


Abstract
The PDBTM database (available at http://pdbtm.enzim.hu), the first comprehensive and up-to-date transmembrane protein selection of the Protein Data Bank, was launched in 2004. The database was created and has been continuously updated by the TMDET algorithm that is able to distinguish between transmembrane and non-transmembrane proteins using their 3D atomic coordinates only. The TMDET algorithm can locate the spatial positions of transmembrane proteins in lipid bilayer as well. During the last 8 years not only the size of the PDBTM database has been steadily growing from ∼400 to 1700 entries but also new structural elements have been identified, in addition to the well-known α-helical bundle and β-barrel structures. Numerous ‘exotic’ transmembrane protein structures have been solved since the first release, which has made it necessary to define these new structural elements, such as membrane loops or interfacial helices in the database. This article reports the new features of the PDBTM database that have been added since its first release, and our current efforts to keep the database up-to-date and easy to use so that it may continue to serve as a fundamental resource for the scientific community.
INTRODUCTION
Transmembrane proteins play an important role in the living cells for energy production, regulation and metabolism. The fact that half of present-day drugs have some effect on transmembrane proteins (1,2) also underlines their biological importance. Furthermore, ∼25% of the human genome might code transmembrane proteins (3), which means about 5–6000 structures. Due to the structural and physiochemical properties of these proteins, the experimental techniques for structure determination are not so straightforward. As a consequence, the proportion of transmembrane and globular proteins in the Protein Data Bank (PDB) (4) database is <2% according to the PDBTM database (5,6). Hence, the PDBTM database was created in 2004 to collect these cases. The PDBTM database was the first to address the problems of transmembrane protein structures in the PDB database, namely the fact that these proteins cannot be identified using the annotation in the PDB’s entries. Therefore, a new method was needed, which is based on only the 3D coordinates to identify transmembrane segments and does not require additional information. Moreover, since one of the most important environments, the double lipid layer, is not part of the solved atomic structures due to the experimental difficulties of structure determination, theoretical methods are required to determine the orientations of the transmembrane proteins relative to the lipid bilayer. We developed a method, called TMDET (7), which addresses and solves the above-mentioned problems. Since then several transmembrane databases have become available on the Internet, utilizing different theoretical algorithms and techniques, and serving different purposes. For the sake of comparability, let us briefly summarize the main properties of such databases.
The OPM (8) contains a well-structured classification of membrane proteins. The orientation of the protein relative to the membrane normal is defined by minimizing its transfer energy (ΔGtransfer) from water to the lipid bilayer with respect to the shift along the bilayer normal, hydrophobic thickness, rotation angle and tilt angle (9). Some missing side-chain atoms are added and the structure of residues at the water–lipid interface is adjusted. The results of these calculations are used to transform the atomic coordinates of integral membrane proteins in a way that the membrane normal be parallel with the z-axis. In the OPM database, the transformed coordinate files contain membrane planes too, which are represented by dummy oxygen and nitrogen atoms. The topology data about transmembrane proteins are also given in the OPM database, i.e. what part of the proteins face to the cytosolic space and what part to the extra-cytosolic one.
The CGDB (10) database contains the final system coordinates of coarse-grained simulation-relaxed transmembrane protein structures in bilayer and their analysis from the aspect of protein–lipid interaction. This database has the most sophisticated model in terms of physics, as it utilizes a previously developed high-throughput computational approach to perform the coarse-grained simulations. There are two other analogous databases which are more specific: the KDB is for K-channels (http://sbcb.bioch.ox.ac.uk/kdb/) and the OMPDB is a set of outer membrane proteins obtained by full-atom simulations (11). These databases contain indispensable information on dynamic aspects and stability.
One of the most reliable database of membrane proteins is the membrane proteins of known structure (Mpstruct, http://blanco.biomol.uci.edu/membrane_proteins_xtal.html), which is regularly updated. In this, membrane proteins are classified using a simpler classification scheme than the one used by the OPM. Although the OPM and the PDBTM contain information about the membrane orientation of proteins and about the classification of sequence segments, the Mpstruct does not.
