


Abstract
PROSITE (http://prosite.expasy.org/) consists of documentation entries describing protein domains, families and functional sites, as well as associated patterns and profiles to identify them. It is complemented by ProRule a collection of rules, which increases the discriminatory power of these profiles and patterns by providing additional information about functionally and/or structurally critical amino acids. PROSITE signatures, together with ProRule, are used for the annotation of domains and features of UniProtKB/Swiss-Prot entries. Here, we describe recent developments that allow users to perform whole-proteome annotation as well as a number of filtering options that can be combined to perform powerful targeted searches for biological discovery. The latest version of PROSITE (release 20.85, of 30 August 2012) contains 1308 patterns, 1039 profiles and 1041 ProRules.
INTRODUCTION
PROSITE is a resource for the identification and annotation of conserved regions in protein sequences. These regions are identified using two types of signatures: generalized profiles (weight matrices) that describe protein families and modular protein domains and patterns (regular expressions) that describe short sequence motifs often corresponding to functionally or structurally important residues (1). PROSITE signatures are linked to annotation rules, or ProRules, which define protein sequence annotations (such as active site and ligand-binding residues) and the conditions under which they apply (for example requiring specific amino acid residues) (2). ProRule is used for the annotation of protein families, domains and sequence features in UniProtKB/Swiss-Prot, the manually curated section of the UniProt KnowledgeBase (3), and currently provides annotation for >75% of the 1054 domains to be found there (release 2012_08, 5 September 2012). Part of the information stored in ProRule (e.g. active and binding sites, disulfide bonds) is also accessible to the ScanProsite user. PROSITE provides extensive documentation for each signature including information on nomenclature, function, sequence features, pointers to 3D structure(s), protein architectures in which the signature is found, its taxonomic distribution and important literature references (1). PROSITE signatures, ProRules and PROSITE documentation can be accessed from our website at http://prosite.expasy.org/ (4). PROSITE signatures are also made available through InterPro (http://www.ebi.ac.uk/interpro/index.html), an integrated database of protein signatures used for the classification and annotation of proteins and genomes (5). Through InterPro users can combine PROSITE classifications with those provided by other InterPro consortium members. Since our last report in the NAR database issue (6), PROSITE has increased the number of available signatures to 1308 patterns and 1039 profiles, which are associated with 1041 ProRules and 1650 documentation entries.
NEW DEVELOPMENTS: SCANPROSITE
The ScanProsite tool (http://prosite.expasy.org/scanprosite/) allows users to search protein sequences against all PROSITE signatures, and to search for matches to defined PROSITE signatures in the UniProtKB and PDB databases (4). To enhance the utility and flexibility of PROSITE searches, we have modified the ScanProsite tool in a number of ways.
Proteome annotation by ScanProsite
The original implementation of ScanProsite allowed users to search only a limited number of sequences against the entire library of PROSITE signatures. We have since relaxed this restriction and now offer users the possibility to upload complete proteome sets in FASTA format to the PROSITE server (subject to a size limitation of 16 Mb, which is sufficient for the majority of proteomes). A unique identifier is assigned to each uploaded set of protein sequences and is returned to the user as a reference for use in subsequent searches. The identifier remains valid for 1 month, allowing users to perform multiple analyses on the same set of sequences, if desired. These analyses are performed on the high-performance cluster of the Vital-IT facility (http://www.vital-it.ch/). It is possible to perform combinatorial scans (see below), and users can perform searches against their own defined sequence patterns.
To demonstrate this application, we annotated the complete proteome sequence of the fire ant Solenopsis invicta at the ScanProsite server (7). The Official Gene Set of S. invicta is predicted to encode 16 569 canonical protein sequences. These were uploaded to the ScanProsite server in FASTA format and run against all PROSITE motifs, including both patterns and profiles. The entire process took <30 min. A total of 14 562 hits to 1248 distinct PROSITE signatures were found in 5496 protein sequences, giving total coverage at the protein level of ∼33% for this organism (Table 1). Users wishing to obtain higher coverage may of course combine the classification and annotation from PROSITE with that provided by other annotation tools and pipelines.
Table 1.
Results of the ScanProsite search of the 16 569 predicted Solenopsis invicta proteins against the complete set of PROSITE patterns and profiles
| Patterns a | Profiles | |
|---|---|---|
| Total number of PROSITE signature matches in all proteins | 4903 | 9664 |
| Number of distinct proteins matching PROSITE signatures | 2696 | 4349 |
| Number of distinct PROSITE signatures matched | 626 | 622 |
| Number of proteins annotated with one or more functional sites | 520 | 1693 |
| Total number of functional sites annotated | 744 | 7022 |
| Number of distinct PROSITE signatures providing annotation for functional sites | 74 | 148 |
| Total number of detected domains annotated with functional sites | 606 | 3397 |
aPattern hits are validated by automatically generated ‘miniprofiles’ that assign a status to pattern matches (8).
