


Abstract
The HemaExplorer (http://servers.binf.ku.dk/hemaexplorer) is a curated database of processed mRNA Gene expression profiles (GEPs) that provides an easy display of gene expression in haematopoietic cells. HemaExplorer contains GEPs derived from mouse/human haematopoietic stem and progenitor cells as well as from more differentiated cell types. Moreover, data from distinct subtypes of human acute myeloid leukemia is included in the database allowing researchers to directly compare gene expression of leukemic cells with those of their closest normal counterpart. Normalization and batch correction lead to full integrity of the data in the database. The HemaExplorer has comprehensive visualization interface that can make it useful as a daily tool for biologists and cancer researchers to assess the expression patterns of genes encountered in research or literature. HemaExplorer is relevant for all research within the fields of leukemia, immunology, cell differentiation and the biology of the haematopoietic system.
INTRODUCTION
Haematopoiesis is the developmental process by which all blood cells are formed. The haematopoietic system is organized in a hierarchical manner with the haematopoietic stem cell (HSC) residing at the apex (Figure 1). HSCs have the ability to self-renew for life and to differentiate into intermediate progenitor cells that subsequently generate the mature cells of the blood stream and immune system (3). Thus, HSCs are ultimately responsible for the constant and life-long generation of intermediate progenitors and terminally differentiated cells. Genetic and epigenetic aberrations may lead to a block in normal haematopoietic differentiation, which may result in the development of Acute Myeloid Leukemia (AML), which is an aggressive blood cancer.
Figure 1.

A model of murine haematopoiesis (1). Dotted line represents self-renewal. Dashed line indicates the separation of MkE lineage from the lymphoid and the remaining myoloid populations as shown by Adolfsson et al. (2). Abbreviations are: LT-HSC, long-term haematopoietic stem cell; ST-HSC, short-term haematopoietic stem cell; LMPP, lymphoid-primed multipotential progenitors; CLP, common lymphoid progenitor cells; CMP, common myeloid progenitor cell; MEP, megakaryocyte-erythroid progenitor cell; preGMP, pre-granulocyte monocyte progenitors; GMP, granulocyte monocyte progenitors; MkEP, megakaryocyte erythroid precursors; MkP, megakaryocyte precursor; PreCFU-E, pre-colony-forming unit erythroid cells; CFU-E, colony-forming unit erythroid cells; NK cells, natural killer cells.
Given the general interest in haematopoiesis and its associated malignancies such as AML, a number of databases now addresses questions related to these fields, a majority of which is focused on mRNA expression data. There are multiple tools available online that provides a quick overview of the expression of single genes in a selection of tissues as well as some sorted cells. The requirement for purified sorted cell populations in the study of haematopoiesis makes the curation of the database as well as the availability of a reasonable selection of haematopoietic cells important. Examples of general and non-curated databases with focus on representing gene expression based on mRNA microarrays are Gene expression Atlas (GXA) (4), GeneNote (5) and BioGPS (6).
GXA provides a count table of over and under expression of a query gene in a wide range of cell types and experimental setups, presently counting 3476 experiments of various types. The count numbers are based on differential expression within each microarray dataset in a selection of publicly available microarray data. GeneNote provides a bar chart with a continuous ratio scale axis, displaying expression intensities pre-processed and scaled to a common mean in a global normalization. The database contains a coherent collection of expression profiles in 28 healthy human tissues types. BioGPS provides a summary of single genes including a chart of gene expression based on a single microarray dataset. The variety of cell or tissue types depends on the selected dataset.
Databases dedicated to research in the haematopoietic system and haematopoietic disorders includes Leukemia Gene Atlas (7), BloodExpress (8) and Haematopoietic Fingerprint (9). The Leukemia Gene atlas comprises 20 datasets from Leukemia research experiments; 13 of those sets are gene expression profiles (GEPs). The database provides the possibility of running a number of standardized gene expression analysis methods on a selected dataset, including value sorted bar plots of expression.
BloodExplorer is a database of 271 microarray-based experiments and provides a list of datasets where a query gene is ‘present’ in terms of a significant P-value and presence on the microarray chip.