There are several other databases collecting transmembrane proteins and some of their properties (12–16): (i) the MPDB (12) is a relational database of structural and functional information on integral, anchored and peripheral membrane proteins and peptides derived from the literature and from the PDB database. It provides various search parameters (protein characteristics, structure determination methods, crystallization techniques, detergents, temperature, ‘pH’, authors, etc.) and records are linked to the PDB, the Pfam (13) or the PubMed. It is a weekly updated database following the PDB weekly updates. In addition, the MPDB provides different statistics about the sources and the detergents used in crystallization, as well as about applied expression systems, among other data. (ii) The TMFunction (14) is a collection of >2900 experimentally observed functional residues in membrane proteins. Each entry in the TMFunction database includes the numerical values for the parameters IC50, V(max), relative activity of mutants with respect to wild-type protein, binding affinity and dissociation constant. (iii) The Transporter Classification Database (15) is a web accessible, curated, relational database containing sequence, classification, structural, functional and evolutionary information about transport systems from a variety of living organisms.
In the PDBTM database, we collect all transmembrane proteins for which structures have been solved so far; we check and if necessary correct their biologically active oligomer form given in PDB files, define their membrane orientation and set their transmembrane segments, membrane re-entrant loops and interfacial helices (IFHs).
NEW FEATURES OF THE PDBTM DATABASE
Although the main architecture of the TMDET algorithm has not been changed, several extensions have been added to the basic algorithm to enhance the usability and reliability of our database. The need for the new features is the consequence of the development this scientific field has experienced. We have enhanced the database to include those structural elements, which were not known or were rarely represented when the database was created. These are IFHs and re-entrant regions (loop, hairpin and re-entrant coil) (17). These and some other new features will be discussed in the following sections.
Correcting biomatrices
The biological form of the protein usually does not correspond to the molecule, which is present in the asymmetric unit. Therefore, the symmetry operations, which need to be applied to generate the active oligomer form, are displayed in the PDB file in the BIOMOLECULE section as a matrix transformation, called biomatrix. The oligomer form usually is defined by the authors or is calculated by theoretical calculations using PQS (18) or PISA (19). Both of these algorithms have been developed to determine the quaternary structure of globular proteins, therefore they may fail when applied to transmembrane proteins. We have found several files, where the crystals contain the biologically active oligomer form, but the BIOMOLECULE records are set improperly (e.g. 2atk, 2jk5, 2zld) and those, where the crystals contain oligomer forms that do not exist in the membrane. These latter cases cannot be recognized by the above-mentioned methods. Most frequently they are subunits with anti-parallel orientation in a homo-dimer transmembrane protein, which were discussed in our original article (5). The usage of inappropriate biomatrices occasionally leads to the inaccurate definitions of the orientation of membrane proteins relative to the membrane. In some cases, it could be a ∼20° or a larger difference between monomer and oligomer forms.
