

Abstract
APOBEC3 (A3) proteins are virus restriction factors that provide intrinsic immunity against infections by viruses like HIV-1 and MMTV. A3 proteins are inducible by inflammatory stimuli such as LPS and IFNα via mechanisms that are not yet fully defined. Using genetic and pharmacological studies on C57BL/6 mice and cells, we show that IFNα and LPS induce A3 via different pathways independent of each other. IFNα positively regulates mA3 mRNA expression through IFNαR•PKC•STAT1 and negatively regulates mA3 mRNA expression via IFNαR•MAPKs signaling pathways. Interestingly, LPS shows some variation in its regulatory behavior. While LPS mediated positive regulation of mA3 mRNA occurs through TLR4•TRIF•IRF3•PKC, it negatively modulates mA3 mRNA via TLR4•MyD88•MAPK signaling pathways. Additional studies on human PBMCs reveal that PKC differentially regulates IFNα and LPS induction of human A3A, A3F, and A3G mRNA expression. In summary, we have identified important signaling targets downstream of IFNαR and TLR4 that mediate A3 mRNA induction by both LPS and IFNα. Our results provide new insights into the signaling targets that could be manipulated to enhance intracellular store of A3 and potentially enhance A3 anti-viral function in the host.
Keywords: APOBEC3, interferon alpha, interferon beta, interferon alpha receptor, interferon regulatory factor 3, lipopolysaccharide, myeloid differentiation primary response gene (88), TIR-domain-containing adapter-inducing interferon-β, protein kinase C delta, Toll-like receptor 4, MAPK, ERK, JNK, P38
Introduction
APOBEC3 (A3) family of proteins is cellular DNA cytidine deaminases that function to inhibit the replication of different viruses, such as retrotransposons, DNA viruses, and retroviruses (1–9). Different vertebrate hosts species have varying number of A3 proteins. The mouse and the human A3 proteins are the most studied with the mouse genome encoding 1 A3 gene (mA3), while the human genome encodes 7 A3 (A3A, A3B, A3C, A3D/E, A3F, A3G, and A3H) genes (10). Amongst all the human A3s, A3F and A3G are the most potent anti-HIV-1 proteins and the most studied. Although A3F and A3G prevent efficient HIV-1 replication, the HIV-1 viral infectivity factor protein (Vif) recruits host cellular partners such as Cullin5-containing E3 ubiquitin ligase to degrade these proteins through a proteasome-dependent mechanism (11–17). Thus, Vif prevents A3F and A3G incorporation into virions, leading to productive HIV infection. However, in the absence of Vif, A3F and A3G are incorporated into viral core and upon infection of new target cells, deaminate deoxycytidines to deoxyuridines in single-stranded viral cDNA during reverse transcription. This process results in hypermutation of the viral genome and attenuation of virus replication (18–22).
A3 proteins are expressed in many cell types, and expression is induced by hormones (23), and cytokines particularly interferon alpha (24–32) and cytokine inducers, such as the bacterial lipopolysaccharide (LPS) (29). Interferons represent a group of pleiotropic cytokines that exhibit different biological activities, including antiviral activities on host cells and tissues (33). Type I (IFNα and IFNβ) and type II Interferon (IFNγ) constitute the major factors that shape the host’s antiviral response. While IFNγ signals through the IFNγ receptor complex, (33), all the Type-I IFNs mediate their effects through the type I IFN receptor (IFNαR). Upon ligand binding, the two Janus kinases, TYK2 and JAK1 are activated (34), followed by phosphorylation and activation of Signal Transducers and Activators of Transcription STAT1 and STAT2 (35). Phosphorylated STAT1 and STAT2 associate with IRF9 to form the interferon-stimulated gene factor-3 (ISGF3) complex. ISGF3 binds specifically to the IFNα-stimulated response element (ISRE), resulting in gene transcription (36), followed by trans-activation of a plethora of interferon-stimulated genes such as A3. Like the IFNαR, Toll-like receptor 4 (TLR4) is a pattern-recognition receptor (PRRs) that triggers innate immune responses (37).
TLR4 is essential for potent LPS signaling. LPS is a well-known pathogen-associated molecular pattern (38). Binding of LPS to its receptor complex (TLR4/MD-2) activates two distinct adapters namely MyD88 and TRIF pathways (39), orchestrating a signaling cascade that results in the activation and nuclear translocation of interferon regulatory factor 3 (IRF3) and nuclear factor kappa B (NF-κB). These events lead to the production of cytokines such as IFNβ, TNFα (40) and the induction of antiviral genes. Interestingly, activation of IRF3 pathway has been implicated in restricting murine herpes virus replication (41). Indeed, LPS-mediated dimerization of TLR4 results in the recruitment of MyD88 which in turn mediates activation of ERK, p38, and JNK (42).
While IFNα and LPS activate the JAK-STAT and TLR4 pathways, other pathways such as the Mitogen-activated protein kinase (MAPK) are also essential for IFNα and/or LPS-dependent biological responses. Indeed, MAPK pathways are differentially activated by IFNα (43–46) and LPS (47–49). Activation of MAPK pathways has been reported to contribute to some human diseases (49–50) and regulation of gene expression (51–52). Included in the MAPK signaling pathways are ERK, JNK, and p38 MAPK; all of which are activated by IFNα and LPS and could play a role in A3 regulation. Up-regulation of A3 mRNA could be achieved by stimulation with LPS (29) and IFNα (26,53). The mechanistic details involved in this A3 up-regulation have not yet been well defined. IFNα has been shown to induce human Apobec3G (hA3G) in STAT1 dependent (54) or independent (25) manners. Here we query the pathways of IFNα and LPS induction of mA3 and investigate whether LPS induced mA3 is dependent on IFNα. We show that both IFNα and LPS induce mA3 via different signaling pathways independent of each other.
Material and Methods
Ethics statement
All experiments involving mice were performed in accordance to NIH guidelines, the Animal Welfare Act, and US federal law. The experiments were approved by the University of Iowa Animal Care and Use Committee (IACUC). Mice were housed according to the policies of the Institutional Animal Care and Use Committee of the University of Iowa.
Animals
C57BL/6 mice were purchased from Jackson laboratory. IFNAR−/− mice were generously provided by Dr. John Harty, University of Iowa and maintained in our colony.
Cell lines
Raw cells were obtained from ATCC. TLR4−/−, MyD88−/− TRIF−/−, and IRF3−/− immortalized macrophages and their wild-type counterparts were kindly provided by Dr. Jerrold Weiss of University of Iowa and Dr. Kate Fitzgerald of University of Massachusetts and have been previously described (55). Briefly, splenic macrophages derived from TLR4−/−, MyD88−/−, TRIF−/−, IRF3−/−, and WT mice on C57/BL6 background were cloned in soft-agar cultures in the presence of M-CSF. Single cell colony was picked and grown in M-CSF containing medium. Supernatants obtained from NIH-3T3 fibroblasts infected with J2 recombinant retrovirus were used to infect the macrophages overnight. Infected macrophages were washed and grown in fresh media containing M-CSF.
Cell cultures
Generation of bone marrow derived MΦs and DCs were previously described (29,56). Briefly, mature macrophages and DCs were obtained from bone marrow stem cells of C57BL/6 mice after 7 days in ex vivo culture at 37°C with 5% CO2 in RPMI 1640 media supplemented with 10% fetal bovine serum, 100U/ml penicillin, 100ug/ml streptomycin and 0.05mM 2-mercaptoethanol along-with either 20ng/ml of recombinant M-CSF for MΦs or GM-CSF for DCs. TLR4, MyD88, TRIF null macrophages, and their wild-type counterparts were grown in DMEM media containing 10% fetal bovine serum, 100ug/ml of sodium pyruvate and in some cases 10ug/ml of ciprofloxacin at 37°C with 5% CO2. Raw cells were grown in RPMI media containing 10% fetal bovine serum, 100U/ml penicillin and 100ug/ml streptomycin.
Isolation of murine PBMCs
Murine PBMCs were obtained from whole blood following treatment of blood with red blood cell (RBC) lysis buffer (Sigma Aldrich). Briefly, PBMCs were treated with RBC lysis buffer for 5 minutes at room temperature. Cells were washed with PBS using centrifugation speeds of 1500 rpm for 10 minutes before culturing in RPMI 1640 media containing 10% fetal bovine serum, 100U/ml penicillin and 100ug/ml streptomycin at 37°C with 5% CO2.
Isolation of human PBMCs
Peripheral blood received from healthy donor was diluted in PBS and subjected to centrifugation at 2000 rpm on Ficoll gradient for 20 min at room temperature. Mononuclear cell layer was separated from the gradient and washed with PBS. Isolated PBMCs were then cultured in RPMI containing 10% fetal bovine serum, 100U/ml of penicillin, 100ug/ml of streptomycin for up to 24 hours at 37°C with 5% CO2.
Antibodies and other reagents
Anti-Interferon alpha and beta antibodies were from Abcam. Endotoxin-free recombinant IFNα and IFNβ were purchased from Miltenyi Biotech. Bacterial LPS, Rottlerin, actinomycin-D, cycloheximide, polymyxin-B and ciprofloxacin were purchased from Sigma Aldrich. TAK-242 or CLI-095 was from Invivogen. M-CSF and GM-CSF were from R&D systems. SYBR green reagent is from (Qiagen) and TaqMan reagent is from ABI. RNALater was obtained from Ambion. SB 203580 and PD 98059 were CalBiochem while SP 600125 was from EMDMillipore.
Gamma irradiation and adoptive transfer
Irradiation and adoptive transfers were carried out as previously described (23). Briefly, 5-week old IFNαR−/− and IFNαR+/+ mice on C57Bl/6 background (n=5) were irradiated with 9-Gy (total impairment of bone marrow function) at the Radiation and Free Radical Research Core (University of Iowa). Four hours after irradiation, mice were injected sub-retinally with 106bone marrow cells. Chimeric mice were examined for bone marrow reconstitution after 4 weeks later by flow cytometry and genotyping.
Administration of LPS and IFNα/IFNβ to mice
LPS (200μg), endotoxin-free recombinant IFNα (10,000U) and endotoxin-free recombinant IFNβ (14,100U) were administered to mice in a total volume of 200 μl via the intra-peritoneal (IP) route.
Chemical Inhibitors, LPS, IFNα/β, and antibody neutralization experiments
Raw cells were treated with LPS (1 μg/ml) or IFNα (1000 units/ml) for 6 or 4hrs respectively followed by addition of fresh media with or without actinomycin-D (1 μg/ml) for up to 15 hours. For inhibition of protein synthesis, raw cells were treated with 10 μg/ml of cycloheximide for 30 minutes at 37°C before adding LPS (1 μg/ml) or IFNα (1000 units/ml) for up to indicated time periods. Treatment of cell with Polymyxin was carried out by treating cells with polymyxin B (50 μg/ml) or TAK-242 (1 μg/ml) for 10 minutes at 37’C before addition of LPS (1 μg/ml) or IFNα for indicated period of time. Inhibition of PKC activity was achieved by pre-treating cells with Rottlerin (0.5 up to 50 μM) as indicated on the figure for 1 hour at 37°C before addition of LPS (1ug/ml) or IFNα (1000 units/ml) for indicated period of time. IFNα neutralizing antibody was used at a concentration of 1ug while IFNβ neutralizing antibody was used at 0.1 to 5μg depending on experiments. Cells treated with neutralizing antibody were incubated for 24 hours followed by stimulation with the appropriate stimulant. Inhibitors of ERK (10μM), JNK (90 nM), and P38 (50 nM), were added to cells and incubated for 15 minutes followed by the addition of the appropriate stimulant as indicated on the figures.
Nucleic acid isolation and PCR
Total RNA was isolated from cells using RNeasy Mini Kit (Qiagen, Inc.). For cDNA synthesis, equal micrograms of RNA were reverse-transcribed with high capacity cDNA reverse transcription Kit (Applied Biosystems, ABI) using Veriti 96-well thermal cycler (ABI). Real-time PCR was performed with the cDNA using ABI 7500 FAST thermal cycler (ABI). The following TaqMan assays were from ABI: A3A (Hs 00377444), A3B (Hs 00358981), A3C (Hs 00826074), A3D/E (Hs 00537163), A3F (Hs 00736570), A3G (Hs 00222415), A3H (Hs 00419665), Human GAPDH (Hs 99999905), and Rodent GAPDH (4308313). The following primer pairs were used: mSTAT1(57), mSTAT2(58), GAPDH(59–60), mA3(2–3,23,29,61–62), mIFNα(63), and mIFNβ(64).