Haematopoietic Fingerprints database contains data from a mouse dataset with 20 samples, and can provide bar charts of gene expression in haematopoietic stem cells and mature populations of erythrocytes, granulocytes, monocytes, NK cells, activated and naive T cells, and B cells. The database also provides an overview of expression across the chromosomes for each cell type.
The time and expenses necessary for sorting cells drastically reduces the availability of data for investigation of haematopoiesis, as compared to whole tissue samples. This is particularly the case for stem and early progenitor cell populations. None of the present databases integrates and compare GEPs from haematopoietic cells derived from several labs and experiments. Furthermore, there are no publicly available databases with the possibility of visualizing gene expression in a range of sorted cells from both human and murine haematopoietic system, just like a direct comparison with human AML in such a normal context presently will require extensive computational or experimental work form the researcher.
Here, we present the first publically available fully integrated and curated database of GEPs of sorted cells from stem, progenitor and mature populations important for haematopoietic development in both mouse and human. Compared to the present databases, this method provides a far more complete view of expression in the haematopoietic system. The interface allows for easy visualization of the expression of single genes as well as tools to assess correlation of two genes across multiple cell types. Furthermore the database includes bone marrow from four karyotypic distinct AML subtypes and provides two options for investigating malignant haematopoiesis compared to the normal context.
MATERIALS AND METHODS
The database is built from a curated selection of publicly available datasets containing GEPs from sorted cell populations. The database integrates several Affymetrix platforms (HG-U133A, HG-U133B and HG-U133 Plus 2; Mouse Genome 430 2.0). For the human data the overlapping probes across the platforms populate the database. The mouse database is solely based on the Mouse Genome 430 2.0 platform. Data were separated into batches divided by platform and laboratory. The batches were normalized by robust multi-array average (RMA) using the affy package for R (10) and corrected for batch effects using ComBat (11). Combat is an empirical Bayes (EB) method that: First, standardizes the data by a gene-wise least-square. Secondly, estimates EB batch effect parameters by the method of moments using Gaussian and inverse gamma as prior distributions. Lastly, the data are adjusted using the calculated batch effect estimators. This procedure leaves all data in the database directly comparable between samples within the human and murine part of the database, respectively.
From the normalized and batch corrected data a new dataset was build containing fold changes between human AML and the closest normal counterpart, as identified by projecting data to a reduced space defined by principle component analysis and deciding Euclidian distance by k-nearest neighbour method. (Rapin et al., manuscript in preparation). The model parameters are constant for the present set of normal sorted cells, and addition of future AMLs will not influence the existing samples.
In order to handle gene aliases a dictionary of gene aliases was constructed from NCBI {ftp://ftp.ncbi.nlm.nih.gov/gene/DATA/} and The HUGO Gene Nomenclature Committee (HGNC) {www.genenames.org}. Ambiguous gene aliases were not included when constructing the dictionary.
The database structure is a combination of Python pickle and HDF5 files. The main data handling and graphic construction were written in Python NumPy and R called from Python via the rpy2 interface (12). Figure 2 shows a diagram of the database structure and query handling of single gene (ERG) lookup (13).
Figure 2.

Diagram of database construction and output from query. Top-down: The database is build from publicly available data, normalized by RMA and batch corrected by ComBat (11). Right-to-left: Queries are sent to gene alias directory, and relevant gene expression data are retrieved from the repository. Plot is generated from gene expression data together with figure text and list of abbreviations used in the plot and formatted in to a result page.
The database is continuously updated with new data as it becomes available. The present selection has been focused on including as little batch effect as possible, and thus introducing as few platforms and data sources as possible.
RESULTS AND DISCUSSION
The HemaExplorer has an easy-to-use interface with a single query field and the possibility to select between several data sources. At present, the database has available: human normal hematopoietic system, human AML, human normal hematopoietic system + human AML, human AML compared with closest normal counterpart, and mouse haematopoietic system. Cell types and data source are listed in Table 1. The database provides plot of expression for query genes and accepts unambiguous gene aliases. In the case of ambiguous gene aliases a deep link to genecards.org (24) with such a query is constructed on the interface level, to allow the user to identify the relevant gene symbol.
