


Abstract
HEXEvent (http://hexevent.mmg.uci.edu) is a new database that permits the user to compile genome-wide exon data sets of human internal exons showing selected splicing events. User queries can be customized based on the type and the frequency of alternative splicing events. For each splicing version of an exon, an ESTs count is given, specifying the frequency of the event. A user-specific definition of constitutive exons can be entered to designate an exon exclusion level still acceptable for an exon to be considered as constitutive. Similarly, the user has the option to define a maximum inclusion level for an exon to be called an alternatively spliced exon. Unlike other existing splicing databases, HEXEvent permits the user to easily extract alternative splicing information for individual, multiple or genome-wide human internal exons. Importantly, the generated data sets are downloadable for further analysis.
INTRODUCTION
The process of pre-mRNA splicing is essential for the expression of most metazoan genes. It is carried out by the spliceosome that catalyzes the removal of non-coding intronic sequences and concatenates remaining exons to form mature mRNAs (1). Of the approximately 25 000 genes encoded by the human genome (2), >90% are believed to produce transcripts that are alternatively spliced (3,4). The process of alternative splicing results in the production of multiple mRNA isoforms from a single pre-mRNA and thereby significantly enriches the proteomic diversity of higher eukaryotic organisms. Major splice events include exon skipping (also referred to as cassette exon events), exons with alternative 3′- and/or 5′-splice sites, intron retention, mutually exclusive exons, as well as alternative first and last exons. Given the complexity of higher eukaryotic genes and the relatively low conservation of splice sites, the precision of the splicing machinery is impressive. Defects in splicing lead to many human genetic diseases (5–7) and splicing mutations in a number of genes involved in growth control have been implicated in multiple types of cancer (8–12).
The vast majority of alternative splicing events are biased toward high (>80%) or low (<20%) inclusion levels (Figure 1). This distribution is not only observed for alternative splicing events defined by Expressed Sequence Tag (ESTs), but also for alternative splicing events derived from deep sequencing data (13). Interestingly, alternatively spliced exons with high inclusion levels (>90%) display physical characteristics indistinguishable from constitutive exons (13), even when using machine learning techniques (our unpublished data). Thus, an exon that is associated with an extremely rare alternative splicing event may behave much more like a constitutive exon. These observations, in combination with the demonstration that even constitutive exons display low levels of alternative splicing (4), challenge the definition of constitutive and alternative exons. Yet, all genome-wide analyses of alternative splicing carried out in the past have relied on comparing sets of alternatively spliced exons with constitutive exons based on simple yes/no decisions, thus potentially introducing large errors. HEXEvent permits the user to define the inclusion level required for an exon to be considered as alternatively or constitutively spliced. This definition can range from the strictest to more relaxed constitutive splicing interpretations.
Figure 1.
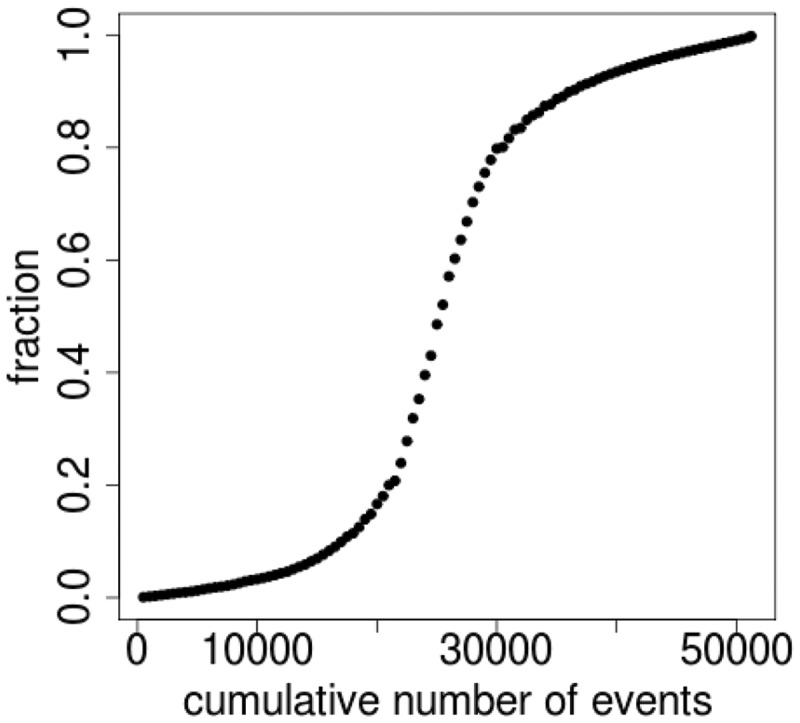
Cassette exon inclusion levels determined from HEXEvent. The plot shows the relationship between exon inclusion levels and the cumulative number of events.