We aimed to identify and correct problems, which can be associated with biomatrices and leads to incorrect oligomers. Therefore, we developed a new algorithm, which uses homologous protein structures to generate biomatrices for proteins with inappropriate biomatrix in the PDB. The outline of the protocol is as follows. Protein structures having only one chain without any biomatrix annotation (or only the identity matrix is given in the biomatrix records) are selected in one pool, whereas those which have only one chain and a biomatrix were stored in an other pool. Then a BLAST search is performed against the sequences of the second pool for each sequence of the first one. The protein with the highest hit is used as a candidate and if the sequential similarity is >90%, then the query structure will be superimposed on the candidate using TM-align (20) algorithm. TM-align gives the transformation (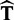 ), which turns
), which turns 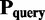 to
to  formally:
formally:
| (1) |
Assuming that there are 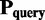 and
and  identical monomer structures with different absolute coordinates and the corresponding biomatrices are
identical monomer structures with different absolute coordinates and the corresponding biomatrices are 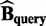 and
and 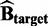 , then we get:
, then we get:
| (2) |
Replacing 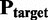 with
with 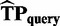 on the bases of Equation (1), in Equation (2), we obtain:
on the bases of Equation (1), in Equation (2), we obtain:
| (3) |
Hence
| (4) |
| (5) |
We have checked the accuracy of this procedure by applying it on those entries, which are homo-oligomer molecules and have correct BIOMOLECULE record. The PDBTM database contains 318 such entries. After sequence filtering to 90% identity, we got 57 entries. We could generate biomatrices for 43 entries using homologous protein structures. After calculating the coordinates using these newly generated biomatrices, we calculate the root mean square deviation (RMSD) between the original and computed coordinates. The RMSD values of 40 out of the 43 entries were <1 Å (avg: 0.38 ± 0.20 Å), while the worst alignment produced a 3.3 Å RMSD.
In cases, when the crystal contains the correct oligomer form, but this is not given in the BIOMOLECULE record, we supply the correct crystallographic symmetry transformation. Altogether, the biomatrices of 34 entries have been corrected. The largest tilt angle difference between the corrected and uncorrected original forms was found in the case of 2w0f, a potassium-channel KcsA–Fab complex with tetraoctylammonium. In the PDB file, it appears as a monomer (after applying the given biomatrix transformation), but its active form is tetramer. The angle deviation was 23° and the region borders moved up to four residues. We have found similar angle deviation in the OPM database as well. The largest tilt angle deviation, 19° in the OPM database, can be found between 1py6 and 1m0l. 1py6 is a monomeric protein in the PDB, while 1m0l is a homo-trimer of the same bacteriorhodopsin.
Membrane re-entrant loops
Membrane re-entrant loops with both ends facing the same side of the membrane were first detected in the late 90 s (21) in the case of the cardiac Na+/Ca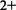 exchanger. Later it was shown that several other channel-like transmembrane proteins contain this type of structural element, e.g. aquaporins (22), potassium channels (23), chloride channels (24), etc. (Figure 1). We have developed a new algorithm as an extension of the TMDET to detect these structural elements using only the 3D atomic coordinates of given transmembrane proteins and the transformation matrices produced by the TMDET algorithm, by searching sequence segments having both end on the same side of the membrane, and diving into the membrane with at least 6 Å (measured from the membrane–water interface). This algorithm can detect any type of re-entrant loops (e.g. helix–loop–coil, coil–loop–helix, coil–loop–coil), but the database currently does not contain these pieces of information. Currently, there are 258 proteins in the PDBTM database, which contain one or more re-entrant loops.
exchanger. Later it was shown that several other channel-like transmembrane proteins contain this type of structural element, e.g. aquaporins (22), potassium channels (23), chloride channels (24), etc. (Figure 1). We have developed a new algorithm as an extension of the TMDET to detect these structural elements using only the 3D atomic coordinates of given transmembrane proteins and the transformation matrices produced by the TMDET algorithm, by searching sequence segments having both end on the same side of the membrane, and diving into the membrane with at least 6 Å (measured from the membrane–water interface). This algorithm can detect any type of re-entrant loops (e.g. helix–loop–coil, coil–loop–helix, coil–loop–coil), but the database currently does not contain these pieces of information. Currently, there are 258 proteins in the PDBTM database, which contain one or more re-entrant loops.
Figure 1.

Loops (coloured in orange) in 1h6i, a refined structure of human aquaporin (22).
Interfacial helices
Another newly implemented structural class is IFHs that are α-helices laying in the membrane–water interface parallel to the membrane plane (Figure 2). They have various structural roles, for example, they are responsible for the regulation of channel gating in both the KirBac 1.1 inward rectifying potassium channel (25) and the MscS mechanosensitive channel (26), while in photosystem I, IFHs appear to shield cofactors from the aqueous phase (27).