Table 1.
Cell types and sources of data currently in the HemaExplorer database
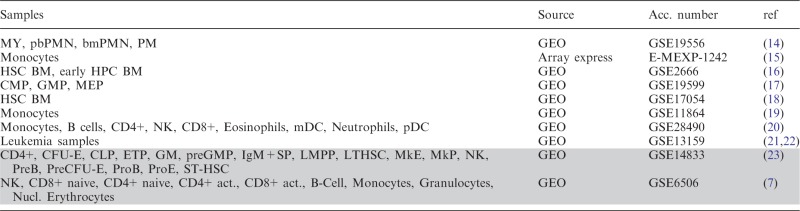 |
Grey-shaded cells contain mouse data and non-shaded cells contain human data.
Abbreviations are: MY, myelocytes; pbPMN, polymorphonuclear cells from peripheral blood; bmPMN, polymorphonuclear cells from bone marrow; PM, promyelocytes; HSC BM, haematopoietic stem cells from bone marrow; early HPC BM, early haematopoietic progenitor cells from bone marrow; CMP, common myeloid progenitor cells; GMP, granulocyte macrophage progenitor cells; MEP, megakaryocyte-erythroid progenitor cells; CD4+, CD4 positive T-cells; CD8+, CD8 positive T-cells; mDC, myeloid dendritic cells; pDC, plasmacytoid dendritic cells; CFU-E, colony-forming unit erythroid cells; CLP, common lymphoid progenitor cells; ETP, early thymic progenitor cells; preGM, pre granulocyte monocyte progenitor cells; GMP, granulocyte monocyte progenitor cells; IgM+SP, IgM positive spleen cells; LMPP, lymphoid-primed multipotential progenitor cells; LT-HSC, long-term haematopoietic stem cells; MkE, megakaryocyte erythroid cells; MkP, megakaryocyte precursor cells; NK, natural killer cells; preB, pre-B cells; PreCFU-E, pre-colony-forming unit erythroid cells; ProB, B-cell progenitor cells; ProE, erythroid progenitor cells; ST-HSC, short-term haematopoietic stem cell.
Currently the interface allows visualization of expression of single genes in haematopoietic cells and any two genes can also be plotted in a pair-wise correlation plot. There is a possibility to include expression in bulk bone marrow of four karyotypes of AML: AML with t(8;21), AML with t(15;17), AML with inv(16)/t(16;16) and AML with t(11q23)/MLL. Furthermore, the interrelationship between these four AML karyotypes can be investigated in a plot displaying their fold change with respect to the closest normal counterpart. In an advanced mode the user can select which of the available haematopoietic cell types to be displayed in the output plot, be it single genes, or two genes for a correlation plot.
Database
The overall integrity of the database is confirmed by dimensionality reduction technique (principle component analysis), as well as correlation analysis of expression profiles by hierarchical clustering, visualized in heatmaps (Supplementary Figure S1 and S2). Both validation techniques show a separation of cell types into well-defined clusters, and reveal no remaining batch effects. The plots are generated using the probe set with the highest mean expression, but normalized expression for all probes annotated to the query gene can be downloaded as a flat file on the result page.
Example of correlation plot between two genes
The possibility of visualizing correlation between two genes makes it possible to identify genes that show similar expression across cell types. Furthermore, this feature enables researchers to visualize characterizing gene pairs with expression patterns that may separate cell populations into distinct clusters and could therefore potentially improve cell sorting strategies.
In Figure 3, an example of a FACS analysis for murine haematopoietic cells is depicted together with expression correlation plots from HemaExplorer. Gene expression in the correlation plots corresponds to the gene that encodes the surface marker used to gate cell populations in the FACS sorting strategy, performed according to Pronk et al. (1). FACS data and gates in Figure 3A and B represent external biological samples from which there are no expression data in the database, and serves illustrative purposes only.
Figure 3.