To carry out large-scale or genome-wide analyses on exons of a certain type, extensive exon data sets of that alternative splicing type are needed. ASPicDB (14) is a recently published alternative splicing database that reports a list of exons within a certain region or gene. However, this list is not made available for download. While the University of California, Santa Cruz (UCSC) Genome Browser offers sets of all alternatively spliced exons for download (15), those sets are missing three important pieces of information: (i) no set of constitutively spliced exons is available; (ii) inclusion and/or usage levels are not assigned to exons or splice sites; and (iii) all sets have a splice event centric view, meaning they list all exons that show a certain splicing event, but do not list all splicing events for an individual exon. Finally, neither ASPicDB nor the UCSC Genome Browser allow user-specific definitions for constitutive and alternative exons. In contrast, HEXEvent allows the user to tailor splicing categorization based on exon inclusion levels. In addition, the queried data is downloadable for further analysis regardless of whether it covers individual or genome-wide sets of internal human exons. Finally, all known alternative splicing events per exon are reported in the output file.
DATABASE GENERATION, CONTENT AND DEFINITIONS
HEXEvent is a new database, which assists the user in compiling genome-wide exon data sets. Its exon information is based on known mRNA isoforms as defined by the UCSC Genome Browser (GRCh37/hg19) (15) as well as available EST information. For each internal exon in the human genome, the number of EST events that include or exclude the queried exon or an alternative version of it was computed. Based on this information, inclusion/exclusion levels of each exon as well as usage frequencies of alternative splice sites are defined. The major splice version of each exon was defined, whereas all minor splice variants are indicated as alternative splicing events of that exon. HEXEvent includes information about exon skipping/inclusion and alternative 3′ and/or 5′-splice sites. At this stage, we do not include information on intron retention events because HEXEvent is currently based on EST information. Due to the short length of the ESTs, intron retention information would be biased toward short retained introns.
If a new splice variant was found only in ESTs, but it was not included in the UCSC isoform list, we accepted it as a real new splice alternative (indicated as ‘onlyEST’ instead of assigning a gene name), if it showed the canonical splice sites (GU at the 5′-splice site, AG at the 3′-splice site). Although this splice site requirement might reduce the identification of new alternative splicing events associated with the minor spliceosome, it significantly reduces the number of false positives. Seemingly, alternative versions of an exon where the annotated splice sites differ only by 1 or 2 nt were combined to a single version of the exon because the addition of one or two more nucleotides rarely defines an alternative splice site (16).
For each alternative splice version of an exon, the number of supporting ESTs is given. For cassette exons, the number of ESTs including and not including the exon is specified. For exons with one or more alternative splice sites, the location of the alternative sites are reported as well as the number of ESTs supporting them.
The basic EST counts as defined in Table 1 are represented as follows:
ccount: the number of ESTs including the exon with the major coordinates;
calt3: number of ESTs including the exon with an alternative 3′-splice site;
calt5: number of ESTs including the exon with an alternative 5′-splice site;
calt3+5: number of ESTs including the exon with both; an alternative 3′-splice site as well as an alternative 5′-splice site; and
cskip: number of ESTs excluding the exon
Based on these numbers several values are calculated for each exon.
The constitutive level evaluates the inclusion of the major version of the exon and compares it to the number of occurrences of any alternative event. The constitutive level is defined as:
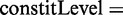
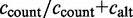 . The alternative count
. The alternative count  includes all counts of possible alternative events,
includes all counts of possible alternative events, 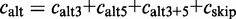 .
.The inclusion level compares the presence of the exon and any alternative version of it with its exclusion. The inclusion level is defined as:
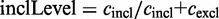 . The inclusion count
. The inclusion count 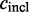 equals the sum of ESTs showing the exon with major coordinates
equals the sum of ESTs showing the exon with major coordinates 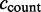 plus the number of ESTs showing the exon with an alternative 3′-splice site
plus the number of ESTs showing the exon with an alternative 3′-splice site 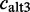 , with an alternative 5′-splice site
, with an alternative 5′-splice site 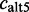 or with an alternative 3′- and 5′-splice site
or with an alternative 3′- and 5′-splice site 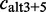 (
(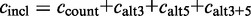 , see Table 1). The exclusion count
, see Table 1). The exclusion count 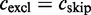 equals the number of ESTs supporting exon skipping.