Figure 2.

IFH (coloured in green) in 1e7p, a quinol-fumarate reductase from Wolinella succinogenes (28).
A further extension of the TMDET algorithm contains a subroutine which identifies these regions. First, we collect α-helical regions not in the membrane, and longer than four residues, and calculate the tilt angle relative to the membrane plane and the distance from the membrane–water boundary. The algorithm uses two threshold parameters: the distance (<9 Å) from the membrane–water boundary and the tilt angle (<30°). As a result of this extension, we have identified IFHs in 851 proteins.
THE NEW USER INTERFACE OF THE PDBTM
The homepage of the upgraded version of the PDBTM database utilizes the Wt C++ Web Toolkit (http://www.webtoolkit.eu/wt) programming library and the OpenAstexViewer (29) to visualize transmembrane protein structures highlighted with different colours for the different region types to make the structure even more informative. We have recently created a complex web application for investigating protein 3D structures and residue–residue interactions (30), where both the Wt and the OpenAstexViewer have been successfully utilized.
The PDBTM entry viewer
The layout of the PDBTM molecule viewer can be seen in Figure 3. The navigation bar (Figure 3A) contains an up-to-date list of IDs of current transmembrane protein structures in the PDBTM database. The arrows serve for the navigation in this list. The previous structure viewer has been replaced with the OpenAstexViewer (29). The colouring of the 3D structure (Figure 3B) and sequence (Figure 3C) is identical in order to help users to find sequence segments more easily in the 3D structure. These two widgets are connected through signals, so by clicking on any sequence regions (except the grey-coloured ones, which represent sequence without solved structure), the representation of the corresponding residues in the structure viewer turns from cartoon to sphere.
Figure 3.

The PDBTM entry viewer. (A) The navigation bar which is always visible for the sake of comfortable and instant navigation. Using the arrows one can navigate to the first entry, step back, step forward or jump to the end. (B) The structure viewer (29), using the same colours as in the sequence box. (C) Sequence box, containing the chain selector and the sequence of the actual protein chain. (D) File download section, where the user can download or simply view the original and the transformed PDB files as well as PDBTM XML files. (E) Cross-reference links to the RCSB PDB and PDBsum (31) databases.
Users can download or simply view the original and the transformed PDB files as well as the PDBTM XML files (Figure 3D), which describe the regions of the structure, chain sequences and all the necessary information to build up the transformed PDB structure from the original one.
Advanced search system
The web server allows users to perform various types of search in the database. Some ordinary, frequently used search requests have already been implemented, but users can also query custom requests, either in a form field or by using the address line of the browser. This latest feature enables the users to refer to their query results as a constantly updated list by bookmarking the given query. The search results can be browsed or downloaded as a whole in various file formats. For more detailed description visit the manual of the PDBTM (http://pdbtm.enzim.hu/?_=/help/manual).
CONCLUSION
The PDBTM database is a comprehensive, up-to-date and continuously updated transmembrane protein database. As of today, it contains >1700 entries whose regions are classified into structural elements such as transmembrane helices, transmembrane beta segments, membrane re-entrant loops or IFHs. The flexible search method makes data mining easier for bioinformaticians who are interested in transmembrane proteins and their structures. All kinds of feedback and advice are most welcome, as they will help us to improve and to satisfy the diverse demands of users more fully.
FUNDING
Hungarian Scientific Research Fund (OTKA) [NK100482 and K104586]; ‘Lendület’ Program of the Hungarian Academy of Sciences (to G.E.T.). Funding for open access charge: ‘Lendület’ Program of the Hungarian Academy of Sciences.
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
Comments on the article by Mónika Fuxreiter and László Benke and on the manual of the PDBTM database by Bálint Mészáros are gratefully acknowledged. We would like to express our gratitude for the help of Koen Deforche and István Reményi in the development of PDBTM.
