRECTIONS
To facilitate the identification and assessment of molecular substructural replacements, we have developed and released the SwissBioisostere database. Via a web interface, accessible at http://www.swissbioisostere.ch, we provide medicinal chemists with a valuable idea generator that is supported by historical data and a platform to aid research on individual molecular replacements. To keep the database up to date with the latest information, it is planned to perform updates at regular intervals. As SwissBioisostere is one of the components of the online tools developed by the Molecular Modelling group of the Swiss Institute of Bioinformatics in Lausanne, we plan to integrate a possibility to crosstalk with other web services already available, e.g. SwissDock (25) (http://www.swissdock.ch), or under development, to provide an even better service to drug discovery researchers.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Sections 1 and 2, Supplementary Tables 1 and 2 and Supplementary References [26–29].
FUNDING
Funding for open access charge: Swiss Institute of Bioinformatics.
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
The authors thank Serge Christmann-Franck, Antoine Daina and Alexander Klenner for valuable feedback during the interface design phase. Friedrich Rippmann is thanked for constructive comments on the manuscript. M.W. thanks Merck Serono S.A. for a PhD fellowship.
REFERENCES
- 1.Paul SM, Mytelka DS, Dunwiddie CT, Persinger CC, Munos BH, Lindborg SR, Schacht AL. How to improve R&D productivity: the pharmaceutical industry’s grand challenge. Nature Rev. Drug Disc. 2010;9:203–214. doi: 10.1038/nrd3078. [DOI] [PubMed] [Google Scholar]
- 2.Langmuir I. The structure of atoms and the octet theory of valence. Proc. Natl Acad. Sci. USA. 1919;5:252–259. doi: 10.1073/pnas.5.7.252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Grimm H. On the systematic arrangement of chemical compounds from the perspective of research on atomic composition; and on some challenges in experimental chemistry. Naturwissenschaften. 1929;17:557–564. [Google Scholar]
- 4.Erlenmeyer H, Basel A, Leo M. Über Pseudoatome. Helv. Chim. Acta. 1932;15:1171–1186. [Google Scholar]
- 5.Friedman H. Influence of isosteric replacements upon biological activity. Washington D.C. Natl Acad. Sci. 1951;206:295. [Google Scholar]
- 6.Thornber C. Isosterism and molecular modification in drug design. Chem. Soc. Rev. 1979;8:563–580. [Google Scholar]
- 7.Patani GA, LaVoie EJ. Bioisosterism: a rational approach in drug design. Chem. Rev. 1996;96:3147–3176. doi: 10.1021/cr950066q. [DOI] [PubMed] [Google Scholar]
- 8.Lima L, Barreiro E. Bioisosterism: a useful strategy for molecular modification and drug design. Curr. Med. Chem. 2005;12:23–49. doi: 10.2174/0929867053363540. [DOI] [PubMed] [Google Scholar]
- 9.Meanwell NA. Synopsis of some recent tactical application of bioisosteres in drug design. J. Med. Chem. 2011;54:2529–2591. doi: 10.1021/jm1013693. [DOI] [PubMed] [Google Scholar]
- 10.BIOSTER Database. Accelrys Inc. San Diego, CA: [Google Scholar]
- 11.Leach AG, Jones HD, Cosgrove DA, Kenny PW, Ruston L, MacFaul P, Wood JM, Colclough N, Law B. Matched molecular pairs as a guide in the optimization of pharmaceutical properties; a study of aqueous solubility, plasma protein binding and oral exposure. J. Med. Chem. 2006;49:6672–6682. doi: 10.1021/jm0605233. [DOI] [PubMed] [Google Scholar]