equals the number of ESTs supporting exon skipping.The usage level of the 3′-splice site represents the usage ratio of the major 3′-splice site and any used 3′-splice site. It is defined as:
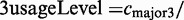
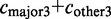 . The number of ESTs showing the usage of the major 3′-splice site
. The number of ESTs showing the usage of the major 3′-splice site 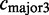 equals the sum of ESTs including the major version of the exon
equals the sum of ESTs including the major version of the exon 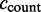 and the number of ESTs including the exon with an alternative 5′-splice site but the same 3′-splice site calt5 (
and the number of ESTs including the exon with an alternative 5′-splice site but the same 3′-splice site calt5 (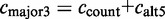 ). In contrast, the number of ESTs not showing the exon with its major 3′-splice site
). In contrast, the number of ESTs not showing the exon with its major 3′-splice site 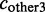 equals the sum of ESTs that show the exon either only with an alternative 3′-splice site
equals the sum of ESTs that show the exon either only with an alternative 3′-splice site 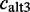 or with mutually occurring 3′- and 5′-splice sites calt3+5 (
or with mutually occurring 3′- and 5′-splice sites calt3+5 (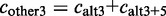 ).
).The usage level of the 5′-splice site is defined analogously to the usage level of the 3′-splice site as:
 .
.
Based on the input, exons are filtered according to the user settings and a customized set of exons will be extracted and prepared for download. A summary of the basic workflow of the creation of the HEXEvent database is shown in Figure 2.
Table 1.
Definition of the columns in the output format of HEXEvent for a randomly chosen exon
| No. | Column name | Example | Description |
|---|---|---|---|
| 1 | chromo | chrX | Reference sequence chromosome name |
| 2 | strand | + | + or − for strand |
| 3 | start | 101 854 639 | First position of the exon (0-based) |
| 4 | end | 101 854 775 | Last position of the exon (1-based) |
| 5 | count | 15 | Number of ESTs that include the exon as given in columns [chromo], [strand], [start], and [end] |
| 6 | alt3 | 10 | Number of ESTs that include the exon with an alternative 3′-splice site |
| 7 | alt5 | 0 | Number of ESTs that include the exon with an alternative 5′-splice site |
| 8 | alt3+5 | 2 | Number of ESTs that include the exon with an alternative 3′ and an alternative 5′-splice site simultaneously |
| 9 | skip | 0 | Number of ESTs in which the exon is skipped |
| 10 | constitLevel | 0.556 | Constitutive level of the exon (=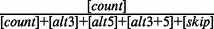 ) ) |
| 11 | inclLevel | 1.000 | Inclusion level of the exon (= 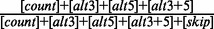 ) ) |
| 12 | 3usageLevel | 0.556 | Usage level of the major 3′-splice site of the exon 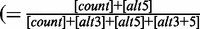 ) ) |
| 13 | 5usageLevel | 0.926 | Usage level of the major 5′-splice site of the exon 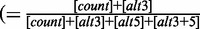 ) ) |
| 14 | alt3singleCount | 10 | Number(s) of ESTs for different alternative 3′- splice site |
| 15 | alt3singleLoc | 101 854 633 | Location(s) of alternative 3′-splice sites |
| 16 | alt5singleCount | 0 | Number(s) of ESTs for different alternative 5′-splice site |
| 17 | alt5singleLoc | # | Location(s) of alternative 5′-splice sites |
| 18 | alt3and5singleCount | 2 | Number(s) of ESTs for different alternative 3′- and 5′-splice sitecombinations |
| 19 | alt3and5singleLocName | 101 854 633–101 854 787 | Location(s) of alternative 3′ and 5′-splice site combinations |
| 20 | OnlyESTexonCount | 0 | Number of ESTs in which the exon is overlapped by an alternative version of it, that is not included in the human isoform list of the UCSC Genome Browser yet, but has at least one EST supporting it |
| 21 | OnlyESTexons | # | Location(s) of alternative version(s) of the exon, that is/are not included in the human isoform list of the UCSC Genome Browser yet, but has/have at least one EST supporting it |
| 22 | genename | ARMCX5 | Name of the gene the exon is part of, if no gene name was assignedyet, it is indicated by ‘onlyEST’ |
The first four columns describe the location of the exon, whereas columns 5–9 give EST counts for inclusion (as given in columns 1–4), alternative splice site usage and exclusion of the exon. Column 10 specifies the constitutive level of the exon. Here, the grade of being constitutive is calculated by comparing the occurrence of the exon as specified in columns 1–4 with all other alternative events. In column 11, the inclusion level of the exon is given. Here, inclusion is calculated as the sum of ESTs showing the exon with the coordinates given in columns 1–4 and EST counts for alternative version of the exons showing an alternative 3′- and/or 5′-splice site, whereas exclusion is represented by the number of ESTs having this exon skipped. Columns 12 and 13 show the usage level of the major 3′-splice site and the major 5′-splice site (as given in columns 3 and 4 or columns 4 and 3 when on the negative strand), respectively. The usage level of the major 3′-splice site of the exon is calculated as the ratio of the number of ESTs showing this 3′-splice site, i.e. all ESTs showing the exons as given in columns 1–4 as well as all ESTs showing the exon with an alternative 5′-splice site, and the number of ESTs that include the exon with any splice site. The usage level of the major 5′-splice site is calculated analogously. The location of all alternative 3′-splice sites of the exon can be found in column 15, whereas the EST counts for each single one are given in column 14. Respective entries can be found in columns 16 and 17 for alternative 5′-splice sites, as well as in columns 18 and 19 for mutually occurring 3′- and 5′-splice sites. The EST count and location of the new versions of the exon that have EST evidence but are not confirmed events in the UCSC Genome Browser yet, are shown in columns 20 and 21. The location is given in the form ‘chromosomeSTRANDstart-end’. The last column shows the name of the gene the exon is part of. If none was assigned yet, ‘onlyEST’ is specified.