Statistical analysis
Statistical analysis was done to determine significance between the experimental groups using paired two-tailed Student’s t test, and a p value less than 0.05 was considered significant. Error bars represent standard errors.
Results
Different inflammatory mediators enhance mA3 mRNA in Dendritic cells and Macrophages
Various inflammatory mediators have been shown to induce both human and mouse A3 mRNA (26,29,53). To set the stage for our study, we examined the ability of IFNα and LPS to induce mA3 in RAW 264.7 cells, bone marrow derived DCs (BMDCs), and bone marrow derived macrophages, MΦs (BMDM). Following 24 hour stimulation of cells with PBS, IFNα or LPS, cells were collected and examined for mA3 mRNA by qPCR. Results show that upon stimulation with IFNα or LPS, mA3 mRNA significantly increased (Figures 1A to 1D). The increase in mA3 was seen in each cell type with each stimulus. These results are in agreement with previous published work which showed that IFNα and LPS upregulated mA3 mRNA (29).
Figure 1. Kinetics of mA3 mRNA induction by IFNα and LPS.

(A to D) Bone marrow derived macrophages (BMDM), bone marrow derived dendritic cells (BMDC), and RAW cells were stimulated for 4 hours with recombinant endotoxin-free IFNα (1000 units/ml), 6 hours with LPS (1μg/ml), or vehicle. RNA was extracted from cells and reverse transcribed. The resulting cDNA was used to examine level of mA3 transcript by qPCR. (E to H) RAW cells and BMDM were stimulated with 1000 units/ml IFNα, 1μg/ml LPS or vehicle. Cells were collected at the times indicated on the figures and used for RNA extraction and cDNA synthesis followed by qPCR analysis of mA3 mRNA. For all experiments, data is normalized to GAPDH and presented as fold change relative to vehicle treated cells. Error bars are standard error, * is significance with p value less than 0.05 and ** is significance with p value less than 0.01. Experiments were repeated at least three different times with similar results.
IFNα and LPS induce mA3 mRNA with similar kinetics in Macrophages
To assess the kinetics of IFNα and LPS-mediated mA3 mRNA induction, we stimulated RAW cells or BMDM with IFNα or LPS and collected cells at the times indicated on the figure for RNA extraction and mA3 mRNA analysis by qPCR. With the extended presence of IFNα (Figures 1E and 1F) or LPS (Figures 1G and 1H) in culture medium, mA3 mRNA levels in RAW cells significantly increased. Induction with both stimulants was maximal between 4 and 6 hours, and remained elevated over baseline for at least 24 hours. Similar time-dependent induction of mA3 mRNA expression by IFNα and LPS was seen in each cell type examined (Figures 1E to 1H).
In vivo induction of mA3 mRNA by IFNα
While IFNα enhances mA3 mRNA in cultured cells, mA3 mRNA induction by IFNα has not been demonstrated in vivo. Previously, we showed that LPS enhances the expression of mA3 mRNA in vivo with a resultant lower virus load (29). Here, we show that both IFNα and LPS enhance mA3 mRNA levels in vivo. Intraperitoneal administration of IFNα or LPS increased the transcription of mA3 gene in PBMCs 5 to 7 fold compared to control mice that received PBS (Figure 2A). This result is the first demonstration that administration of exogenous IFNα to mice increases the level of endogenous APOBEC3 mRNA and supports our previous finding that LPS induced mA3 mRNA in vivo.
Figure 2. IFNα induces mA3 in a receptor dependent manner in vivo.

(A) Age and weight-matched C57BL/6 mice (n=5) were injected with 10,000U of endotoxin-free recombinant IFNα, 200μg of LPS, or PBS in 200μl volume via the intraperitoneal (IP) route. Twenty four hours later, mice were bled to obtain PBMCs which was used for RNA extraction, cDNA synthesis and mA3 mRNA examination by qPCR (B) Age and weight-matched IFNαR−/− and IFNαR+/+ mice (n=3) on C57BL/6 background were bled to obtain PBMCs. RNA was extracted from cell followed by cDNA synthesis and qPCR to measure levels of mA3. (C and D) Age and weight-matched IFNαR−/− and IFNαR+/+ mice (n=5) on C57BL/6 background were given 10,000 units of endotoxin-free recombinant IFNα, 14,100U of endotoxin-free recombinant IFNβ, or PBS via the IP route. Twenty four hours later, mice were bled to obtain PBMCs. PBMCs were used for RNA extraction and cDNA synthesis, followed by examination of mA3 transcript levels by qPCR. (E and F) Age and weight-matched IFNαR−/− and IFNαR+/+ mice (n=5) were lethally irradiated with 9-Gy gamma rays at the Radiation and Free Radical Research Core of the University of Iowa. Four hours after irradiation, mice were injected sub-retinally with bone marrow stem cells as follows: IFNαR−/− received stem cells from IFNαR+/+ donors while IFNαR+/+ were given stem cells from IFNαR−/− donors. Four weeks after reconstitution, mice were bled for PBMCs. A portion of PBMCs were used for genotyping. In addition, DNA from tail snips obtained from donor and recipient mice were also subjected to genotyping PCR. (G and H) PBMCs was obtained donor or recipient IFNαR−/− and IFNαR+/+ mice described above. Cells were stimulated with 1000U/ml of endotoxin-free recombinant IFNα for 4 hours or vehicle followed by extraction of RNA, cDNA synthesis and examination of mA3 mRNA levels. (I) Female IFNαR−/− and male IFNαR+/+ mice on C57BL/6 background were crossed to obtain the F1 generation (IFNαR+/−). Tail snips from IFNαR−/−, IFNαR+/+, IFNαR+/− were obtained and used for DNA extraction followed by genotyping PCR to determine genotype. (J and K) Age and weight-matched IFNαR−/−, IFNαR+/+, and IFNαR+/− mice (n=5) were given 10,000U of endotoxin-free recombinant IFNα or vehicle via the IP route. Twenty four hours later, mice were bled to obtain PBMCs which was used to examine mA3 mRNA levels by qPCR following RNA extraction and cDNA synthesis. All qPCR data were normalized to GAPDH and presented as fold change relative to vehicle treated cells. Error bars are standard error, * is significance with p value less than 0.05 and ** is significance with p value less than 0.01. Experiments were repeated at least three different times with similar results.
IFNα receptor-deficient cells and mice express reduced levels of mA3 mRNA and do not support mA3 mRNA induction by IFNα
As IFNα efficiently induces mA3 in vivo, we predicted that mice lacking IFNα receptor (IFNαR−/−) would express less mA3 mRNA than their wild type counterparts. To test this, PBMCs from IFNAR−/− and wild-type (WT) mice were subjected to mA3 mRNA analysis by qPCR following RNA extraction and cDNA synthesis. Basal level of mA3 mRNA was significantly lower in IFNAR−/− mice compared to WT controls (Figure 2B), indicating that some level of IFNα signaling via the receptor is required to maintain basal mA3 mRNA in vivo. We further examined if IFNAR−/− mice will respond to IFNα or IFNβ-mediated mA3 mRNA induction. IFNAR−/− or WT mice were stimulated with IFNα (Figure 2C) or IFNβ (Figure 2D) and their PBMCs examined for mA3 mRNA expression. Whereas IFNα or IFNβ enhanced mA3 mRNA in WT mice, mA3 mRNA level did not increase above basal level in the absence of IFNαR (Figures 2C and 2D), suggesting that IFNαR is required to regulate IFNα/β-dependent mA3 mRNA induction in vivo.
Next, we used adoptive transfer experiments (Figure 2E) to further examine the requirement for IFNαR signaling in mediating mA3 mRNA expression and induction. We adoptively transferred bone marrow stem cells obtained from WT mice into lethally gamma irradiated IFNAR−/− hosts. Similarly bone marrow stem cells derived from IFNAR−/− mice were adoptively introduced into lethally gamma irradiated WT hosts (Figure 2E). Four weeks later, mice were bled to obtain PBMCs. A portion of PBMCs and corresponding tail snips from mice were used to determine PBMC and host genotypes. Results show that the host genotype (tail genotype) is different from the PBMCs genotype in recipient mice while donor tail and PBMCs genotypes are similar (Figure 2F); confirming that the adoptive transfer experiment worked. PBMCs from these mice were stimulated with IFNα or PBS for 24 hours. Upon examination of mA3 mRNA levels, we observed lower level of mA3 mRNA in PBS treated IFNAR−/− compared to WT control mice and no enhancement in IFNα treated IFNAR−/− compared to WT treated control mice (Figure 2G). However, there was significant enhancement of mA3 mRNA by IFNα in IFNAR−/− mice that received stem cells from WT mice (Figure 2H – orange bar) compared to WT mice that received stem cells from IFNAR−/− mice (Figure 2H – blue bar), indicating that mA3 mRNA induction by IFNα partially requires both cell-intrinsic IFNAR and environmental signals.
A single IFNαR allele is sufficient but not optimal to mediate mA3 mRNA induction by IFNα
To establish the importance of IFNAR in mA3 induction we then proceeded to restore one allele of the IFNAR (Figure 2I) by generating IFNαR+/− (F1) through mating of WT and IFNαR−/− mice and asked if one IFNαR allele is capable of mediating mA3 induction. PBMCs from age-matched IFNαR−/−, IFNαR+/− and WT mice were used to determine basal mA3 mRNA expression level. We observed a significant increase in basal level of mA3 mRNA in IFNαR+/−compared to IFNαR−/− (Figure 2J), indicating that restoration of one copy of IFNαR is sufficient to maintain basal mA3 mRNA level comparable to WT. Next, we examined mA3 mRNA following a 24 hour PBS or IFNα treatment of PBMCs. IFNα increased mA3 mRNA level in IFNAR+/− and WT but not in IFNα−/− PBMCs (Figure 2K). Although, IFNα induced mA3 mRNA in IFNα+/− PBMCs, the level was lower than that seen in WT PBMCs. These results indicate that IFNαR-mediated mA3 mRNA induction phenotypes as a dominant autosomal allele in F1 progeny. Taken together, these genetic data strongly suggest that mA3 mRNA expression is dependent on the presence of at least one copy of the IFNα receptor.
Activation of STAT1 is necessary for IFNα-mediated mA3 induction
Since mA3 induction by IFNα is IFNαR dependent and both STAT1 and STAT2 have been shown to mediate human A3G induction (25,54), we postulate that activation of STAT1 and/or STAT2 plays a role in mediating IFNα dependent induction of mA3 mRNA. To test our postulation, we pretreated immortalized WT BMDM with IFNα neutralizing antibody or vehicle followed by stimulation with IFNα and examined STAT1, STAT2, and mA3 mRNA level, as well as STAT1 and STAT2 phosphorylation. Our results show that mA3 mRNA is induced by IFNα (Figure 3A) and that this induction requires Serine727 phosphorylation of STAT1 with little or no change in Tyrosine689 phosphorylation of STAT2 (Figure 3B). Additionally, IFNα neutralizing antibody blocked IFNα-mediated mA3 mRNA induction (Figure 3A), STAT1 and STAT2 phosphorylation (Figure 3B) as well as their mRNA induction (Figure 3C). We also show that IFNα enhanced mA3 (Figure 3D), STAT1 (Figure 3E), and STAT2 (Figure 3F) transcripts in WT cells but not in IFNαR−/− cells further arguing that IFNαR is required for IFNα mediated STAT1/2 dependent mA3 induction.
Figure 3. STAT activation is required for optimal induction of mA3 by IFNα.

Immortalized WT BMDM cells on C57BL/6 background were pre-treated with vehicle or anti-IFNα neutralizing antibody for 24 hours. Cells were stimulated with IFNα or left unstimulated for 30 minutes or 4 hours depending on experiment. RNA was extracted from cells, followed by cDNA synthesis and examination of (A and C) mA3, STAT1, and STAT2 mRNA. The remaining portion was used for Western blot examination of STAT1 and STAT2 phosphorylation (B). GAPDH was used as loading control. Primary BMDM from WT and IFNαR−/− mice on C57BL/6 background was left unstimulated or stimulated with IFNα for 4 hours. Cells were used for RNA extraction, cDNA synthesis, and qPCR examination of mA3, STAT1, and STAT2 transcripts (D to F). QPCR data were normalized to GAPDH and presented as fold change relative to vehicle treated cells. Error bars are standard error, * is significance with p value less than 0.05 and ** is significance with p value less than 0.01. Experiments were repeated at least three different times with similar results.