Combinatorial search
In parallel to this work, we have developed and implemented a number of search options that enhance the power and flexibility of ScanProsite. The first of these allows users to search for specific combinations of signatures. This feature may be useful in fine-grained functional inference, allowing users to search a given set of sequences for instances of domains (profiles) that are associated with particular functional residues (patterns) or to search for specific combinations of domains that may confer particular functions (9,10). PROSITE descriptors are combined using the logical operators ‘and’, ‘or’ and ‘not’, with parentheses used to define the priority in which the operators are applied (Figure 1). Users can also define their own sequence patterns and combine them with existing PROSITE signatures. This may allow the further discrimination of particular domain variants or subfamilies that are not yet covered by existing PROSITE signatures (2).
Figure 1.
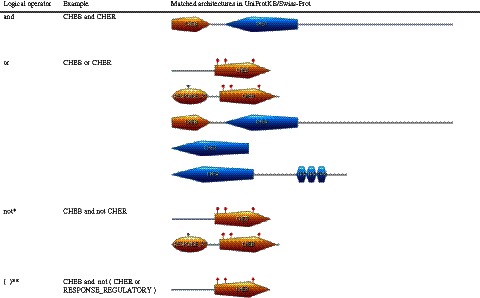
The use of logical operators in ScanProsite. The PROSITE profiles used are PS50122 (CHEB), PS50123 (CHER) and PS50110 (RESPONSE_REGULATORY). The matched architectures correspond to the following UniProtKB/Swiss-Prot entries: Q02998 (YH19_RHOCA), A1SMR4 (CHEB_NOCSJ), P31758 (FRZG_MYXXA), P31759 (FRZF_MYXXA) and A1VZQ6 (CHER_CAMJJ). Single-asterisk symbol denotes that ‘not’ has to be used with another operator (‘and’ or ‘or’). Double-asterisk symbol denotes that parentheses have to be preceded and followed by a space.
Targeted search with filters
The results of PROSITE searches on UniProtKB can be further restricted using a variety of filtering options. Users can limit the results to only those proteins that derived from one or more taxa, according to the taxonomic classification of UniProtKB (http://www.uniprot.org/taxonomy/), at any desired level in the taxonomy. Taxonomic information is found in the ‘OC’ and ‘OS’ line(s) of the UniProtKB flat file. Users can also limit their results to only those proteins having a particular name (be it the recommended name or alternative name), which can be a general class of protein such as ‘protease.’ Such nomenclature information is found in the ‘DE’ line(s) of the UniProtKB flat file. Users can also limit their results to only those proteins that are expressed in one of 56 adult tissues, using data from the Bgee resource (http://bgee.unil.ch/bgee/bgee), a database of gene expression and evolution (11). This particular filter is applicable to proteins of Homo sapiens, Mus musculus, Xenopus laevis and Danio rerio. Finally, users can also limit their results to only those proteins having a certain size or within a certain size range. Together, these filters allow users to combine prior biological knowledge with specific sequence features (or combinations of them) in order to perform very powerful targeted searches.
We illustrate a typical application of these search options using the alkylglycerol mono-oxygenase of M. musculus as an example (12). Prior to the identification of the sequence encoding this enzyme, a limited amount of information was available regarding its biological and biochemical characteristics. We used this information to identify a number of possible candidate sequences for experimental validation. It was known that this enzyme, along with nitric oxide synthase and aromatic amino acid hydroxylase, required tetrahydrobiopterin and iron to be active. The enzyme was also known to have similar iron-binding characteristics to aromatic amino acid hydroxylase, suggesting a role for histidine residues in this process (13). The protein was known to be present in brain and liver, and its size was estimated at between 400 and 650 amino acids (14). We used this information to perform a restricted search of the murine proteome using a degenerate pattern corresponding to the two iron-coordinating histidines of aromatic amino acid hydroxylase (H–X(3,5)–H). This reduced the list of UniProtKB protein entries matching this motif from over 1000 to only 31, corresponding to 22 genes. Following manual inspection of these sequences, we excluded a number of previously characterized proteins that were unlikely to be responsible for the specified activity, including transcription factors, transporters and enzymes. The remaining set of 16 proteins constituted a reasonable number of candidate sequences for experimental investigation. One of these was found to possess alkylglycerol mono-oxygenase activity, and this is described in UniProtKB/Swiss-Prot entry Q8BS35.
CONCLUSION
PROSITE provides a resource for the identification and annotation of conserved regions in protein sequences, covering protein families, domains and motifs. We will continue to develop new PROSITE profiles and ProRules as new proteins, domains and functions are characterized. We describe here improvements to ScanProsite that permit PROSITE to be applied by users for whole-proteome annotation, as well as a number of options that allow very fine-grained searches including prior biological knowledge. Our current software developments are addressed at further enhancing the speed of ScanProsite for improved proteome annotation. To achieve this, the original code of pfsearch is being rewritten and optimized to efficiently use modern multi-core processors and an heuristic implemented for further speed enhancements. This work will be described in a forthcoming publication (L. Cerutti and T. Schuepbach, personal communication).