Figure 2.

Workflow during the creation of HEXEvent. We downloaded the UCSC Genes track, the spliced ESTs track, as well as the human mRNAs track from the UCSC Genome Browser. Using all three data sets, we extracted all known versions of human internal exons. An EST count was assigned to each version of each exon, specifying inclusion and exclusion levels. In a last step, overlapping exons were combined and indicated as alternative versions of each other.
USAGE
To use the HEXEvent database, characteristics of the queried exons have to be specified by the user. These specifications include defining which types of alternative splicing events should be investigated and designating an inclusion level to classify constitutive and alternative exons. Additionally, the user is asked to specify whether exons are allowed to show alternative splicing events other than the selected ones. For each queried exon, HEXEvents reports the genome location, the type(s) of alternative splicing and their location, as well as the number of ESTs in support of the events. An EST count is given for each alternative version.
Input
All input selections are used to specify the type of exons the user is interested in. The basic input can be a genomic region, a list of genes or the whole genome. Second, the type of alternative splicing the user is interested in needs to be defined. The user can choose among options including all exon types, only constitutive exons, or any combination of cassette, alternative 3′-splice site, alternative 5′-splice site and simultaneously alternative 3′- and 5′-splice sites exons. Third, the definition of a constitutive exon can be specified, i.e. the user decides what exon constitutive level (as defined in ‘Database Generation, Content and Definitions’ section) is acceptable for an exon to still be called constitutive. Furthermore, the user can restrict the set of alternatively spliced exon events by defining an upper inclusion level, which, for instance, is useful when analyzing only low-inclusion exons. Analogously, the user has the option to restrict the 3′- and/or 5′-splice site usage level. Fourth, the user chooses whether selected alternative splicing events should be unique or can occur in combination with other splicing events. To do so, the user will be asked whether selected exon types should be ‘strict’. Here, a ‘non-strict’ exon definition means that the exon has to show at least one of the selected types of alternative splicing, but it may also be associated with any of the not selected types. In contrast, when a strict exon definition is chosen, the exon has to be involved in at least one of the selected alternative splicing types, but must not show any of the not selected types. The database will compile a final list of exons based on these input parameters to be displayed in the browser or to be downloaded and saved to a file.
Output
The output of each query will be a table of all requested exons showing the frequencies of their known splicing events. Depending on the user choice, the results will be displayed in the browser or they will be written to a downloadable text file. All output columns are described in Table 1.
Example applications
In an example application of the HEXEvent database, we are interested in all exons of the gene ARMCX4 and all their known splicing events. To get this information, the gene name needs to be specified in the input mask of the database. Furthermore, an interest in all types of exons, i.e. all alternative events, needs to be selected. The refinement of the exon type as well as a strict or non-strict exon definition has no effect on the output because we requested all exons, no matter what type. Selecting all exons overrides any other selections made. The output of this query is shown in Table 2.
Table 2.