LPS-mediated IFNβ production induces mA3 mRNA
LPS-mediated TLR4 signaling induces IFNβ production through the activation of IRF3 (65–66). Because IFNβ is one of the initial cytokines induced by LPS, we sought to determine the extent to which IFNβ contributes to LPS-induced mA3 expression. We hypothesized that LPS will mediate mA3 mRNA induction in an IFNβ-dependent and independent manners. We tested our hypothesis by treating immortalized BMDM cells with LPS or endotoxin-free recombinant IFNβ and in some experiments, cells were pretreated with IFNβ neutralizing antibodies followed by stimulation with LPS. Results show an increase in IFNβ mRNA in cells stimulated with LPS (Figure 4A). Treatment of cells with IFNβ induced mA3 mRNA in a dose-dependent manner (Figure 4B) and enhanced STAT1 and STAT2 mRNA expression (Figure 4C) and phosphorylation (Figure 4D). Pretreatment of cells with IFNβ neutralizing antibody significantly reduced mA3 mRNA expression level (Figure 4E) in a dose dependent manner (Figure 4F) and blocked STAT1 and STAT2 mRNA induction (Figure 4G). Examination of TNFα transcript in the same cells shows that LPS induced equal levels of TNFα irrespective of anti-IFNβ treatment (Figure 4H). These data demonstrate that neutralizing IFNβ elicited by LPS blocks LPS-mediated IFNβ-dependent mA3 mRNA induction (Figure 4E).
Figure 4. LPS induces mA3 in Type I IFN dependent and independent manner.

Immortalized WT BMDM cells on C57BL/6 background were pre-treated with vehicle or anti-IFNβ neutralizing antibody for 24 hours. Cells were stimulated with LPS or left unstimulated for 6 hours followed by RNA extraction and cDNA synthesis or protein extraction, and examination of (A, B, C, E, F, G, and H) IFNβ, STAT1, STAT2, TNFα, mA3 mRNA, and (D) STAT1 and STAT2 phosphorylation. Data were normalized to GAPDH and presented as fold change relative to vehicle treated cells. GAPDH was used as protein loading control. Error bars are standard error, * is significance with p value less than 0.05 and ** is significance with p value less than 0.01. Experiments were repeated at least three different times with similar results.
IFNαR-independent regulation of mA3 mRNA induction by LPS
Our previous (29) and current data (Figure 2A) show that the TLR4 agonist - LPS induces mA3 mRNA in vivo. Since TLR4 activation by LPS leads to type I interferon production, it seemed possible that LPS-induced mA3 was secondary to the induction of type I IFN. To test this hypothesis, PBMC from IFNαR−/− mice were stimulated with PBS, IFNα, or LPS. We observed a significant increase in mA3 mRNA induction in IFNαR−/− upon LPS but not IFNα stimulation (Figure 5A), indicating that LPS-mediated mA3 mRNA is independent of type I IFN receptor (IFNαR). To further compare the effect of LPS in vivo in IFNαR−/− and IFNαR+/+ mice, we administered LPS intra-peritoneally to mice. PBMCs collected from mice 24 hours later were analyzed for mA3 mRNA levels. As expected, levels of mA3 mRNA were increased by LPS in both genetic backgrounds (Figure 5B). Although LPS induced mA3 mRNA in IFNαR−/− mice, the magnitude of induction was greater in mice with functional IFNαR. Thus, the effect of LPS on induction of mA3 mRNA is both IFNαR-independent and -dependent.
Figure 5. LPS induces mA3 in an IFNαR-independent but TLR4-dependent manner.

(A) PBMCs obtained from IFNαR−/− mice were stimulated with 100 units/ml of endotoxin-free recombinant IFNα for 4 hours, 1μg/ml LPS for 6 hours, or vehicle. After stimulation, RNA was extracted from cells followed by cDNA synthesis and examination of mA3 mRNA levels. (B) Age and weight-matched IFNαR−/− and IFNαR+/+ mice (n=5) on C57BL/6 background were bled to obtain PBMCs for pre-stimulation sample. Mice were then given 200μg of LPS via the IP route. Twenty four hours later, mice were bled to obtain PBMCs which was used to examine mRNA expression level by qPCR. (C and D) PBMCs obtained from age and weight-matched IFNαR−/− and IFNαR+/+ mice were stimulated with 1μg/ml of LPS or vehicle for 6 hours or pre-treated with polymixin B (50μg/ml) or vehicle for 10 minutes and stimulated with LPS for 6 hours. After stimulation, cells were used to evaluate mA3 transcript level by qPCR. (E) Immortalized BMDM from C57BL/6 mice were pre-treated with CLI-095 (1μg/ml) for 10 minutes followed by stimulation with 1000 units/ml of endotoxin-free recombinant IFNα, 1μg/ml LPS, or vehicle for 4 or 6 hours. Cells were used for RNA extraction, cDNA synthesis and qPCR analysis of mA3 mRNA expression level. (F) Immortalized BMDM from WT and TLR4−/− mice in C57BL/6 background were stimulated with 1000 units/ml of endotoxin-free recombinant IFNα, 1μg/ml LPS, or vehicle for 4 or 6 hours followed by RNA extraction. RNA was reverse transcribed and cDNA used for qPCR analysis of mA3 mRNA levels. (G) Primary BMDM was obtained from bone marrow stem cells of C3H/HeN and C3H/HeJ mice. Cells were treated with endotoxin-free recombinant IFNα (1000 units/ml), LPS (1μg/ml), or vehicle for 4 or 6 hours respectively followed by RNA extraction, cDNA synthesis and qPCR analysis of mA3 mRNA levels. (H to L) Immortalized BMDM obtained from WT, MyD88−/−, and TRIF−/− mice in C57BL/6 background were stimulated with 1000 units/ml of endotoxin-free recombinant IFNα, 1μg/ml LPS, or vehicle for 4 or 6 hours followed by RNA extraction. RNA was reverse transcribed and cDNA used for qPCR analysis of (H, I, J, and L) mA3 mRNA levels and (K) TNFα transcripts. Data are normalized to GAPDH and presented as fold change relative to vehicle, LPS, or WT cells. Error bars are standard error, * is significance with p value less than 0.05 and ** is significance with p value less than 0.01. Experiments were repeated at least three different times with similar results.
Next, we hypothesized that LPS mediated mA3 mRNA induction is independent of activation of the IFNαR. PBMCs from WT and IFNαR−/− mice were pre-treated with PBS or polymyxin B (a compound that blocks TLR4 signaling by binding endotoxin) for 10 minutes followed by addition of LPS. Six hours later, cells were collected and examined for mA3 mRNA expression levels. LPS enhanced mA3 mRNA in PBMCs from both IFNαR−/− and WT mice (Figure 5C). The presence of polymyxin B in the PBMC culture blocked LPS signaling and thus blocked mA3 mRNA induction (Figure 5D). These data confirmed IFNαR-dependent and – independent induction of mA3 mRNA by LPS.
To determine whether IFNα-dependent mA3 mRNA induction occurs independent of TLR4 mediated signaling, we blocked both ligand-dependent and -independent signaling of TLR4 by pretreating BMDM with the TLR4 signaling inhibitor TAK-242 also known as CLI-095 (67) for 10 minutes followed by stimulation with IFNα or LPS. Results show that while TAK-242 blocked TLR4-mediated LPS signaling and inhibited mA3 mRNA induction, IFNα-dependent mA3 mRNA induction is independent of TAK-242 (Figure 5E). To validate this pharmacological data, BMDM from TLR4−/− and WT mice were stimulated with IFNα or LPS followed by examination of mA3 transcripts. IFNα enhanced mA3 mRNA in the presence and absence of TLR4 but with better efficiency in WT cells, while LPS enhanced mA3 mRNA expression only in WT cells (Figure 5F).
Next, we used BMDM obtained from bone marrow stem cells of TLR4-sufficient C3H/HeN (HeN) and TLR4-defective C3H/HeJ (HeJ) mice to validate TLR4 dependent LPS-mediated mA3 induction. As observed in figure 4F, IFNα induced mA3 in both HeN and HeJ mice with slightly better induction in HeN mice (Figure 5G), however, in HeJ but not HeN mice, LPS did not enhance mA3 mRNA level (Figure 5G). Thus our pharmacological and genetic data suggest that both IFNαR and TLR4 regulate mA3 mRNA induction, each substantially independent of the other. It is not clear why mA3 mRNA level is slightly higher in WT cells (Figures 5F and 5G); however, studies have shown that IFNα induces the expression of TLRs 2, 3, 4, 5, 7, 8, and MyD88 in PBMCs and liver cells (68). Additionally, TLR4 activation involves the induction of an autocrine loop resulting in the production and signaling of endogenous IFNβ. It is therefore possible that in cells with intact TLR4, TLR4 signaling could induce the expression of inflammatory genes including IFNβ and IFNα response genes by autocrine-acting IFNβ – resulting in higher levels of IFNα response genes, such as A3 in as shown in Figures 5F and 5G.
TRIF regulates LPS•TLR4-mediated mA3 mRNA induction
TLR4 ligation by LPS results in signal transduction events that involve two different sets of adapter proteins downstream of TLR4 – one includes the myeloid differentiation primary response gene 88 (MyD88) and the other including Toll-IL-1-receptor domain-containing adaptor-inducing IFN-β (TRIF). To define the relative roles of these two signaling pathways in LPS-triggered TLR4-dependent mA3 mRNA expression, WT, MyD88−/−, or TRIF−/− BMDM were stimulated with PBS, IFNα or LPS. Following stimulation, IFNα-triggered mA3 mRNA induction was observed both in MyD88−/− and TRIF−/− cells. In addition, mA3 mRNA induction by LPS is dependent on MyD88, as there was a subtle difference in mA3 mRNA levels when comparing vehicle and LPS treated MyD88−/− cells (Figure 5H). A significant difference in LPS-mediated induction of mA3 was observed when LPS-treated MyD88−/− cells were compared with WT cells (Figure 5J). In contrast, mA3 gene transcription shows a complete dependence on TRIF. LPS significantly induced mA3 mRNA in WT and MyD88−/− cells (Figure 5J) but not in TRIF−/− cells (Figures 5I and 5J). As a control, we examined the level of TNFα transcripts and observed that LPS induces TNFα normally in TRIF−/− cells (Figure 5K) despite lack of mA3 induction (Figures 5I and 5J). Additionally, IFNα induced mA3 mRNA in TRIF−/− cells similar to the level observed in MyD88−/− cells (Figure 5L) but failed to induce TNFα in these cells (Figure 5K). These genetic studies demonstrate that induction of mA3 mRNA by LPS requires functional TLR4 and TRIF.
IRF3 is required for LPS but not IFNα mediated mA3 mRNA induction
To further define the signal transduction pathways that regulate mA3 mRNA induction, we tested whether the transcription factor -IRF3 is necessary for IFNα or LPS mediated mA3 mRNA induction. As shown in Figure 6A, IFNα induces mA3 mRNA in the absence of IRF since no difference in mA3 mRNA expression was observed in WT and IRF3−/− cells stimulated with IFNα. In contrast, mA3 mRNA levels increased in WT cells upon LPS stimulation but LPS treatment of IRF3−/− cells results in significant suppression of mA3 mRNA (Figure 6B). To ensure that IRF3−/− cells are responsive to LPS, we evaluated level of TNFα present in IFNα and LPS treated cells compared to cells treated with the vehicles. We show that despite the inability of LPS to induce mA3 mRNA in IRF3−/− cells, LPS but not IFNα significantly enhanced TNFα mRNA in WT and IRF3−/− cells (Figure 6C). Indeed, level of TNFα transcript in IRF3−/− cells stimulated with LPS was about 40 fold higher than the level in WT cells. These data indicate that LPS-mediated induction of mA3 mRNA is dependent on IRF3.
Figure 6. Requirement for IRF3 in LPS mediated mA3 mRNA expression.