(A–D) FACS analysis of a subpopulation of murine bone marrow cells depicted together with HemaExplorer gene expression correlation plots. Correlation between gene expression is shown for genes encoding the markers used in the FACS plot shown to the right. (A) FACS gating strategy for separating GMP from CFU-E, preCFU-E and preGM cells. (B) FACS gating strategy for separating CFU-E, preCFU-E and preGM cells. (E) Single gene lookup in the HemaExplorer of the expression of BMI-1 in AML normal sorted cell populations from the myeloid compartment. (F) Single gene lookup in the HemaExplorer of the VEGFA gene. The dataset is human AML compared with the closest healthy myeloid differentiation stage and presents bone marrow samples from AML patients. Abbreviations are: HSC_BM, haematopoietic stem cells from bone marrow; early HPC_BM, haematopoietic progenitor cells from bone marrow; CMP, common myeloid progenitor cell; GMP, granulocyte monocyte progenitors; MEP, megakaryocyte-erythroid progenitor cell; PM_BM, promyelocyte from bone marrow; MY_BM, myelocyte from bone marrow; PMN_BM, polymorphonuclear cells from bone marrow; PMN_PB, polymorphonuclear cells from peripheral blood; AMLI_ETO, AML with t(8;21); APL, AML with t(15;17).
The comparison between a FACS sorting strategy and the expression correlation plots from HemaExplorer shows similar clustering of cell types.
Example of single gene lookup
In Figure 3E, a single gene lookup for BMI-1 is depicted. The BMI-1 gene is known for its role in proliferation, senescence and self-renewal under various conditions (25–27). The expression plot for BMI-1 shows high expression for immature cells, gradually decreasing for cell types found in later stages of the myeloid haematopoietic differentiation pathway. BMI-1 expression in the four AML karyotypes included in the database has levels comparable to the more immature stages, suggesting that the bulk of cells in leukemic bone marrow tends to be less mature and expresses more stem-like phenotype.
Example of comparison between human AML and the closest normal counterpart
In Figure 3F, a single gene lookup is depicted for the query gene VEGFA (vascular endothelial growth factor A). The plot shows the fold change between the AML and the closest normal counterpart. The plot shows clear difference in mRNA expression of VEGFA between the two groups: AML with t(8;21) (AMLI_ETO) and AML with t(15;17) (APL) on one side and AML with inv(16)/t(16;16) and AML with t(11q23)/MLL on the other side. This, and similar distinct separations of AML karyotypes when they are compared to their closest healthy counterpart, can provide researchers with valuable research targets then investigating differences between gene expression in discrete AML subtypes relative to normal cells.
In conclusion, the HemaExplorer is a curated normalized and batch corrected database of GEPs in normal and malignant haematopoiesis in human and mouse. The easy-to-use interface allows for an easy lookup of the expression levels of a gene and for the correlation of expression between pairs of genes. Full integration and comparability of data collected from several sources extents the scope, and possible overview of single mRNA expression in haematopoiesis, compared to the present public available databases. Moreover, the HemaExplorer contains four karyotypes of human AML that can be put in context of the normal haematopoietic system, when visualizing gene expression. Therefore, HemaExplorer will be of use to researchers within the fields of leukemia, immunology, cell differentiation and the biology of the haematopoietic system as a powerful easy-accessible tool for the assessment of gene expression.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Figures 1 and 2.
FUNDING
Danish Research Council for Strategic Research; NovoNordisk Foundation. Funding for open access charge: Rigshospitalet.
Conflict of interest statement. None declared.