REFERENCES
- 1.Overington JP, Al-Lazikani B, Hopkins AL. How many drug targets are there? Nat. Rev. Drug Discov. 2006;5:993–996. doi: 10.1038/nrd2199. [DOI] [PubMed] [Google Scholar]
- 2.Parrill AL. Crystal structures of a second G protein-coupled receptor: triumphs and implications. ChemMedChem. 2008;3:1021–1023. doi: 10.1002/cmdc.200800070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fagerberg L, Jonasson K, von Heijne G, Uhlén M, Berglund L. Prediction of the human membrane proteome. Proteomics. 2010;10:1141–1149. doi: 10.1002/pmic.200900258. [DOI] [PubMed] [Google Scholar]
- 4.Rose PW, Beran B, Bi C, Bluhm WF, Dimitropoulos D, Goodsell DS, Prlic A, Quesada M, Quinn GB, Westbrook JD, et al. The RCSB Protein Data Bank: redesigned web site and web services. Nucleic Acids Res. 2011;39:D392–D401. doi: 10.1093/nar/gkq1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tusnády GE, Dosztányi Z, Simon I. Transmembrane proteins in the Protein Data Bank: identification and classification. Bioinformatics. 2004;20:2964–2972. doi: 10.1093/bioinformatics/bth340. [DOI] [PubMed] [Google Scholar]
- 6.Tusnády GE, Dosztányi Z, Simon I. PDB_TM: selection and membrane localization of transmembrane proteins in the protein data bank. Nucleic Acids Res. 2005;33:D275–D278. doi: 10.1093/nar/gki002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tusnády GE, Dosztányi Z, Simon I. TMDET: web server for detecting transmembrane regions of proteins by using their 3D coordinates. Bioinformatics. 2005;21:1276–1277. doi: 10.1093/bioinformatics/bti121. [DOI] [PubMed] [Google Scholar]
- 8.Lomize MA, Pogozheva ID, Joo H, Mosberg HI, Lomize AL. OPM database and PPM web server: resources for positioning of proteins in membranes. Nucleic Acids Res. 2012;40:D370–D376. doi: 10.1093/nar/gkr703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lomize AL, Pogozheva ID, Lomize MA, Mosberg HI. Positioning of proteins in membranes: a computational approach. Protein Sci. 2006;15:1318–1333. doi: 10.1110/ps.062126106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chetwynd AP, Scott KA, Mokrab Y, Sansom MSP. CGDB: a database of membrane protein/lipid interactions by coarse-grained molecular dynamics simulations. Mol. Membr. Biol. 2008;25:662–669. doi: 10.1080/09687680802446534. [DOI] [PubMed] [Google Scholar]
- 11.Tsirigos KD, Bagos PG, Hamodrakas SJ. OMPdb: a database of beta-barrel outer membrane proteins from Gram-negative bacteria. Nucleic Acids Res. 2011;39:D324–D331. doi: 10.1093/nar/gkq863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Raman P, Cherezov V, Caffrey M. The Membrane Protein Data Bank. Cell. Mol. Life Sci. 2006;63:36–51. doi: 10.1007/s00018-005-5350-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Punta M, Coggill PC, Eberhardt RY, Mistry J, Tate J, Boursnell C, Pang N, Forslund K, Ceric G, Clements J, et al. The Pfam protein families database. Nucleic Acids Res. 2012;40:D290–D301. doi: 10.1093/nar/gkr1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gromiha MM, Yabuki Y, Suresh MX, Thangakani AM, Suwa M, Fukui K. TMFunction: database for functional residues in membrane proteins. Nucleic Acids Res. 2009;37:D201–D204. doi: 10.1093/nar/gkn672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Saier MH, Ming RY, Keith N, Dorjee GT, Charles E. The Transporter