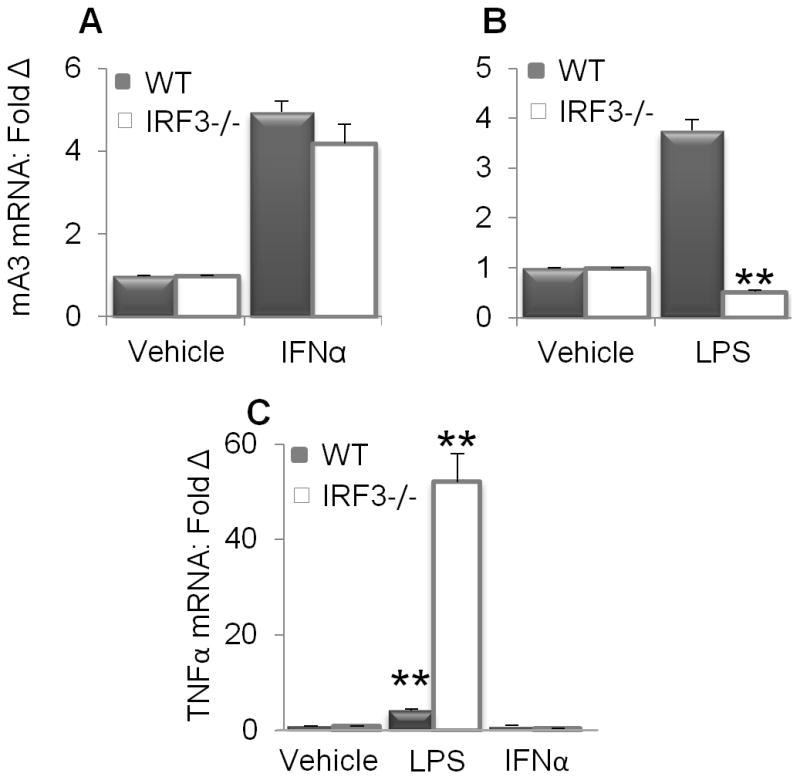
Immortalized IRF3−/− and WT BMDM on C57BL/6 background were stimulated with 1000 units/ml of endotoxin-free recombinant IFNα for 4 hours, 1μg/ml LPS for 6 hours, or vehicle. Cells were used for RNA extraction, cDNA synthesis and examination of (A and B) mA3 mRNA levels (C) TNFα mRNA levels by qPCR. Data were normalized to GAPDH and presented as fold change relative to vehicle treated cells. Error bars are standard error, * is significance with p value less than 0.05 and ** is significance with p value less than 0.01. Experiments were repeated at least three different times with similar results.
PKC- is a regulator of mA3 gene transcription
Since TRIF and IRF3 mediate mA3 mRNA induction by LPS we further dissected the mechanisms of mA3 mRNA induction to identify other factors involved in this process. Previous studies have indicated Protein kinase C α/β as mediators of IFNα induction of A3G (25,69). PKC is a family of protein kinase enzymes known to play a role in transducing signals that regulate the function of other proteins. PKCs are divided into 3 subclasses namely conventional PKCs (cPKC; α, βI, βII, and γ), the novel PKC isoforms (nPKC; δ, ε, η, and θ), and the atypical PKC isoforms (aPKCs; ζ and ι/λ) (70–71). Thus, we examined the effect of PKC on mA3 mRNA induction by stimulating WT BMDM with IFNα or LPS followed by addition of Rottlerin - a PKC inhibitor (25,72) in the medium in the presence or absence of IFNα or LPS. Evaluation of mA3 mRNA revealed that Rottlerin was unable to block mA3 mRNA once mA3 is already induced by IFNα (Figure 7A) or LPS (Figure 7B) irrespective of continuous or discontinuous stimulation by IFNα or LPS. Previous studies indicated Rottlerin as a blocker of type I interferon signaling (73) and IFNα-mediated A3G mRNA in Hep3B liver cells (25). Since inhibition of PKC with Rottlerin is unable to block mA3 mRNA induction once mRNA synthesis is initiated, we hypothesized that blocking PKC prior to the addition of IFNα will block mA3 mRNA induction. Hence we pre-treated WT BMDM with PBS or different doses of Rottlerin followed by IFNα or LPS stimulation. Interestingly, Rottlerin blocked IFNα and LPS mediated mA3 mRNA in a dose dependent manner (Figure 7C). Using the 6μM concentration of Rottlerin, we repeated experiment and showed that Rottlerin greatly diminished IFNα and LPS mediated STAT1 and STAT2 mRNA (Figure 7D) similar to the effect seen on mA3 mRNA (Figure 7E).
Figure 7. PKC is involved in IFNα and LPS-mediated mA3 induction.

(A and B) Immortalized wild-type BMDM on C57BL/6 background were stimulated with 1000 units/ml of endotoxin-free recombinant IFNα for 4 hours, 1μg/ml LPS for 6 hours, or vehicle. Cells were washed and medium containing 6μM of the PKC inhibitor Rottlerin was added with or without IFNα or LPS. Cells were cultured for additional 4 or 6 hours and then used for RNA extraction, cDNA synthesis and examination of mA3 mRNA levels by qPCR. (C ) Immortalized wild-type BMDM on C57BL/6 background were pre-treated with different concentrations (0.5, 6, 10, 20, 50 μM) of Rottlerin as indicated on the figure or vehicle for 1 hour followed by stimulation with 1000 units/ml of endotoxin-free recombinant IFNα for 4 hours, 1μg/ml LPS for 6 hours, or vehicle. Cells were used for RNA extraction, cDNA synthesis and qPCR examination of mA3 mRNA levels. (D and E) Immortalized wild-type BMDM on C57BL/6 background were pre-treated with Rottlerin (6μM) or vehicle for 1 hour followed by stimulation with 1000 units/ml of endotoxin-free recombinant IFNα for 4 hours, 1μg/ml LPS for 6 hours, or vehicle. Cells were used for RNA extraction, cDNA synthesis and qPCR examination of STAT1, STAT2, and mA3 mRNA levels. (F) Immortalized IRF3−/− BMDM on C57BL/6 background were pre-treated with Rottlerin (6μM) or vehicle for 1 hour followed by stimulation with 1000 units/ml of endotoxin-free recombinant IFNα for 4 hours, 1μg/ml LPS for 6 hours, or vehicle. Cells were used for RNA extraction, cDNA synthesis and qPCR examination of mA3 mRNA levels. Data were normalized to GAPDH and presented as fold change relative to vehicle treated cells. Error bars are standard error, * is significance with p value less than 0.05 and ** is significance with p value less than 0.01. Experiments were repeated at least three different times with similar results.
Next, we examined the involvement of PKC in regulating mA3 mRNA induction through IRF3. Rottlerin reduced mA3 mRNA induction by IFNα but not LPS in the absence of IRF3 (Figure 7F). The IRF3 Rottlerin data further suggest that LPS-mediated induction of mA3 requires a different set of signaling molecules than IFNα-mediated induction of mA3 mRNA.
MAPK family members ERK, JNK, and P38 negatively regulates mA3 mRNA expression
The TLR4 ligand LPS activates MAPK family members - including ERK, p38, and JNK (74). Activation of these MAPKs participates in inflammatory cytokines induction (47,74) which in turn could induce the expression of anti-viral genes. To identify targets downstream of TLR4 adaptor proteins – MyD88 and TRIF that are involved in the regulation of mA3 expression, we used pharmacological inhibitors of the MAPK family of proteins ERK, JNK, and P38 to pretreat primary BMDM obtained from WT and IFNαR−/− mice prior to stimulation with IFNα and LPS. Results show that ERK, JNK, and P38 are negative regulators of IFNα and LPS mediated mA3 mRNA induction (Figures 8A and 8B). As expected, treatment of BMDM obtained from IFNαR−/− revealed that MAPK-dependent IFNα-mediated mA3 mRNA induction requires IFNαR (Figure 8C) but MAPK-dependent LPS-mediated mA3 mRNA induction is independent of the IFNαR (Figure 8D). Indeed, negative regulation of LPS-dependent mA3 mRNA expression by ERK, JNK, and P38 is independent of the IFNαR because blocking ERK, JNK, and P38 in IFNαR−/− BMDM greatly enhanced mA3 mRNA (Figure 8D). Although MAPK inhibitors augmented mA3 mRNA induction by LPS in WT and IFNαR−/− cells, these inhibitors only enhanced LPS-mediated IFNβ mRNA induction in WT cells (Figure 8E) but not in IFNαR−/−(Figure 8F). These data further points to IFNαR-independent MAPK-dependent LPS-mediated mA3 mRNA induction.
Figure 8. MAPK family members are negative regulators of mA3 in primary macrophages.

(A, B, and E) primary IFNαR WT BMDM (C, D, and F) primary IFNαR−/− BMDM on C57BL/6 background were left untreated or pre-treated with inhibitors of ERK (10μM), JNK (90 nM), and P38 (50 nM) for 15 minutes at 37°C followed by stimulation with 1000 units/ml of endotoxin-free recombinant IFNα for 4 hours, 1μg/ml LPS for 6 hours, or vehicle. Cells were used for RNA extraction, cDNA synthesis and qPCR evaluation of (A, B, C, and D) mA3 mRNA levels or (E and F) IFNβ mRNA. (G ) Immortalized wild-type and MyD88−/− BMDM on C57BL/6 background were left untreated or pre-treated with inhibitors of ERK (10μM), JNK (90 nM), and P38 (50 nM) for 15 minutes at 37°C followed by stimulation with 1000 units/ml of endotoxin-free recombinant IFNα for 4 hours, 1μg/ml LPS for 6 hours, or vehicle. Cells were used for RNA extraction, cDNA synthesis and qPCR examination of mA3 mRNA levels. Data were normalized to GAPDH and presented as fold change relative to vehicle treated cells. Error bars are standard error, * is significance with p value less than 0.05 and ** is significance with p value less than 0.01. The numbers on figure E denote mRNA values. Experiments were repeated at least three different times with similar results.
Since MAPK signaling plays a crucial role in mediating MyD88-dependent induction of gene expression (47,74–75), we hypothesized that MAPK-dependent regulation of LPS-mediated mA3 mRNA induction will require MyD88. Hence, we used pharmacological inhibitors of ERK, JNK, and P38 to pretreat WT and MyD88−/− BMDM prior to stimulation with LPS. Results show that inhibition of ERK, JNK, or P38 enhanced mA3 mRNA expression following stimulation with LPS in WT cells but not in MyD88−/− cells (Figure 8G), suggesting that LPS augments mA3 mRNA induction via the MyD88•MAPK signaling pathway. Although LPS can activate MAPK in MyD88−/− cells with delayed kinetics (76) we did not observe an increase in LPS-mediated mA3 mRNA expression in these cells (Figure 8G), perhaps because our experiments were performed within 6 hours of LPS induction. These genetic and pharmacological data infer that ERK, JNK, and P38 are negative regulators of IFNα and LPS dependent mA3 mRNA expression in macrophages.
LPS but not IFNα-mediated induction of mA3 mRNA requires de novo mRNA synthesis
To examine the functional regulation and mechanistic differences between IFNα and LPS-mediated mA3 induction, we first estimated the half-life of mA3 mRNA by qPCR using total RNA from IFNα stimulated and ActD treated RAW cells. Raw cells were stimulated with IFNα for 4 hours and after washing the cells to remove extracellular IFNα, the cells were further incubated in fresh medium with or without ActD. Cells were collected every hour for 15 hours followed by analysis of mA3 mRNA level. By inhibiting de novo mRNA synthesis and degrading existing transcripts, we determined that IFNα-induced mA3 transcripts remained stable and were maintained with similar kinetics compared to cells treated with IFNα alone where transcription is ongoing (Figure 9A). Indeed, both ActD treated and ActD naïve transcripts showed minimal drop in mA3 mRNA 8 hours following treatment with IFNα alone or IFNα and ActD, indicating that once induced by IFNα, mA3 does not require sustained IFNα signal.
Figure 9. Requirement for de novo mRNA and protein synthesis in mA3 mRNA induction.

(A and B) Raw cells were stimulated with 1000 units/ml of endotoxin-free recombinant IFNα, 1g/ml LPS, or vehicle. Stimulant-containing medium was removed and replaced with fresh medium with or without 1ug/ml Actinomycin D (ActD). Cells were collected every hour for 15 hours as indicated on the figures. Total RNA was extracted and used for cDNA synthesis and cDNA used for examination of level ofmA3 mRNA by qPCR. (C and D) Raw cells were pre-treated with 10μg/ml CHX for 30 minutes followed by stimulation with 1000 units/ml of endotoxin-free recombinant IFNα, 1μg/ml LPS, or vehicle. Cells were collected at the time points indicated on the figure for total RNA was extraction and cDNA synthesis. Level of mA3 transcript was examined by qPCR. In addition, STAT1 and STAT2 mRNA levels were examined. Data is normalized to GAPDH and presented as fold change relative to vehicle treated cells. Error bars are standard error. Experiments were repeated at least three different times with similar results.