Examples of HEXEvent database outputs
| chromo | strand | start | end | count | alt3 | alt5 | alt3+5 | skip | constit Level | incl Level | 3usage Level | 5usage Level | alt3single Count | alt3single Loc | alt5single Count | alt5single Loc | alt3and5single Count | alt3and5 singleLoc | OnlyEST exonCount | OnlyEST exons | genename |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| All exons of gene ARMCX4, all alternative eventsa | |||||||||||||||||||||
| chrX | + | 100 673 897 | 100 673 988 | 15 | 0 | 1 | 0 | 0 | 0.938 | 1.000 | 1.000 | 0.938 | 0 | # | 1 | 100 673 984 | 0 | # | 0 | # | ARMCX4 |
| chrX | + | 100 699 039 | 100 699 143 | 9 | 0 | 0 | 0 | 7 | 0.562 | 0.562 | 1.000 | 1.000 | 0 | # | 0 | # | 0 | # | 0 | # | ARMCX4 |
| chrX | + | 100 700 985 | 100 701 032 | 7 | 0 | 0 | 0 | 5 | 0.583 | 0.583 | 1.000 | 1.000 | 0 | # | 0 | # | 0 | # | 0 | # | ARMCX4 |
| chrX | + | 100 741 009 | 100 741 079 | 33 | 5 | 5 | 1 | 1 | 0.733 | 0.978 | 0.864 | 0.864 | 5 | 100 741 012 | 5 | 100 741 085 | 1 | 100 741 012– 100 741 085b | 0 | # | ARMCX4 |
| chrX | + | 100 742 179 | 100 742 259 | 45 | 1 | 0 | 0 | 0 | 0.978 | 1.000 | 0.978 | 1.000 | 1 | 100 742 191 | 0 | # | 0 | # | 1 | chrX+100 742 179– 100 742 255c | ARMCX4 |
| chrX | + | 100 742 594 | 100 742 678 | 44 | 0 | 0 | 0 | 0 | 1.000 | 1.000 | 1.000 | 1.000 | 0 | # | 0 | # | 0 | # | 0 | # | ARMCX4 |
| chrX | + | 100 743 030 | 100 743 086 | 38 | 7 | 0 | 0 | 0 | 0.844 | 1.000 | 0.844 | 1.000 | 7 | 100 742 994 | 0 | # | 0 | # | 0 | # | ARMCX4 |
| chrX | + | 100 743 430 | 100 744 302 | 5 | 0 | 0 | 0 | 19 | 0.208 | 0.208 | 1.000 | 1.000 | 0 | # | 0 | # | 0 | # | 0 | # | ARMCX4 |
| chrX | + | 100 753 131 | 100 754 353 | 5 | 0 | 2 | 0 | 0 | 0.714 | 1.000 | 1.000 | 0.714 | 0 | # | 2 | 100 753 318 | 0 | # | 0 | # | ARMCX4 |
| chrX | + | 100 759 923 | 100 760 342 | 5 | 0 | 0 | 0 | 0 | 1.000 | 1.000 | 1.000 | 1.000 | 0 | # | 0 | # | 0 | # | 0 | # | ARMCX4 |
| chrX | + | 100 764 345 | 100 764 414 | 3 | 3 | 0 | 0 | 0 | 0.500 | 1.000 | 0.500 | 1.000 | 3 | 100764350 | 0 | # | 0 | # | 0 | # | ARMCX4 |
| chrX | + | 100 764 576 | 100 764 665 | 7 | 0 | 0 | 0 | 0 | 1.000 | 1.000 | 1.000 | 1.000 | 0 | # | 0 | # | 0 | # | 0 | # | ARMCX4 |
| chrX | + | 100 766 011 | 100 766 042 | 7 | 0 | 0 | 0 | 0 | 1.000 | 1.000 | 1.000 | 1.000 | 0 | # | 0 | # | 0 | # | 0 | # | ARMCX4 |
| chrX | + | 100 779 190 | 100 779 419 | 2 | 1 | 0 | 0 | 3 | 0.333 | 0.500 | 0.667 | 1.000 | 1 | 100779272 | 0 | # | 0 | # | 0 | # | ARMCX4 |
| chrX | + | 100 786 630 | 100 786 999 | 3 | 0 | 0 | 0 | 0 | 1.000 | 1.000 | 1.000 | 1.000 | 0 | # | 0 | # | 0 | # | 0 | # | ARMCX4 |
| All exons with an alternative 3′-splice site of gene ARMCX4, no other alternative events allowedd | |||||||||||||||||||||
| chrX | + | 100 742 179 | 100 742 259 | 45 | 1 | 0 | 0 | 0 | 0.978 | 1.000 | 0.978 | 1.000 | 1 | 100 742 191 | 0 | # | 0 | # | 1 | chrX+100 742 179– 100 742 255c | ARMCX4 |
| chrX | + | 100 743 030 | 100 743 086 | 38 | 7 | 0 | 0 | 0 | 0.844 | 1.000 | 0.844 | 1.000 | 7 | 100 742 994 | 0 | # | 0 | # | 0 | # | ARMCX4 |
| chrX | + | 100 764 345 | 100 764 414 | 3 | 3 | 0 | 0 | 0 | 0.500 | 1.000 | 0.500 | 1.000 | 3 | 100 764 350 | 0 | # | 0 | # | 0 | # | ARMCX4 |
| All exons with an alternative 3′-splice site of gene ARMCX4, other alternative events possiblee | |||||||||||||||||||||
| chrX | + | 100 741 009 | 100 741 079 | 33 | 5 | 5 | 1 | 1 | 0.733 | 0.978 | 0.864 | 0.864 | 5 | 100 741 012 | 5 | 100 741 085 | 1 | 100 741 012– 100 741 085b | 0 | # | ARMCX4 |
| chrX | + | 100 742 179 | 100 742 259 | 45 | 1 | 0 | 0 | 0 | 0.978 | 1.000 | 0.978 | 1.000 | 1 | 100 742 191 | 0 | # | 0 | # | 1 | chrX+100 742 179– 100 742 255c | ARMCX4 |
| chrX | + | 100 743 030 | 100 743 086 | 38 | 7 | 0 | 0 | 0 | 0.844 | 1.000 | 0.844 | 1.000 | 7 | 100 742 994 | 0 | # | 0 | # | 0 | # | ARMCX4 |
| chrX | + | 100 764 345 | 100 764 414 | 3 | 3 | 0 | 0 | 0 | 0.500 | 1.000 | 0.500 | 1.000 | 3 | 100 764 350 | 0 | # | 0 | # | 0 | # | ARMCX4 |
| chrX | + | 100 779 190 | 100 779 419 | 2 | 1 | 0 | 0 | 3 | 0.333 | 0.500 | 0.667 | 1.000 | 1 | 100 779 272 | 0 | # | 0 | # | 0 | # | ARMCX4 |
Columns are as defined in Table 1. All ‘hash’ symbol indicate a non-existing value, meaning that there are no alternative 3′- or 5′-splice sites known.