REFERENCES
- 1.Pronk CJH, Rossi DJ, Månsson R, Attema JL, Norddahl GL, Chan CKF, Sigvardsson M, Weissman IL, Bryder D. Elucidation of the phenotypic, functional, and molecular topography of a myeloerythroid progenitor cell hierarchy. Cell Stem Cell. 2007;1:428–442. doi: 10.1016/j.stem.2007.07.005. [DOI] [PubMed] [Google Scholar]
- 2.Adolfsson J, Månsson R, Buza-Vidas N, Hultquist A, Liuba K, Jensen CT, Bryder D, Yang L, Borge O-J, Thoren LAM, et al. Identification of Flt3+ lympho-myeloid stem cells lacking erythro-megakaryocytic potential a revised road map for adult blood lineage commitment. Cell. 2005;121:295–306. doi: 10.1016/j.cell.2005.02.013. [DOI] [PubMed] [Google Scholar]
- 3.Orkin SH, Zon LI. Hematopoiesis: an evolving paradigm for stem cell biology. Cell. 2008;132:631–644. doi: 10.1016/j.cell.2008.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kapushesky M, Adamusiak T, Burdett T, Culhane A, Farne A, Filippov A, Holloway E, Klebanov A, Kryvych N, Kurbatova N, et al. Gene Expression Atlas update–a value-added database of microarray and sequencing-based functional genomics experiments. Nucleic Acids Res. 2012;40:D1077–D1081. doi: 10.1093/nar/gkr913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yanai I, Benjamin H, Shmoish M, Chalifa-Caspi V, Shklar M, Ophir R, Bar-Even A, Horn-Saban S, Safran M, Domany E, et al. Genome-wide midrange transcription profiles reveal expression level relationships in human tissue specification. Bioinformatics. 2005;21:650–659. doi: 10.1093/bioinformatics/bti042. [DOI] [PubMed] [Google Scholar]
- 6.Wu C, Orozco C, Boyer J, Leglise M, Goodale J, Batalov S, Hodge CL, Haase J, Janes J, Huss JW, et al. BioGPS: an extensible and customizable portal for querying and organizing gene annotation resources. Genome Biol. 2009;10:R130. doi: 10.1186/gb-2009-10-11-r130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hebestreit K, Gröttrup S, Emden D, Veerkamp J, Ruckert C, Klein H-U, Müller-Tidow C, Dugas M. Leukemia gene atlas – a public platform for integrative exploration of genome-wide molecular data. PLoS One. 2012;7:e39148. doi: 10.1371/journal.pone.0039148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Miranda-Saavedra D, De S, Trotter MW, Teichmann SA, Göttgens B. BloodExpress: a database of gene expression in mouse haematopoiesis. Nucleic Acids Res. 2009;37:D873–D879. doi: 10.1093/nar/gkn854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chambers SM, Boles NC, Lin KK, Tierney MP, Bowman TV, Bradfute SB, Chen AJ, Merchant AA, Sirin O, Weksberg DC, et al. Hematopoietic fingerprints: an expression database of stem cells and their progeny. Cell Stem Cell. 2007;1:578–591. doi: 10.1016/j.stem.2007.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4:249–264. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- 11.Johnson WE, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 2007;8:118–127. doi: 10.1093/biostatistics/kxj037. [DOI] [PubMed] [Google Scholar]
- 12.Gautier L. An intuitive Python interface for Bioconductor libraries demonstrates the utility of language translators. BMC Bioinform. 2010;11(Suppl. 1):S11. doi: 10.1186/1471-2105-11-S12-S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bagger FO, Rapin N, Theilgaard-Mönch K, Kaczkowski B, Jendholm J, Winther O, Porse B. HemaExplorer: a Web server for easy and fast visualization of gene expression in normal and malignant hematopoiesis. Blood. 2012;119:6394–6395. doi: 10.1182/blood-2012-05-427310. [DOI] [PubMed] [Google Scholar]
- 14.Marstrand TT, Borup R, Willer A, Borregaard N, Sandelin A, Porse BT, Theilgaard-Mönch K. A conceptual framework for the identification of candidate drugs and drug targets in acute promyelocytic leukemia. Leukemia. 2010;24:1265–1275. doi: 10.1038/leu.2010.95. [DOI] [PubMed] [Google Scholar]