Next, we investigated whether de novo mRNA synthesis is required for LPS-induced mA3 mRNA. Raw cells were treated with LPS for 6 hours. Cells were washed to remove extracellular LPS and further incubated in fresh medium with or without ActD to facilitate inhibition of de novo mRNA synthesis and degradation of remaining mA3 transcripts. Cells were collected hourly for 15 hours and subjected to RNA extraction, cDNA synthesis, and qPCR examination of mA3 mRNA. While LPS-induced mA3 transcript remained relatively stable for up to 8 hours in the presence of ActD followed by a significant decay, induction of mA3 mRNA by LPS in ActD naïve cells continued and stayed elevated for about 15 hours (Figure 9B). This result suggests that similar to IFNα induced mA3 mRNA, LPS induced mA3 mRNA is stable following DNA transcription arrest for up to 8 hours after transcription arrest. However, unlike the kinetics of IFNα induction of mA3 mRNA which is similar between ActD treated and ActD naïve cells (Figure 9A), the kinetics of LPS induction of mA3 mRNA is different between ActD treated and untreated cells (Figure 9B). Whereas continuous exposure to IFNα beyond 12 hours (4 hours of IFNα stimulation plus 8 hours of chase) does not result in new mA3 mRNA synthesis (Figure 9A), exposure to LPS results in new mA3 transcripts being made for up to 21 hours (6 hours of LPS stimulation plus 15 hours of chase) after LPS addition (Figure 9B).
Together, these data suggest that LPS but not IFNα-induction of mA3 mRNA is sensitive to disruption of de novo mRNA synthesis, suggesting that these two stimulants mediate mA3 mRNA transcription via two different pathways.
Induction of mA3 mRNA by IFNα is dependent on de novo protein synthesis
To further determine whether different mechanisms are involved in IFNα and LPS mediated mA3 mRNA induction, we used Cycloheximide (CHX) chase experiments to evaluate if the synthesis of mA3 mRNA following induction with IFNα requires new protein synthesis. RAW cells were pretreated with PBS or 10μg/ml of CHX for 30 minutes and then stimulated with or without IFNα for the times indicated on the figure. A rapid decay of mA3 mRNA was observed within 2 hours of CHX treatment (Figure 9C – top panel), indicating that mA3 may not be a direct transcriptional target of IFNα because IFNα-induced mA3 expression requires de novo protein synthesis. The rapid decay phase of mA3 in the presence of CHX was followed by a super-induction phase 24 hours later. In contrast, examination of transcripts of STAT1 and STAT2 showed that although IFNα enhanced expression of STAT1 and STAT2 mRNA, their transcripts were super-induced by CHX (Figure 9C – middle and bottom panels). Together, these results suggest that mA3 is an IFNα induced delayed early response gene, whose expression requires in addition to STATs -1 and/or -2 another protein downstream of STATs.
LPS mediates mA3 mRNA induction independent of de novo protein synthesis
LPS increased the steady state level of mA3 within four hours and remained elevated for 24 hours (Figure 9D – top panel). However, pre-incubation of cells with CHX before stimulation with LPS increased mA3 mRNA level further by about 4 fold within 10 to 30 minutes (Figure 9D – top panel). The maximal level of mA3 induced by LPS in the presence of CHX was reached in 30 minutes, followed by a drop 1 hour later, and a significant decay in 24 hours (Figure 9D – top panel), indicating that mA3 is a LPS early response gene. Evaluation of mRNA of STAT1 and STAT2 following CHX treatment and LPS stimulation revealed that CHX had no effect on the transcripts of these genes for at least 5 hours (Figure 9D – middle and bottom panels). These data demonstrate that mA3 is a LPS-induced early response gene that does not require de novo protein synthesis. The super-induction of mA3 and STATs transcription by IFNα or LPS in the presence of protein synthesis inhibitor may be as a result of the decay of labile repressors of mRNA transcription. Together these results further show that different mechanisms are involved in IFNα and LPS-mediated mA3 mRNA regulation.
IFNα and LPS differentially modulate induction of human APOBEC3 genes
Our data so far suggest that IFNα and LPS regulate mA3 gene transcription via different mechanisms. To further demonstrate this concept, we examined the effect of IFNα or LPS in regulating all seven human A3 mRNA expressions in human PBMCs. Previous studies have shown that IFNα induces the expression of different human A3 genes with different magnitudes (28). Upon IFNα or LPS stimulation of PBMCs obtained from healthy donors, we observed that induction of A3A, A3F, and A3G transcripts is time-dependent (Figures 10A to 10C – upper panels). Transcripts of A3A, A3F and A3G were induced as early as 6 hours and remained elevated for up to 24 hours after IFNα stimulation of cells (Figures 10A to 10C - upper panels). IFNα also enhanced mRNA of A3 -B, -C, and –H, but not A3D/E (Figure 10D). Examination of the effect of LPS on human A3 mRNA showed that like IFNα, LPS enhanced A3A, A3F, and A3G mRNA levels by 6 hours, but unlike IFNα, this enhancement was followed by a significant drop in A3F mRNA and suppression in A3A and A3G mRNAs at later times (Figures 10A to 10C – upper panels). Similarly, LPS enhanced A3C, A3D/E, and A3H mRNA but suppressed A3B mRNA (Figure 10D).
Figure 10. LPS and IFNα differentially induce A3A, A3F and A3G.

(A to C – top panels) PBMCs from healthy donors were plated and stimulated with 1000 units/ml of endotoxin-free recombinant human IFNα, 1μg/ml LPS, or vehicle for the times indicated on the figures. Cells were used for RNA extraction and cDNA synthesis. Using the cDNA, transcripts of A3A, A3F, and A3G were examined by qPCR. (A to C – middle panels) PBMCs from healthy donors were plated and pre-treated with Rottlerin (6uM) for 1 hour. Cells were then stimulated with 1000 units/ml of endotoxin-free recombinant human IFNα for 6 or 24 hours, followed by analysis of A3A, A3F, and A3G mRNA levels. (A to C – bottom panels) PBMCs from healthy donors were plated and pre-treated with Rottlerin (6uM) for 1 hour. Cells were then stimulated with 1μg/ml of LPS for 6 or 24 hours, followed by analysis of A3A, A3F, and A3G mRNA levels by qPCR. (D) PBMCs from healthy donors were plated and stimulated with 1000 units/ml of endotoxin-free recombinant human IFNα, 1μg/ml LPS, or vehicle for 24 hours. Cells were used for RNA extraction and cDNA synthesis followed by examination of A3B, A3C, A3D/E, and A3H transcripts by qPCR. Data were normalized to GAPDH and presented as fold change relative to vehicle treated cells. Error bars are standard error, * is significance with p value less than 0.05 and ** is significance with p value less than 0.01. Experiments were repeated four times with PBMCs from different donor and similar results obtained.
To further elucidate the differences in IFNα and LPS mechanism of A3 mRNA induction, we tested whether IFNα-mediated induction of A3A, A3F, and A3G mRNA is dependent on PKC. While IFNα induction of A3G mRNA has been shown to be sensitive to the PKCα/β and PKC inhibitor Rottlerin (25,69), the effect of Rottlerin in the transcription of other human A3 genes has not been demonstrated. We tested whether IFNα-mediated induction of A3A, A3F, and A3G mRNA is sensitive to Rottlerin. We found a significant time-dependent suppression of IFNα induced A3A, A3F and A3G mRNA in the presence of Rottlerin (Figures 10A to 10C middle panels). In contrast, to A3F and A3G, Rottlerin mediated suppression of A3A mRNA was short live. There was a reduction in the transcription of A3A mRNA in the presence of Rottlerin 6 hours after Rottlerin addition but by 24 hours A3A mRNA level increased relative to the 6 hour time point level (Figure 10A middle panel). This result is in line with published report on the effect of Rottlerin on A3G mRNA induction. Our result also extends the importance of PKC in IFNα-mediated induction of A3A and A3F gene transcription.
Next, we examined whether similar signal transduction pathways control LPS-mediated A3A, A3F, and A3G transcription by testing the sensitivity of LPS induced A3 mRNA to Rottlerin. Our data show that while LPS-induced A3F mRNA is highly sensitive to Rottlerin (Figure 10B – bottom panel), induction of A3A and A3G mRNA is less sensitive to Rottlerin but are significantly induced in a time-dependent manner in the presence of Rottlerin (Figures 10A to 10C – bottom panels). Although A3F mRNA is sensitive to Rottlerin, this sensitivity is also time-dependent since we observed a recovery from suppression 24 hours later (Figure 10B – lower panel). These data indicate that PKC mediated signaling events differentially control human A3 mRNA induction by IFNα and LPS, further suggesting that mechanisms of IFNα and LPS-mediated human A3 gene transcription may be distinct from each other. Further studies are on the way to dissect these differences.
Discussion
APOBEC3 (A3) proteins have emerged as new players in innate immunity against viral infections (1–2). In humans and mouse, A3 mRNAs are expressed at varying levels in hematopoietic and non-hematopoietic cells (15,23–24,27–28,62,77). Although A3 proteins are constitutively expressed, the basal levels can be enhanced by various compounds including steroid hormones (23) and inflammatory stimulants such as IFNα and LPS (15,23–24,28–29,62,77). Given the importance of A3 proteins in blocking virus replication (1–2), it is imperative to determine how the expression of these genes are controlled. Understanding the regulatory events that mediate A3 mRNA induction by different inflammatory mediators is of fundamental importance in the eventual use of A3 in anti-viral therapies. Here, we present data demonstrating that the inflammatory mediators IFNα and LPS enhance A3 mRNA expression using different signaling pathways.
Our studies reveal that exogenous IFNα and IFNβ, or LPS-induced IFNβ mediate mA3 mRNA induction; and that mA3 induction requires a functional IFNαR. In mice deficient in IFNαR, basal level of mA3 is significantly lower compared to their IFNαR sufficient counterpart. It is possible that some level of IFNα signaling is necessary for maintenance of mA3 gene transcription. The important role of IFNαR is further supported by our finding that both cell intrinsic IFNαR and environmental cues contribute to mA3 mRNA induction. This is because bone marrow stem cells from IFNαR−/− mice introduced to WT acquired the ability to respond to IFNα and up-regulate mA3 mRNA. However, when WT mice received IFNαR−/− cells, mA3 mRNA was up-regulated to a lesser degree. Additionally, genetic restoration of one IFNαR allele as in IFNαR+/− results in higher basal mA3 mRNA level and induction of mA3 mRNA by IFNα. Given the fact that IFNα restricts virus replication (78–80), and induces high levels of mA3 mRNA which in turn inhibits virus replication (62), our data support the importance of IFNα signaling in activating mA3-based anti-viral response in the host.
Although, IFNαR is critical for enhancing mA3 mRNA levels by IFNα or IFNβ in vivo, LPS is capable of enhancing mA3 mRNA in the absence of IFNαR but through TLR4. Upon TLR4 engagement by LPS, level of mA3 mRNA in IFNαR−/− mice increased albeit lower than the levels in WT counterparts. The LPS-induced TLR4 activation inhibitor Polymyxin B blocked mA3 mRNA induction in IFNαR−/− and WT cells, further supporting that TLR4 engagement is crucial for mA3 transcription in the absence of IFNαR. The identification of an IFNαR independent pathway of mA3 mRNA induction is of significance, particularly in the context of viral infection. Most viruses are known to evade the host immune response by blocking IFNα-mediated anti-viral response (81–84). In this regard, our finding reveals that mA3 anti-viral response could be activated through a different pathway other than the IFNαR pathway, such as the TLR4 pathway to restrict virus replication and contain virus induced pathologies. Additionally, the importance of LPS/TLR4 mediated mA3 mRNA induction cannot be overemphasized. Endogenous LPS produced by bacteria in the gut could help in maintaining basal mA3 mRNA in the host, which in turn could contribute in shaping the host innate anti-viral response.