aOutput files were generated when searching for all internal human exons of the gene ARMCX4 including all of their known alternative splicing events.
bLocation of both alternative splice sites (3′- and 5′-splice site).
cLocation of an alternative version of the exon not found in the UCSC Gene list, but in ESTs. It is given in the format ‘chromosomeSTRANDstart-end’.
dOutput files were generated when searching for internal human exons of the gene ARMCX4 that have an alternative 3′-splice site, but do not show any other alternative event.
eOutput files were generated when searching for internal human exons of the gene ARMCX4 that have an alternative 3′-splice site and possibly show other alternative splicing events.
If the user is only interested in exons of the gene ARMCX4 that have an alternative 3′-splice site, only alternative 3′-splice site exons should be selected in the exon-type specification. If a strict exon-type definition is chosen, exons that have an alternative 3′-splice site, but can also be skipped or have an alternative 5′-splice site will not be shown (Table 2). To show those, a non-strict exon definition should be chosen (Table 2).
In case the user requests only constitutive exons, a constitutive exon needs to be defined. In the most conservative scenario, where the user allows no alternative events to call an exon constitutive, the HEXEvent database will report the five exons shown in Table 3. In contrast, if the user allows up to 5% of the ESTs showing alternative splicing, the database will report one additional exon that shows one ESTs with an alternative 3′-splice site in addition to 45 ESTs including the major version of the exon (Table 3).
Table 3.
Output of constitutive exons of the gene ARMCX4
| chromo | strand | start | end | count | alt3 | alt5 | alt3+5 | skip | constit Level | inclLevel | 3usage Level | 5usage Level | alt3single Count | alt3single Loc | alt5single Count | alt5single Loc | alt3and5single Count | alt3and5single Loc | OnlyESTexon Count | OnlyEST exons | genename |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| All constitutive exons of gene ARMCX4, no alternative eventsa | |||||||||||||||||||||
| chrX | + | 100 742 594 | 100 742 678 | 44 | 0 | 0 | 0 | 0 | 1.000 | 1.000 | 1.000 | 1.000 | 0 | # | 0 | # | 0 | # | 0 | # | ARMCX4 |
| chrX | + | 100 759 923 | 100 760 342 | 5 | 0 | 0 | 0 | 0 | 1.000 | 1.000 | 1.000 | 1.000 | 0 | # | 0 | # | 0 | # | 0 | # | ARMCX4 |
| chrX | + | 100 764 576 | 100 764 665 | 7 | 0 | 0 | 0 | 0 | 1.000 | 1.000 | 1.000 | 1.000 | 0 | # | 0 | # | 0 | # | 0 | # | ARMCX4 |
| chrX | + | 100 766 011 | 100 766 042 | 7 | 0 | 0 | 0 | 0 | 1.000 | 1.000 | 1.000 | 1.000 | 0 | # | 0 | # | 0 | # | 0 | # | ARMCX4 |
| chrX | + | 100 786 630 | 100 786 999 | 3 | 0 | 0 | 0 | 0 | 1.000 | 1.000 | 1.000 | 1.000 | 0 | # | 0 | # | 0 | # | 0 | # | ARMCX4 |
| All constitutive exons of gene ARMCX4, up to 5% alternative events allowedb | |||||||||||||||||||||
| chrX | + | 100 742 179 | 100 742 259 | 45 | 1 | 0 | 0 | 0 | 0.978 | 1.000 | 0.978 | 1.000 | 1 | 100 742 191 | 0 | # | 0 | # | 1 | chrX+100 742 179– 100 742 255 | ARMCX4 |
| chrX | + | 100 742 594 | 100 742 678 | 44 | 0 | 0 | 0 | 0 | 1.000 | 1.000 | 1.000 | 1.000 | 0 | # | 0 | # | 0 | # | 0 | # | ARMCX4 |
| chrX | + | 100 759 923 | 100 760 342 | 5 | 0 | 0 | 0 | 0 | 1.000 | 1.000 | 1.000 | 1.000 | 0 | # | 0 | # | 0 | # | 0 | # | ARMCX4 |
| chrX | + | 100 764 576 | 100 764 665 | 7 | 0 | 0 | 0 | 0 | 1.000 | 1.000 | 1.000 | 1.000 | 0 | # | 0 | # | 0 | # | 0 | # | ARMCX4 |
| chrX | + | 100 766 011 | 100 766 042 | 7 | 0 | 0 | 0 | 0 | 1.000 | 1.000 | 1.000 | 1.000 | 0 | # | 0 | # | 0 | # | 0 | # | ARMCX4 |