Classification Database: recent advances. Nucleic Acids Res. 2009;37:D274–D278. doi: 10.1093/nar/gkn862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gromiha MM, Yabuki Y, Kundu S, Suharnan S, Suwa M. TMBETA-GENOME: database for annotated beta-barrel membrane proteins in genomic sequences. Nucleic Acids Res. 2007;35:D314–D316. doi: 10.1093/nar/gkl805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nugent T, Jones DT. Membrane protein structural bioinformatics. J. Struct. Biol. 2011;179:327–337. doi: 10.1016/j.jsb.2011.10.008. [DOI] [PubMed] [Google Scholar]
- 18.Henrick K. PQS: a protein quaternary structure file server. Trends Biochem. Sci. 1998;23:358–361. doi: 10.1016/s0968-0004(98)01253-5. [DOI] [PubMed] [Google Scholar]
- 19.Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 20.Zhang Y, Skolnick J. TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 2005;33:2302–2309. doi: 10.1093/nar/gki524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Iwamoto T, Nakamura TY, Pan Y, Uehara A, Imanaga I, Shigekawa M. Unique topology of the internal repeats in the cardiac Na+/Ca2+ exchanger. FEBS Lett. 1999;446:264–268. doi: 10.1016/s0014-5793(99)00218-5. [DOI] [PubMed] [Google Scholar]
- 22.de Groot BL, Engel A, Grubmüller H. A refined structure of human aquaporin-1. FEBS Lett. 2001;504:206–211. doi: 10.1016/s0014-5793(01)02743-0. [DOI] [PubMed] [Google Scholar]
- 23.Zhou Y, Morais-Cabral JH, Kaufman A, MacKinnon R. Chemistry of ion coordination and hydration revealed by a K+ channel–Fab complex at 2.0 Å resolution. Nature. 2001;414:43–48. doi: 10.1038/35102009. [DOI] [PubMed] [Google Scholar]
- 24.Dutzler R, Campbell EB, Cadene M, Chait BT, MacKinnon R. X-ray structure of a ClC chloride channel at 3.0 Å reveals the molecular basis of anion selectivity. Nature. 2002;415:287–294. doi: 10.1038/415287a. [DOI] [PubMed] [Google Scholar]
- 25.Doyle DA. Structural themes in ion channels. Eur. Biophys. J. 2004;33:175–179. doi: 10.1007/s00249-003-0382-z. [DOI] [PubMed] [Google Scholar]
- 26.Bass RB, Locher KP, Borths E, Poon Y, Strop P, Lee A, Rees DC. The structures of BtuCD and MscS and their implications for transporter and channel function. FEBS Lett. 2003;555:111–115. doi: 10.1016/s0014-5793(03)01126-8. [DOI] [PubMed] [Google Scholar]
- 27.Jordan P, Fromme P, Witt HT, Klukas O, Saenger W, Krauss N. Three-dimensional structure of cyanobacterial photosystem I at 2.5 Å resolution. Nature. 2001;411:909–917. doi: 10.1038/35082000. [DOI] [PubMed] [Google Scholar]
- 28.Lancaster CR, Gross R, Simon J. A third crystal form of Wolinella succinogenes quinol:fumarate reductase reveals domain closure at the site of fumarate reduction. Eur. J. Biochem. 2001;268:1820–1827. [PubMed] [Google Scholar]
- 29.Hartshorn MJ. AstexViewer: a visualisation aid for structure-based drug design. J. Comput. Aided Mol. Des. 2002;16:871–881. doi: 10.1023/a:1023813504011. [DOI] [PubMed] [Google Scholar]
- 30.Kozma D, Simon I, Tusnády GE. CMWeb: an interactive on-line tool for analysing residue–residue contacts and contact prediction methods. Nucleic Acids Res. 2012;40:W329–W333. doi: 10.1093/nar/gks488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Laskowski RA. PDBsum new things. Nucleic Acids Res. 2009;37:D355–D359. doi: 10.1093/nar/gkn860. [DOI] [PMC free article] [PubMed] [Google Scholar]