We found that the TLR4/TRIF pathway mediates LPS signal and leads to the transcription of mA3 gene since addition of LPS to TRIF−/− cells failed to induce mA3 mRNA compared to levels seen in WT cells. Since TRIF is involved in regulating mA3 transcription, it is possible that TLR3 may also mediate mA3 mRNA induction. Moreover, ligands of TLR3 including viral RNA (85) may equally activate the TLR3-TRIF, RIG-I, or MDA-5 pathway and induce mA3 transcription to mediate viral clearance. Indeed, different RNA species have been shown to induce A3G mRNA in A549 cells (86). Although we found complete dependence of LPS mediated mA3 mRNA induction on the TLR4 adaptor protein TRIF, the impact of the classical TLR4 adaptor MyD88 cannot be over looked. While loss of TRIF results in absolute loss of LPS but not IFNα mediated mA3 mRNA induction, loss of MyD88 had no effect on mA3 mRNA induction by both LPS and IFNα. LPS•TLR4•MyD88 induced mA3 mRNA could be facilitated by pro-inflammatory cytokines, such as TNF-α and IL1 which have been shown to induce human A3G (87). Indeed, we discovered that the Mitogen-activated protein kinases (MAPKs) play central role in regulating mA3 mRNA induction by IFNα and LPS as has been shown for other genes (88–90). Depending on the cell type, stimulant and the target gene, MAPKs could up-regulate (91–93), down-regulate (94–96), or have no effect on regulation of gene expression (97–98).
Pharmacological inhibition of MAPK family members ERK, JNK, and P38 augmented IFNα and LPS-mediated mA3 mRNA expression in BMDM. Although MAPK-dependent IFNα-mediated augmentation of mA3 mRNA requires IFNαR, LPS enhancement of mA3 mRNA upon inhibition of MAPK is dependent on MyD88 but independent of IFNαR. Our data is in line with other published reports that showed the importance of MyD88 in MAPK dependent gene expression (47,74–75). Our finding that inhibition of MAPK family members – ERK, JNK, and P38 in macrophages enhances mA3 mRNA in this cell type is significant especially since activating the MAPK genes could lead to cancer (99–101) or negatively impact the outcome of viral infection, including infection with HIV-1 (102–105).
Downstream of TRIF is the transcription factor IRF3. IRF3 activates host anti-viral programs including ISG54 and ISG56, in an IFN-dependent and -independent manner (106). Our finding that induction of mA3 mRNA expression by LPS is dependent on IRF3 adds mA3 to the list of genes that are regulated by IRF3. Importantly, the fact that IFNα mediated mA3 mRNA induction does not require IRF3 further demonstrates that IFNα and LPS use different signaling mechanisms to regulate mA3 expression.
The involvement of TLR4 in host immune response to viral infections and virus-induced products has been demonstrated. For example, polymorphisms in TLR4 gene have been implicated in susceptibilities to HIV-1 infection (107). Whether these polymorphisms modulate A3 mRNA levels in the host is unknown. In addition, it remains to be tested if mA3 induced by either IFNαR or TLR4 are equally potent in virus restriction. To further determine how IFNα•IFNαR or LPS•TLR4•TRIF•IRF3 pathways differentially induced mA3 mRNA; we demonstrated a role for PKC mediated signaling in these pathways. Both LPS and IFNα-mediated induction of mA3 mRNA is sensitive to PKC inhibition in that the PKC inhibitor Rottlerin blocked mA3 transcription. In IRF3−/− cells IFNα but not LPS mediated mA3 gene transcription is PKC dependent, suggesting that IRF3 is upstream of PKC. Our results indicate that PKC positively regulates both IFNα and LPS-mediated mA3 mRNA induction in WT cells. In other systems, IFNα was shown to up-regulate the expression of TLR signaling molecules MyD88 (108) and engagement of TLR4 directly activate STAT1 in a PKC dependent manner (109), leading to the transcription of ISGs. Down-regulation of PKC leads to decreased activation of NFκB and secretion of TLR-mediated cytokines (110–111).
In agreement with previous studies on A3G, PKC has been shown to be involved in A3G induction (25,69). We therefore extended these findings as we show that IFNα and LPS differentially regulate mRNA levels of all 7 human A3 genes tested. While PKC may act as a positive regulator of IFNα induced A3A, A3F, A3G and LPS induced A3F mRNA in a time-dependent manner, this kinase negatively regulates LPS-induced mRNAs of A3A and A3G, again in a time-dependent manner. These data further indicates that the induction of different human A3 proteins is controlled by different signaling events. Undoubtedly, additional studies are required to establish the precise regulatory signaling molecules and pathways that regulate expression and anti-viral potency of different A3 proteins.
Natural ligands for TLR4 that may lead to IFNα production include microbial products (112–117). Elucidation of the molecular mechanisms of IRF3-mediated downstream signaling events is crucial for understanding TLR mediated biological effects on mA3 mRNA. Whether the IFNαR and TLR4 pathways mediate mA3 mRNA in the host simultaneously remains to be tested. Synergistic induction of mA3 mRNA using PKC pathway by both IFNα and TLR4 ligands in vivo may be a more efficient way for the host to effectively contain viral infection, irrespective of which signaling pathway the virus activates or represses. Importantly, we identified MAPK family members as a pathway that could be blocked to enhance mA3 mRNA expression. Our data are consistent with receptor mediated gene regulation involving IFNαR or TLR4 initiated signaling events. Based on our data we propose a model (Figure 11) for mA3 mRNA induction via TLR4 and IFNαR. In bone marrow derived MΦs, IFNα receptor is necessary for optimal expression and IFNα-mediated induction of mA3 mRNA. IFNα-mediated mA3 mRNA enhancement requires IFNαR and PKC. Pharmacological inhibition of PKC with Rottlerin suppressed IFNα-mediated STAT1, STAT2, and mA3 mRNA induction, while specific inhibitors of ERK, P38, and JNK results in enhancement of IFNαR mediated IFNα induction of mA3 mRNA. Additionally, ligand-activation of TLR4 induces mA3 mRNA through the TRIF•IRF3•PKC pathway. In IFNαR sufficient cells, PKC positively regulates LPS mediated mA3 mRNA induction. Induction of mA3 could also occur through LPS activation of TLR4-mediated IFNβ production in a paracrine manner. Induction of mA3 by TLR4•LPS is negatively regulated by pharmacological inhibitors of ERK, P38, and JNK. These MAP kinases regulate mA3 mRNA induction by LPS via MyD88 independent of IFNαR.
Figure 11. Diagram illustrating the proposed signaling pathways that lead to IFNα or LPS-mediated mA3 mRNA induction.

In bone marrow derived MΦs, IFNα receptor is necessary for optimal expression and IFNα-mediated induction of mA3 mRNA. IFNα-mediated mA3 mRNA enhancement requires IFNαR, PKC and STAT1. Pharmacological inhibition of PKC with Rottlerin suppressed IFNα-mediated STAT1 and mA3 mRNA induction, while specific inhibitors of ERK, P38, and JNK results in enhancement of IFNαR mediated IFNα induction of mA3 mRNA. Additionally, ligand-activation of TLR4 induces mA3 mRNA through the TLR4•TRIF•IRF3•PKC pathway. In IFNαR sufficient cells, PKC positively regulates LPS mediated mA3 mRNA induction. Induction of mA3 could also occur through LPS activation of TLR4-mediated IFNβ production in a paracrine manner. Induction of mA3 by TLR4•LPS is negatively regulated by pharmacological inhibitors of ERK, P38, and JNK. These MAP kinases regulate mA3 mRNA induction by LPS via MyD88 independent of IFNαR. PD 98059, SP 600125, SB 203580, and Rottlerin respectively inhibit ERK, JNK, P38, and PKC in that order.
Evidently, our data demonstrate that two different signaling pathways are involved in IFNα and LPS mediated A3 mRNA induction (Figure 11). Clearly, differences abound in mA3 transcription programs initiated by IFNα or LPS. IFNα-induced mA3 is a delayed-early gene whose induction requires the synthesis of a new protein product but not mRNA. In contrast, LPS induced mA3 is an immediate-early gene and induction of mA3 mRNA by LPS requires de novo mRNA but not protein synthesis. These findings point to mA3 as a gene that is expressed in both immediate and delayed phases depending on the stimulus. The ability of mA3 to respond either as immediate or delayed early gene may have bestowed upon mA3 the ability to rapidly and persistently respond to the presence of viruses or virus induced inflammatory products.
Although, we identified some signaling targets that are involved in mA3 mRNA induction (Figure 11), it is worth noting that other signaling pathways may also exist. In our study, we used mice and cells genetically modified for different genes to illustrate an absolute requirement for IFNαR•PKC•STAT1 or TLR4•TRIF•IRF3•PKC mediated positive regulation and IFNαR•MAPKs or TLR4•MyD88•MAPKs negative regulation of mA3 gene transcription. The dependency of IFNα and LPS on PKC means that there could be an overlapping pathway for mA3 induction by these two stimulants. In the absence of IFNαR, LPS mediated mA3 mRNA induction is independent of PKC. However, in the presence of IFNαR, LPS mediated mA3 mRNA is PKC dependent.
Finally, since viruses including HIV-1 (118–119) have the capability to disrupt innate immune signaling pathways especially that of the IFN system, our study therefore identifies an alternative pathway that links innate recognition to host restriction factor induction. Further studies will examine the molecular basis underlying TLR4•TRIF•IRF3•PKC and TLR4•MyD88•MAPKs regulation of A3. Our findings may therefore have important implications for the development of new therapeutic strategies that will involve direct targeting, activation, or inhibition of identified targets to increase the level of intracellular A3 and enhance inhibition of virus replication.
Acknowledgments
We sincerely thank Dr. John Harty of University of Iowa for providing us with IFNAR null mice and Dr. Kate Fitzgerald of the University of Massachusetts Medical School for kindly providing the IRF3 cells. We are also grateful to Drs. Jeffery Meier and, Jinxiang Yuan of University of Iowa for providing us with MAPK inhibitors. Radiation experiments were made possible by the Radiation and Free Radical Research Core Facility of the University of Iowa and Holden Comprehensive Cancer Center. Our unreserved gratitude goes to Bryson Okeoma for critically reading this manuscript.
Footnotes
This work was supported by NIAID K22 AI 81899-01 grant to CMO.