FUNDING
An FNS project [315230-116864]. PROSITE activities are also supported by the Swiss Federal Government through the Federal Office of Education and Science. Funding for open access charge: Swiss Federal Office of Education and Science.
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
The authors thank Frédéric Bastian for helpful discussions regarding gene expression library databases and for the selection of multi-species tissues that are used in ScanProsite.
REFERENCES
- 1.Sigrist CJA, Cerutti L, Hulo N, Gattiker A, Falquet L, Pagni M, Bairoch A, Bucher P. PROSITE: a documented database using patterns and profiles as motif descriptors. Brief. Bioinform. 2002;3:265–274. doi: 10.1093/bib/3.3.265. [DOI] [PubMed] [Google Scholar]
- 2.Sigrist CJA, De Castro E, Langendijk-Genevaux PS, Le Saux V, Bairoch A, Hulo N. ProRule: a new database containing functional and structural information on PROSITE profiles. Bioinformatics. 2005;21:4060–4066. doi: 10.1093/bioinformatics/bti614. [DOI] [PubMed] [Google Scholar]
- 3.The UniProt Consortium. Reorganizing the protein space at the Universal Protein Resource (UniProt) Nucleic Acids Res. 2012;40:D71–D75. doi: 10.1093/nar/gkr981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.de Castro E, Sigrist CJA, Gattiker A, Bulliard V, Langendijk-Genevaux PS, Gasteiger E, Bairoch A, Hulo N. ScanProsite: detection of PROSITE signature matches and ProRule-associated functional and structural residues in proteins. Nucleic Acids Res. 2006;34:W362–W365. doi: 10.1093/nar/gkl124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hunter S, Jones P, Mitchell A, Apweiler R, Attwood TK, Bateman A, Bernard T, Binns D, Bork P, Burge S, et al. InterPro in 2011: new developments in the family and domain prediction database. Nucleic Acids Res. 2012;40:D306–D312. doi: 10.1093/nar/gkr948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sigrist CJA, Cerutti L, de Castro E, Langendijk-Genevaux PS, Bulliard V, Bairoch A, Hulo N. PROSITE, a protein domain database for functional characterization and annotation. Nucleic Acids Res. 2010;38:D161–D166. doi: 10.1093/nar/gkp885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wurm Y, Wang J, Riba-Grognuz O, Corona M, Nygaard S, Hunt BG, Ingram KK, Falquet L, Nipitwattanaphon M, Gotzek D, et al. The genome of the fire ant Solenopsis invicta . Proc. Natl Acad. Sci. USA. 2011;108:5679–5684. doi: 10.1073/pnas.1009690108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hulo N, Bairoch A, Bulliard V, Cerutti L, Cuche BA, de Castro E, Lachaize C, Langendijk-Genevaux PS, Sigrist CJA. The 20 years of PROSITE. Nucleic Acids Res. 2008;36:D245–D249. doi: 10.1093/nar/gkm977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Levitt M. Nature of the protein universe. Proc. Natl Acad. Sci. USA. 2009;106:11079–11084. doi: 10.1073/pnas.0905029106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Buljan M, Bateman A. The evolution of protein domain families. Biochem. Soc. Trans. 2009;37:751–755. doi: 10.1042/BST0370751. [DOI] [PubMed] [Google Scholar]
- 11.Bastian F, Parmentier G, Roux J, Moretti S, Laudet V, Robinson-Rechavi M. Bgee: integrating and comparing heterogeneous transcriptome data among species. In: Bairoch A, Cohen-Boulakia S, Froidevaux C, editors. Data Integration in the Life Sciences. Vol. 5109. Springer, Berlin: 2008. pp. 124–131. [Google Scholar]
- 12.Watschinger K, Keller MA, Golderer G, Hermann M, Maglione M, Sarg B, Lindner HH, Hermetter A, Werner-Felmayer G, Konrat R, et al. Identification of the gene encoding alkylglycerol monooxygenase defines a third class of tetrahydrobiopterin-dependent enzymes. Proc. Natl Acad. Sci. USA. 2010;107:13672–13677. doi: 10.1073/pnas.1002404107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Watschinger K, Keller MA, Hermetter A, Golderer G, Werner-Felmayer G, Werner ER. Glyceryl ether monooxygenase resembles aromatic amino acid hydroxylases in metal ion and tetrahydrobiopterin dependence. Biol. Chem. 2009;390:3–10. doi: 10.1515/BC.2009.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Soodsma JF, Piantadosi C, Snyder F. Partial characterization of the alkylglycerol cleavage enzyme system of rat liver. J. Biol. Chem. 1972;247:3923–3929. [PubMed] [Google Scholar]