| chrX | + | 100 786 630 | 100 786 999 | 3 | 0 | 0 | 0 | 0 | 1.000 | 1.000 | 1.000 | 1.000 | 0 | # | 0 | # | 0 | # | 0 | # | ARMCX4 |
DISCUSSION
HEXEvent allows users to apply their own definition of alternative/constitutive exons and generates a list of exons matching the input criteria. For each queried exon, HEXEvents reports the genome location, the type(s) of alternative splicing, their location and EST counts supporting each alternative version. Based on these entries, an exon inclusion and splice site usage levels are reported. The HEXEvent database is a valuable tool to customize future bioinformatic analyses of alternative splicing. While HEXEvent is currently based on UCSC Genome Browser and available EST information, we plan to add alternative splicing and exon inclusion data derived from deep sequencing reactions in the very near future. Furthermore, we intend to expand the database to other species.
Comparison with other databases: while the UCSC Genome Browser offers sets of alternatively spliced exons for download (15), those sets do not include inclusion/usage-level information and no set of constitutively spliced exons is made available. Furthermore, all available sets of alternatively spliced exons have a splice event centric view, i.e. they include all exons that show a certain splicing event, but do not list all splicing events for an individual exon. ASPicDB is a database providing information about the splicing pattern of human genes (14). While ASPicDB also reports a list of exons in a certain region or gene, this list cannot be downloaded. In contrast, the lists of exons generated by HEXEvent are downloadable to provide the foundation for subsequent bioinformatic analyses. Thus, HEXEvent is suitable for local single-gene analyses as well as for more complex or genome-wide analyses using downloaded lists. Furthermore, in HEXEvent, users can set their own definition of constitutive and alternative exons by specifying inclusion levels up to which an exon is considered a member of either category. Finally, HEXEvent summarizes alternative splice site versions of the same exons to one entry, thereby specifying all possible alternative splice sites. In comparison to HEXEvent, ASPicDB has one entry per alternative splice site version, which inherently makes the output harder to view and process. While this is a comparison to the most recent alternative splicing database published in NAR, it is worth noting that several other useful databases exist with prior publication date, such as Hollywood (17) or ASD/ASTD (18). Alternative Splicing Database/Alternative Splicing and Transcript Diversity database (ASD/ASTD) was closed in the beginning of 2012. Instead, their features have been integrated in Ensembl (19). These databases are excellent venues to interrogate the splicing patterns of individual genes, especially in light of their excellent accompanying graphics. However, all of the existing databases are limited by (i) not offering the ability to download multiple splicing events at a time and by (ii) not permitting user definitions of alternative splicing. The most useful features of HEXEvent bridge this gap, thus permitting users to custom design genome-wide exon data sets.
AVAILABILITY
This HEXEvent database is freely available at http://hexevent.mmg.uci.edu and open to all users. There is no login requirement.
FUNDING
Funding for open access charge: National Institutes of Health [RO1 GM62287 and R21 CA149548 to K.J.H.]; Postdoc Programme of the German Academic Exchange Service, DAAD (fellowship to A.B.).
Conflict of interest statement. None declared.