References
- 1.Bishop KN, Holmes RK, Sheehy AM, Davidson NO, Cho SJ, Malim MH. Cytidine deamination of retroviral DNA by diverse APOBEC proteins. Curr Biol. 2004;14:1392–1396. doi: 10.1016/j.cub.2004.06.057. [DOI] [PubMed] [Google Scholar]
- 2.Okeoma CM, Lovsin N, Peterlin BM, Ross SR. APOBEC3 inhibits mouse mammary tumour virus replication in vivo. Nature. 2007;445:927–930. doi: 10.1038/nature05540. [DOI] [PubMed] [Google Scholar]
- 3.Low A, Okeoma CM, Lovsin N, de las Heras M, Taylor TH, Peterlin BM, Ross SR, Fan H. Enhanced replication and pathogenesis of Moloney murine leukemia virus in mice defective in the murine APOBEC3 gene. Virology. 2009;385:455–463. doi: 10.1016/j.virol.2008.11.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen H, Lilley CE, Yu Q, Lee DV, Chou J, Narvaiza I, Landau NR, Weitzman MD. APOBEC3A is a potent inhibitor of adeno-associated virus and retrotransposons. Curr Biol. 2006;16:480–485. doi: 10.1016/j.cub.2006.01.031. [DOI] [PubMed] [Google Scholar]
- 5.Holmes RK, Koning FA, Bishop KN, Malim MH. APOBEC3F can inhibit the accumulation of HIV-1 reverse transcription products in the absence of hypermutation. Comparisons with APOBEC3G. J Biol Chem. 2007;282:2587–2595. doi: 10.1074/jbc.M607298200. [DOI] [PubMed] [Google Scholar]
- 6.Muckenfuss H, Hamdorf M, Held U, Perkovic M, Lower J, Cichutek K, Flory E, Schumann GG, Munk C. APOBEC3 proteins inhibit human LINE-1 retrotransposition. J Biol Chem. 2006;281:22161–22172. doi: 10.1074/jbc.M601716200. [DOI] [PubMed] [Google Scholar]
- 7.OhAinle M, Kerns JA, Li MM, Malik HS, Emerman M. Antiretroelement activity of APOBEC3H was lost twice in recent human evolution. Cell Host Microbe. 2008;4:249–259. doi: 10.1016/j.chom.2008.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tan L, Sarkis PT, Wang T, Tian C, Yu XF. Sole copy of Z2-type human cytidine deaminase APOBEC3H has inhibitory activity against retrotransposons and HIV-1. FASEB J. 2009;23:279–287. doi: 10.1096/fj.07-088781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhen A, Wang T, Zhao K, Xiong Y, Yu XF. A single amino acid difference in human APOBEC3H variants determines HIV-1 Vif sensitivity. J Virol. 2010;84:1902–1911. doi: 10.1128/JVI.01509-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Su AI, Wiltshire T, Batalov S, Lapp H, Ching KA, Block D, Zhang J, Soden R, Hayakawa M, Kreiman G, Cooke MP, Walker JR, Hogenesch JB. A gene atlas of the mouse and human protein-encoding transcriptomes. Proc Natl Acad Sci U S A. 2004;101:6062–6067. doi: 10.1073/pnas.0400782101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Conticello SG, Harris RS, Neuberger MS. The Vif protein of HIV triggers degradation of the human antiretroviral DNA deaminase APOBEC3G. Curr Biol. 2003;13:2009–2013. doi: 10.1016/j.cub.2003.10.034. [DOI] [PubMed] [Google Scholar]
- 12.Marin M, Rose KM, Kozak SL, Kabat D. HIV-1 Vif protein binds the editing enzyme APOBEC3G and induces its degradation. Nat Med. 2003;9:1398–1403. doi: 10.1038/nm946. [DOI] [PubMed] [Google Scholar]
- 13.Sheehy AM, Gaddis NC, Malim MH. The antiretroviral enzyme APOBEC3G is degraded by the proteasome in response to HIV-1 Vif. Nat Med. 2003;9:1404–1407. doi: 10.1038/nm945. [DOI] [PubMed] [Google Scholar]
- 14.Yu X, Yu Y, Liu B, Luo K, Kong W, Mao P, Yu XF. Induction of APOBEC3G ubiquitination and degradation by an HIV-1 Vif-Cul5-SCF complex. Science. 2003;302:1056–1060. doi: 10.1126/science.1089591. [DOI] [PubMed] [Google Scholar]
- 15.Stopak K, de Noronha C, Yonemoto W, Greene WC. HIV-1 Vif blocks the antiviral activity of APOBEC3G by impairing both its translation and intracellular stability. Mol Cell. 2003;12:591–601. doi: 10.1016/s1097-2765(03)00353-8. [DOI] [PubMed] [Google Scholar]
- 16.Mehle A, Strack B, Ancuta P, Zhang C, McPike M, Gabuzda D. Vif overcomes the innate antiviral activity of APOBEC3G by promoting its degradation in the ubiquitin-proteasome pathway. J Biol Chem. 2004;279:7792–7798. doi: 10.1074/jbc.M313093200. [DOI] [PubMed] [Google Scholar]
- 17.Yu Y, Xiao Z, Ehrlich ES, Yu X, Yu XF. Selective assembly of HIV-1 Vif-Cul5-ElonginB-ElonginC E3 ubiquitin ligase complex through a novel SOCS box and upstream cysteines. Genes Dev. 2004;18:2867–2872. doi: 10.1101/gad.1250204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Harris RS, Bishop KN, Sheehy AM, Craig HM, Petersen-Mahrt SK, Watt IN, Neuberger MS, Malim MH. DNA deamination mediates innate immunity to retroviral infection. Cell. 2003;113:803–809. doi: 10.1016/s0092-8674(03)00423-9. [DOI] [PubMed] [Google Scholar]
- 19.Lecossier D, Bouchonnet F, Clavel F, Hance AJ. Hypermutation of HIV-1 DNA in the absence of the Vif protein. Science. 2003;300:1112. doi: 10.1126/science.1083338. [DOI] [PubMed] [Google Scholar]
- 20.Mangeat B, Turelli P, Caron G, Friedli M, Perrin L, Trono D. Broad antiretroviral defence by human APOBEC3G through lethal editing of nascent reverse transcripts. Nature. 2003;424:99–103. doi: 10.1038/nature01709. [DOI] [PubMed] [Google Scholar]
- 21.Zhang H, Yang B, Pomerantz RJ, Zhang C, Arunachalam SC, Gao L. The cytidine deaminase CEM15 induces hypermutation in newly synthesized HIV-1 DNA. Nature. 2003;424:94–98. doi: 10.1038/nature01707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yu Q, Chen D, Konig R, Mariani R, Unutmaz D, Landau NR. APOBEC3B and APOBEC3C are potent inhibitors of simian immunodeficiency virus replication. J Biol Chem. 2004;279:53379–53386. doi: 10.1074/jbc.M408802200. [DOI] [PubMed] [Google Scholar]
- 23.Okeoma CM, Huegel AL, Lingappa J, Feldman MD, Ross SR. APOBEC3 proteins expressed in mammary epithelial cells are packaged into retroviruses and can restrict transmission of milk-borne virions. Cell Host Microbe. 2010;8:534–543. doi: 10.1016/j.chom.2010.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Peng G, Lei KJ, Jin W, Greenwell-Wild T, Wahl SM. Induction of APOBEC3 family proteins, a defensive maneuver underlying interferon-induced anti-HIV-1 activity. J Exp Med. 2006;203:41–46. doi: 10.1084/jem.20051512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sarkis PT, Ying S, Xu R, Yu XF. STAT1-independent cell type-specific regulation of antiviral APOBEC3G by IFN-alpha. J Immunol. 2006;177:4530–4540. doi: 10.4049/jimmunol.177.7.4530. [DOI] [PubMed] [Google Scholar]
- 26.Stopak KS, Chiu YL, Kropp J, Grant RM, Greene WC. Distinct patterns of cytokine regulation of APOBEC3G expression and activity in primary lymphocytes, macrophages, and dendritic cells. J Biol Chem. 2007;282:3539–3546. doi: 10.1074/jbc.M610138200. [DOI] [PubMed] [Google Scholar]
- 27.Ying S, Zhang X, Sarkis PT, Xu R, Yu X. Cell-specific regulation of APOBEC3F by interferons. Acta Biochim Biophys Sin (Shanghai) 2007;39:297–304. doi: 10.1111/j.1745-7270.2007.00275.x. [DOI] [PubMed] [Google Scholar]
- 28.Koning FA, Newman EN, Kim EY, Kunstman KJ, Wolinsky SM, Malim MH. Defining APOBEC3 expression patterns in human tissues and hematopoietic cell subsets. J Virol. 2009;83:9474–9485. doi: 10.1128/JVI.01089-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Okeoma CM, Low A, Bailis W, Fan HY, Peterlin BM, Ross SR. Induction of APOBEC3 in vivo causes increased restriction of retrovirus infection. J Virol. 2009;83:3486–3495. doi: 10.1128/JVI.02347-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bonvin M, Achermann F, Greeve I, Stroka D, Keogh A, Inderbitzin D, Candinas D, Sommer P, Wain-Hobson S, Vartanian JP, Greeve J. Interferon-inducible expression of APOBEC3 editing enzymes in human hepatocytes and inhibition of hepatitis B virus replication. Hepatology. 2006;43:1364–1374. doi: 10.1002/hep.21187. [DOI] [PubMed] [Google Scholar]
- 31.Zhang W, Zhang X, Tian C, Wang T, Sarkis PT, Fang Y, Zheng S, Yu XF, Xu R. Cytidine deaminase APOBEC3B interacts with heterogeneous nuclear ribonucleoprotein K and suppresses hepatitis B virus expression. Cell Microbiol. 2008;10:112–121. doi: 10.1111/j.1462-5822.2007.01020.x. [DOI] [PubMed] [Google Scholar]
- 32.Xu R, Zhang X, Zhang W, Fang Y, Zheng S, Yu XF. Association of human APOBEC3 cytidine deaminases with the generation of hepatitis virus B x antigen mutants and hepatocellular carcinoma. Hepatology. 2007;46:1810–1820. doi: 10.1002/hep.21893. [DOI] [PubMed] [Google Scholar]
- 33.Pestka S, Langer JA, Zoon KC, Samuel CE. Interferons and their actions. Annu Rev Biochem. 1987;56:727–777. doi: 10.1146/annurev.bi.56.070187.003455. [DOI] [PubMed] [Google Scholar]
- 34.Platanias LC, Uddin S, Colamonici OR. Tyrosine phosphorylation of the alpha and beta subunits of the type I interferon receptor. Interferon-beta selectively induces tyrosine phosphorylation of an alpha subunit-associated protein. J Biol Chem. 1994;269:17761–17764. [PubMed] [Google Scholar]
- 35.Darnell JE, Jr, Kerr IM, Stark GR. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science. 1994;264:1415–1421. doi: 10.1126/science.8197455. [DOI] [PubMed] [Google Scholar]
- 36.Qureshi SA, Salditt-Georgieff M, Darnell JE., Jr Tyrosine-phosphorylated Stat1 and Stat2 plus a 48-kDa protein all contact DNA in forming interferon-stimulated-gene factor 3. Proc Natl Acad Sci U S A. 1995;92:3829–3833. doi: 10.1073/pnas.92.9.3829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Akira S, Sato S. Toll-like receptors and their signaling mechanisms. Scand J Infect Dis. 2003;35:555–562. doi: 10.1080/00365540310015683. [DOI] [PubMed] [Google Scholar]
- 38.Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X, Birdwell D, Alejos E, Silva M, Galanos C, Freudenberg M, Ricciardi-Castagnoli P, Layton B, Beutler B. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282:2085–2088. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- 39.Yamamoto M, Sato S, Hemmi H, Sanjo H, Uematsu S, Kaisho T, Hoshino K, Takeuchi O, Kobayashi M, Fujita T, Takeda K, Akira S. Essential role for TIRAP in activation of the signalling cascade shared by TLR2 and TLR4. Nature. 2002;420:324–329. doi: 10.1038/nature01182. [DOI] [PubMed] [Google Scholar]
- 40.May MJ, Ghosh S. Signal transduction through NF-kappa B. Immunol Today. 1998;19:80–88. doi: 10.1016/s0167-5699(97)01197-3. [DOI] [PubMed] [Google Scholar]
- 41.Doyle S, Vaidya S, O’Connell R, Dadgostar H, Dempsey P, Wu T, Rao G, Sun R, Haberland M, Modlin R, Cheng G. IRF3 mediates a TLR3/TLR4-specific antiviral gene program. Immunity. 2002;17:251–263. doi: 10.1016/s1074-7613(02)00390-4. [DOI] [PubMed] [Google Scholar]
- 42.Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol. 2004;4:499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- 43.Uddin S, Majchrzak B, Woodson J, Arunkumar P, Alsayed Y, Pine R, Young PR, Fish EN, Platanias LC. Activation of the p38 mitogen-activated protein kinase by type I interferons. J Biol Chem. 1999;274:30127–30131. doi: 10.1074/jbc.274.42.30127. [DOI] [PubMed] [Google Scholar]
- 44.Arora T, Floyd-Smith G, Espy MJ, Jelinek DF. Dissociation between IFN-alpha-induced anti-viral and growth signaling pathways. J Immunol. 1999;162:3289–3297. [PubMed] [Google Scholar]
- 45.Zhao LJ, Hua X, He SF, Ren H, Qi ZT. Interferon alpha regulates MAPK and STAT1 pathways in human hepatoma cells. Virol J. 2011;8:157. doi: 10.1186/1743-422X-8-157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chi H, Barry SP, Roth RJ, Wu JJ, Jones EA, Bennett AM, Flavell RA. Dynamic regulation of pro- and anti-inflammatory cytokines by MAPK phosphatase 1 (MKP-1) in innate immune responses. Proc Natl Acad Sci U S A. 2006;103:2274–2279. doi: 10.1073/pnas.0510965103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dong C, Davis RJ, Flavell RA. MAP kinases in the immune response. Annu Rev Immunol. 2002;20:55–72. doi: 10.1146/annurev.immunol.20.091301.131133. [DOI] [PubMed] [Google Scholar]