- 15.Wildenberg ME, van Helden-Meeuwsen CG, van de Merwe JP, Drexhage HA, Versnel MA. Systemic increase in type I interferon activity in Sjögren’s syndrome: a putative role for plasmacytoid dendritic cells. Eur. J. Immunol. 2008;38:2024–2033. doi: 10.1002/eji.200738008. [DOI] [PubMed] [Google Scholar]
- 16.Eckfeldt CE, Mendenhall EM, Flynn CM, Wang T-F, Pickart MA, Grindle SM, Ekker SC, Verfaillie CM. Functional analysis of human hematopoietic stem cell gene expression using zebrafish. PLoS Biol. 2005;3:e254. doi: 10.1371/journal.pbio.0030254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Andersson A, Edén P, Olofsson T, Fioretos T. Gene expression signatures in childhood acute leukemias are largely unique and distinct from those of normal tissues and other malignancies. BMC Med. Genom. 2010;3:6. doi: 10.1186/1755-8794-3-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Majeti R, Becker MW, Tian Q, Lee T-LM, Yan X, Liu R, Chiang J-H, Hood L, Clarke MF, Weissman IL. Dysregulated gene expression networks in human acute myelogenous leukemia stem cells. Proc. Natl Acad. Sci. USA. 2009;106:3396–3401. doi: 10.1073/pnas.0900089106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hu X, Chung AY, Wu I, Foldi J, Chen J, Ji JD, Tateya T, Kang YJ, Han J, Gessler M, et al. Integrated regulation of Toll-like receptor responses by Notch and interferon-gamma pathways. Immunity. 2008;29:691–703. doi: 10.1016/j.immuni.2008.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Allantaz F, Cheng DT, Bergauer T, Ravindran P, Rossier MF, Ebeling M, Badi L, Reis B, Bitter H, D’Asaro M, et al. Expression profiling of human immune cell subsets identifies miRNA-mRNA regulatory relationships correlated with cell type specific expression. PLoS One. 2012;7:e29979. doi: 10.1371/journal.pone.0029979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kohlmann A, Kipps TJ, Rassenti LZ, Downing JR, Shurtleff SA, Mills KI, Gilkes AF, Hofmann W-K, Basso G, Dell’orto MC, et al. An international standardization programme towards the application of gene expression profiling in routine leukaemia diagnostics: the Microarray Innovations in LEukemia study prephase. Br. J. Haematol. 2008;142:802–807. doi: 10.1111/j.1365-2141.2008.07261.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Haferlach T, Kohlmann A, Wieczorek L, Basso G, Kronnie GT, Béné M-C, De Vos J, Hernández JM, Hofmann W-K, Mills KI, et al. Clinical utility of microarray-based gene expression profiling in the diagnosis and subclassification of leukemia: report from the International Microarray Innovations in Leukemia Study Group. J. Clin. Oncol. 2010;28:2529–2537. doi: 10.1200/JCO.2009.23.4732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Di Tullio A, Vu Manh TP, Schubert A, Månsson R, Graf T. CCAAT/enhancer binding protein alpha (C/EBP(alpha))-induced transdifferentiation of pre-B cells into macrophages involves no overt retrodifferentiation. Proc. Natl Acad. Sci. USA. 2011;108:17016–17021. doi: 10.1073/pnas.1112169108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Safran M, Dalah I, Alexander J, Rosen N, Iny Stein T, Shmoish M, Nativ N, Bahir I, Doniger T, Krug H, et al. GeneCards Version 3: the human gene integrator. Database. 2010;2010:baq020. doi: 10.1093/database/baq020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lessard J, Sauvageau G. Bmi-1 determines the proliferative capacity of normal and leukaemic stem cells. Nature. 2003;423:255–260. doi: 10.1038/nature01572. [DOI] [PubMed] [Google Scholar]
- 26.Park I, Qian D, Kiel M, Becker MW, Pihalja M, Weissman IL, Morrison SJ, Clarke MF. Bmi-1 is required for maintenance of adult self-renewing haematopoietic stem cells. Nature. 2003;423:302–305. doi: 10.1038/nature01587. [DOI] [PubMed] [Google Scholar]
- 27.Wang Y, Guan Y, Wang F, Huang A, Wang S, Zhang YA. Bmi-1 regulates self-renewal, proliferation and senescence of human fetal neural stem cells in vitro. Neurosci. Lett. 2010;476:74–78. doi: 10.1016/j.neulet.2010.04.006. [DOI] [PubMed] [Google Scholar]