- 12.Gaulton A, Bellis LJ, Bento AP, Chambers J, Davies M, Hersey A, Light Y, McGlinchey S, Michalovich D, Al-Lazikani B, et al. ChEMBL: a large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2011;40:D1100–D1107. doi: 10.1093/nar/gkr777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Warner DJ, Griffen EJ, St-Gallay SA. WizePairZ: a novel algorithm to identify, encode, and exploit matched molecular pairs with unspecified cores in medicinal chemistry. J. Chem. Inf. Model. 2010;50:1350–1357. doi: 10.1021/ci100084s. [DOI] [PubMed] [Google Scholar]
- 14.Hussain J, Rea C. Computationally efficient algorithm to identify matched molecular pairs (MMPs) in large data sets. J. Chem. Inf. Model. 2010;50:339–348. doi: 10.1021/ci900450m. [DOI] [PubMed] [Google Scholar]
- 15.Griffen E, Leach AG, Robb GR, Warner DJ. Matched molecular pairs as a medicinal chemistry tool. J. Med. Chem. 2011;54:7739–7750. doi: 10.1021/jm200452d. [DOI] [PubMed] [Google Scholar]
- 16.Nicola G, Liu T, Gilson MK. Public domain databases for medicinal chemistry. J. Med. Chem. 2012;55:6987–7002. doi: 10.1021/jm300501t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pipeline Pilot. Accelrys Inc. San Diego, CA: [Google Scholar]
- 18. Stereochemical information was removed from atoms that were not considered true stereo atoms, e.g. non-chiral C drawn with wedges. No stereo information has been added for atoms that were perceived as chiral, but not annotated as such in the original compound.
- 19.Sayle R, Delaney J. Canonicalization And Enumeration Of Tautomers. 1999. EuroMUG99, Cambridge, UK. [Google Scholar]
- 20.Steinbeck C, Han Y, Kuhn S, Horlacher O, Luttmann E, Willighagen E. The chemistry development kit (CDK): an open-source java library for chemo- and bioinformatics. J. Chem. Inf. Comp. Sci. 2003;43:493–500. doi: 10.1021/ci025584y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. The canonicalization procedure also considers the order of attachment point IDs to ensure the correct mapping/direction in the case of multiple cuts.
- 22.Papadatos G, Alkarouri M, Gillet VJ, Willett P, Kadirkamanathan V, Luscombe CN, Bravi G, Richmond NJ, Pickett SD, Hussain J, et al. Lead optimization using matched molecular pairs: inclusion of contextual information for enhanced prediction of hERG inhibition, solubility, and lipophilicity. J. Chem. Inf. Model. 2010;50:1872–1886. doi: 10.1021/ci100258p. [DOI] [PubMed] [Google Scholar]
- 23.Bemis GW, Murcko MA. The properties of known drugs 1. Molecular frameworks. J. Med. Chem. 1996;39:2887–2893. doi: 10.1021/jm9602928. [DOI] [PubMed] [Google Scholar]
- 24.Holliday JD, Jelfs SP, Willett P, Gedeck P. Calculation of intersubstituent similarity using R-group descriptors. J. Chem. Inf. Comp. Sci. 2003;43:406–411. doi: 10.1021/ci025589v. [DOI] [PubMed] [Google Scholar]
- 25.Grosdidier A, Zoete V, Michielin O. SwissDock, a protein-small molecule docking web service based on EADock DSS. Nucleic Acids Res. 2011;39:W270–W277. doi: 10.1093/nar/gkr366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. The R Project for Statistical Computing, Version 2.15.1.
- 27.Ghose AK, Viswanadhan VN, Wendoloski JJ. Prediction of hydrophobic (lipophilic) properties of small organic molecules using fragment methods: an analysis of alogP and clogP methods. J. Phys. Chem. A. 1998;102:3762–3772. [Google Scholar]
- 28.Ghose AK, Crippen GM. Atomic physicochemical parameters for three-dimensional-structure-directed quantitative structure-activity relationships. 2. modeling dispersive and hydrophobic interactions. J. Chem. Inf. Comp. Sci. 1986;9:80–90. doi: 10.1021/ci00053a005. [DOI] [PubMed] [Google Scholar]
- 29.Gasteiger J, Marsili M. A new model for calculating atomic charges in molecules. Tetrahedron Lett. 1978;19:3181–3184. [Google Scholar]