REFERENCES
- 1.Hertel KJ. Combinatorial control of exon recognition. J. Biol. Chem. 2008;283:1211–1215. doi: 10.1074/jbc.R700035200. [DOI] [PubMed] [Google Scholar]
- 2.International Human Genome Sequencing Consortium. Finishing the euchromatic sequence of the human genome. Nature. 2004;431:931–945. doi: 10.1038/nature03001. [DOI] [PubMed] [Google Scholar]
- 3.Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, Kingsmore SF, Schroth GP, Burge CB. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008;456:470–476. doi: 10.1038/nature07509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fox-Walsh KL, Hertel KJ. Splice-site pairing is an intrinsically high fidelity process. Proc. Natl Acad. Sci. USA. 2009;106:1766–1771. doi: 10.1073/pnas.0813128106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Krawczak M, Reiss J, Cooper DN. The mutational spectrum of single base-pair substitutions in mRNA splice junctions of human genes: causes and consequences. Hum. Genet. 1992;90:41–54. doi: 10.1007/BF00210743. [DOI] [PubMed] [Google Scholar]
- 6.Cartegni L, Chew SL, Krainer AR. Listening to silence and understanding nonsense: exonic mutations that affect splicing. Nat. Rev. Genet. 2002;3:285–298. doi: 10.1038/nrg775. [DOI] [PubMed] [Google Scholar]
- 7.Faustino NA, Cooper TA. Pre-mRNA splicing and human disease. Genes Dev. 2003;17:419–437. doi: 10.1101/gad.1048803. [DOI] [PubMed] [Google Scholar]
- 8.Carstens RP, Eaton JV, Krigman HR, Walther PJ, Garcia-Blanco MA. Alternative splicing of fibroblast growth factor receptor 2 (FGF-R2) in human prostate cancer. Oncogene. 1997;15:3059–3065. doi: 10.1038/sj.onc.1201498. [DOI] [PubMed] [Google Scholar]
- 9.Mercatante D, Kole R. Modification of alternative splicing pathways as a potential approach to chemotherapy. Pharmacol. Ther. 2000;85:237–243. doi: 10.1016/s0163-7258(99)00067-4. [DOI] [PubMed] [Google Scholar]
- 10.Xu Q, Lee C. Discovery of novel splice forms and functional analysis of cancer-specific alternative splicing in human expressed sequences. Nucleic Acids Res. 2003;31:5635–5643. doi: 10.1093/nar/gkg786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang Z, Lo HS, Yang H, Gere S, Hu Y, Buetow KH, Lee MP. Computational analysis and experimental validation of tumor-associated alternative RNA splicing in human cancer. Cancer Res. 2003;63:655–657. [PubMed] [Google Scholar]
- 12.Brinkman BMN. Splice variants as cancer biomarkers. Clin. Biochem. 2004;37:584–594. doi: 10.1016/j.clinbiochem.2004.05.015. [DOI] [PubMed] [Google Scholar]
- 13.Shepard PJ, Choi E-A, Busch A, Hertel KJ. Efficient internal exon recognition depends on near equal contributions from the 3′ and 5′ splice sites. Nucleic Acids Res. 2011;39:8928–8937. doi: 10.1093/nar/gkr481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Martelli PL, D’Antonio M, Bonizzoni P, Castrignanò T, D’Erchia AM, D’Onorio De Meo P, Fariselli P, Finelli M, Licciulli F, Mangiulli M, et al. ASPicDB: a database of annotated transcript and protein variants generated by alternative splicing. Nucleic Acids Res. 2011;39:D80–D85. doi: 10.1093/nar/gkq1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dreszer TR, Karolchik D, Zweig AS, Hinrichs AS, Raney BJ, Kuhn RM, Meyer LR, Wong M, Sloan CA, Rosenbloom KR, et al. The UCSC Genome Browser database: extensions and updates 2011. Nucleic Acids Res. 2012;40:D918–D923. doi: 10.1093/nar/gkr1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dou Y, Fox-Walsh KL, Baldi PF, Hertel KJ. Genomic splice-site analysis reveals frequent alternative splicing close to the dominant splice site. RNA. 2006;12:2047–2056. doi: 10.1261/rna.151106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Holste D, Huo G, Tung V, Burge CB. HOLLYWOOD: a comparative relational database of alternative splicing. Nucleic Acids Res. 2006;34:D56–D62. doi: 10.1093/nar/gkj048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Koscielny G, Le Texier V, Gopalakrishnan C, Kumanduri V, Riethoven JJ, Nardone F, Stanley E, Fallsehr C, Hofmann O, Kull M, et al. ASTD: The Alternative Splicing and Transcript Diversity database. Genomics. 2009;93:213–220. doi: 10.1016/j.ygeno.2008.11.003. [DOI] [PubMed] [Google Scholar]
- 19.Flicek P, Amode MR, Barrell D, Beal K, Brent S, Carvalho-Silva D, Clapham P, Coates G, Fairley S, Fitzgerald S, et al. Ensembl 2012. Nucleic Acids Res. 2012;40:D84–D90. doi: 10.1093/nar/gkr991. [DOI] [PMC free article] [PubMed] [Google Scholar]