- 48.Dumitru CD, Ceci JD, Tsatsanis C, Kontoyiannis D, Stamatakis K, Lin JH, Patriotis C, Jenkins NA, Copeland NG, Kollias G, Tsichlis PN. TNF-alpha induction by LPS is regulated posttranscriptionally via a Tpl2/ERK-dependent pathway. Cell. 2000;103:1071–1083. doi: 10.1016/s0092-8674(00)00210-5. [DOI] [PubMed] [Google Scholar]
- 49.Chi H, Flavell RA. Acetylation of MKP-1 and the control of inflammation. Sci Signal. 2008;1:pe44. doi: 10.1126/scisignal.141pe44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Johnson GL, Lapadat R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science. 2002;298:1911–1912. doi: 10.1126/science.1072682. [DOI] [PubMed] [Google Scholar]
- 51.Lawrence MC, McGlynn K, Naziruddin B, Levy MF, Cobb MH. Differential regulation of CHOP-10/GADD153 gene expression by MAPK signaling in pancreatic beta-cells. Proc Natl Acad Sci U S A. 2007;104:11518–11525. doi: 10.1073/pnas.0704618104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Guo B, Inoki K, Isono M, Mori H, Kanasaki K, Sugimoto T, Akiba S, Sato T, Yang B, Kikkawa R, Kashiwagi A, Haneda M, Koya D. MAPK/AP-1-dependent regulation of PAI-1 gene expression by TGF-beta in rat mesangial cells. Kidney Int. 2005;68:972–984. doi: 10.1111/j.1523-1755.2005.00491.x. [DOI] [PubMed] [Google Scholar]
- 53.Kreisberg JF, Yonemoto W, Greene WC. Endogenous factors enhance HIV infection of tissue naive CD4 T cells by stimulating high molecular mass APOBEC3G complex formation. J Exp Med. 2006;203:865–870. doi: 10.1084/jem.20051856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chen H, Wang LW, Huang YQ, Gong ZJ. Interferon-alpha Induces High Expression of APOBEC3G and STAT-1 in Vitro and in Vivo. Int J Mol Sci. 2010;11:3501–3512. doi: 10.3390/ijms11093501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Roberson SM, Walker WS. Immortalization of cloned mouse splenic macrophages with a retrovirus containing the v-raf/mil and v-myc oncogenes. Cell Immunol. 1988;116:341–351. doi: 10.1016/0008-8749(88)90236-5. [DOI] [PubMed] [Google Scholar]
- 56.Mehta H, Glogauer M, Becart S, Altman A, Coggeshall KM. Adaptor protein SLAT modulates Fcgamma receptor-mediated phagocytosis in murine macrophages. J Biol Chem. 2009;284:11882–11891. doi: 10.1074/jbc.M809712200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hammer KD, Yum MY, Dixon PM, Birt DF. Identification of JAK-STAT pathways as important for the anti-inflammatory activity of a Hypericum perforatum fraction and bioactive constituents in RAW 264.7 mouse macrophages. Phytochemistry. 2010;71:716–725. doi: 10.1016/j.phytochem.2010.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Khorooshi R, Babcock AA, Owens T. NF-kappaB-driven STAT2 and CCL2 expression in astrocytes in response to brain injury. J Immunol. 2008;181:7284–7291. doi: 10.4049/jimmunol.181.10.7284. [DOI] [PubMed] [Google Scholar]
- 59.Okeoma CM, Shen M, Ross SR. A novel block to mouse mammary tumor virus infection of lymphocytes in B10. BR mice. J Virol. 2008;82:1314–1322. doi: 10.1128/JVI.01848-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jones PH, Mehta HV, Maric M, Roller RJ, Okeoma CM. Bone marrow stromal cell antigen 2 (BST-2) restricts mouse mammary tumor virus (MMTV) replication in vivo. Retrovirology. 2012;9:10. doi: 10.1186/1742-4690-9-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jones PH, Mehta HV, Okeoma CM. A novel role for APOBEC3: Susceptibility to sexual transmission of murine acquired immunodeficiency virus (mAIDS) is aggravated in APOBEC3 deficient mice. Retrovirology. 2012;9:50. doi: 10.1186/1742-4690-9-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Okeoma CM, Petersen J, Ross SR. Expression of murine APOBEC3 alleles in different mouse strains and their effect on mouse mammary tumor virus infection. J Virol. 2009;83:3029–3038. doi: 10.1128/JVI.02536-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Muller U, Steinhoff U, Reis LF, Hemmi S, Pavlovic J, Zinkernagel RM, Aguet M. Functional role of type I and type II interferons in antiviral defense. Science. 1994;264:1918–1921. doi: 10.1126/science.8009221. [DOI] [PubMed] [Google Scholar]
- 64.Auerbuch V, Brockstedt DG, Meyer-Morse N, O’Riordan M, Portnoy DA. Mice lacking the type I interferon receptor are resistant to Listeria monocytogenes. J Exp Med. 2004;200:527–533. doi: 10.1084/jem.20040976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Oshiumi H, Matsumoto M, Funami K, Akazawa T, Seya T. TICAM-1, an adaptor molecule that participates in Toll-like receptor 3-mediated interferon-beta induction. Nat Immunol. 2003;4:161–167. doi: 10.1038/ni886. [DOI] [PubMed] [Google Scholar]
- 66.Yamamoto M, Sato S, Mori K, Hoshino K, Takeuchi O, Takeda K, Akira S. Cutting edge: a novel Toll/IL-1 receptor domain-containing adapter that preferentially activates the IFN-beta promoter in the Toll-like receptor signaling. J Immunol. 2002;169:6668–6672. doi: 10.4049/jimmunol.169.12.6668. [DOI] [PubMed] [Google Scholar]
- 67.Kawamoto T, Ii M, Kitazaki T, Iizawa Y, Kimura H. TAK-242 selectively suppresses Toll-like receptor 4-signaling mediated by the intracellular domain. Eur J Pharmacol. 2008;584:40–48. doi: 10.1016/j.ejphar.2008.01.026. [DOI] [PubMed] [Google Scholar]
- 68.Lanford RE, Guerra B, Lee H, Chavez D, Brasky KM, Bigger CB. Genomic response to interferon-alpha in chimpanzees: implications of rapid downregulation for hepatitis C kinetics. Hepatology. 2006;43:961–972. doi: 10.1002/hep.21167. [DOI] [PubMed] [Google Scholar]
- 69.Rose KM, Marin M, Kozak SL, Kabat D. Transcriptional regulation of APOBEC3G, a cytidine deaminase that hypermutates human immunodeficiency virus. J Biol Chem. 2004;279:41744–41749. doi: 10.1074/jbc.M406760200. [DOI] [PubMed] [Google Scholar]
- 70.Kikkawa U, Ogita K, Ono Y, Asaoka Y, Shearman MS, Fujii T, Ase K, Sekiguchi K, Igarashi K, Nishizuka Y. The common structure and activities of four subspecies of rat brain protein kinase C family. FEBS Lett. 1987;223:212–216. doi: 10.1016/0014-5793(87)80291-0. [DOI] [PubMed] [Google Scholar]
- 71.Carpenter D, Jackson T, Hanley MR. Protein kinase Cs. Coping with a growing family. Nature. 1987;325:107–108. doi: 10.1038/325107a0. [DOI] [PubMed] [Google Scholar]
- 72.Gschwendt M, Muller HJ, Kielbassa K, Zang R, Kittstein W, Rincke G, Marks F. Rottlerin, a novel protein kinase inhibitor. Biochem Biophys Res Commun. 1994;199:93–98. doi: 10.1006/bbrc.1994.1199. [DOI] [PubMed] [Google Scholar]
- 73.Shoenfelt JL, Fenton MJ. TLR2- and TLR4-dependent activation of STAT1 serine phosphorylation in murine macrophages is protein kinase C-delta-independent. J Endotoxin Res. 2006;12:231–240. doi: 10.1179/096805106X102219. [DOI] [PubMed] [Google Scholar]
- 74.Takeda K, Kaisho T, Akira S. Toll-like receptors. Annu Rev Immunol. 2003;21:335–376. doi: 10.1146/annurev.immunol.21.120601.141126. [DOI] [PubMed] [Google Scholar]
- 75.Gorina R, Font-Nieves M, Marquez-Kisinousky L, Santalucia T, Planas AM. Astrocyte TLR4 activation induces a proinflammatory environment through the interplay between MyD88-dependent NFkappaB signaling, MAPK, and Jak1/Stat1 pathways. Glia. 2011;59:242–255. doi: 10.1002/glia.21094. [DOI] [PubMed] [Google Scholar]
- 76.Shepherd EG, Zhao Q, Welty SE, Hansen TN, Smith CV, Liu Y. The function of mitogen-activated protein kinase phosphatase-1 in peptidoglycan-stimulated macrophages. J Biol Chem. 2004;279:54023–54031. doi: 10.1074/jbc.M408444200. [DOI] [PubMed] [Google Scholar]
- 77.Pion M, Granelli-Piperno A, Mangeat B, Stalder R, Correa R, Steinman RM, Piguet V. APOBEC3G/3F mediates intrinsic resistance of monocyte-derived dendritic cells to HIV-1 infection. J Exp Med. 2006;203:2887–2893. doi: 10.1084/jem.20061519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Baca-Regen L, Heinzinger N, Stevenson M, Gendelman HE. Alpha interferon-induced antiretroviral activities: restriction of viral nucleic acid synthesis and progeny virion production in human immunodeficiency virus type 1-infected monocytes. J Virol. 1994;68:7559–7565. doi: 10.1128/jvi.68.11.7559-7565.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Krown SE, Lee JY, Lin L, Fischl MA, Ambinder R, Von Roenn JH. Interferon-alpha2b with protease inhibitor-based antiretroviral therapy in patients with AIDS-associated Kaposi sarcoma: an AIDS malignancy consortium phase I trial. J Acquir Immune Defic Syndr. 2006;41:149–153. doi: 10.1097/01.qai.0000194237.15831.23. [DOI] [PubMed] [Google Scholar]
- 80.Goujon C, Malim MH. Characterization of the alpha interferon-induced postentry block to HIV-1 infection in primary human macrophages and T cells. J Virol. 2010;84:9254–9266. doi: 10.1128/JVI.00854-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Colamonici OR, Domanski P, Sweitzer SM, Larner A, Buller RM. Vaccinia virus B18R gene encodes a type I interferon-binding protein that blocks interferon alpha transmembrane signaling. J Biol Chem. 1995;270:15974–15978. doi: 10.1074/jbc.270.27.15974. [DOI] [PubMed] [Google Scholar]
- 82.Basler CF, Mikulasova A, Martinez-Sobrido L, Paragas J, Muhlberger E, Bray M, Klenk HD, Palese P, Garcia-Sastre A. The Ebola virus VP35 protein inhibits activation of interferon regulatory factor 3. J Virol. 2003;77:7945–7956. doi: 10.1128/JVI.77.14.7945-7956.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Foy E, Li K, Wang C, Sumpter R, Jr, Ikeda M, Lemon SM, Gale M., Jr Regulation of interferon regulatory factor-3 by the hepatitis C virus serine protease. Science. 2003;300:1145–1148. doi: 10.1126/science.1082604. [DOI] [PubMed] [Google Scholar]
- 84.Lin R, Noyce RS, Collins SE, Everett RD, Mossman KL. The herpes simplex virus ICP0 RING finger domain inhibits IRF3- and IRF7-mediated activation of interferon-stimulated genes. J Virol. 2004;78:1675–1684. doi: 10.1128/JVI.78.4.1675-1684.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature. 2001;413:732–738. doi: 10.1038/35099560. [DOI] [PubMed] [Google Scholar]
- 86.Pauli EK, Schmolke M, Hofmann H, Ehrhardt C, Flory E, Munk C, Ludwig S. High level expression of the anti-retroviral protein APOBEC3G is induced by influenza A virus but does not confer antiviral activity. Retrovirology. 2009;6:38. doi: 10.1186/1742-4690-6-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wang YJ, Wang X, Zhang H, Zhou L, Liu S, Kolson DL, Song L, Ye L, Ho WZ. Expression and regulation of antiviral protein APOBEC3G in human neuronal cells. J Neuroimmunol. 2009;206:14–21. doi: 10.1016/j.jneuroim.2008.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Guan Z, Buckman SY, Springer LD, Morrison AR. Both p38alpha(MAPK) and JNK/SAPK pathways are important for induction of nitric-oxide synthase by interleukin-1beta in rat glomerular mesangial cells. J Biol Chem. 1999;274:36200–36206. doi: 10.1074/jbc.274.51.36200. [DOI] [PubMed] [Google Scholar]
- 89.Hua LL, Zhao ML, Cosenza M, Kim MO, Huang H, Tanowitz HB, Brosnan CF, Lee SC. Role of mitogen-activated protein kinases in inducible nitric oxide synthase and TNFalpha expression in human fetal astrocytes. J Neuroimmunol. 2002;126:180–189. doi: 10.1016/s0165-5728(02)00055-3. [DOI] [PubMed] [Google Scholar]
- 90.Lahti A, Jalonen U, Kankaanranta H, Moilanen E. c-Jun NH2-terminal kinase inhibitor anthra(1,9-cd)pyrazol-6(2H)-one reduces inducible nitric-oxide synthase expression by destabilizing mRNA in activated macrophages. Mol Pharmacol. 2003;64:308–315. doi: 10.1124/mol.64.2.308. [DOI] [PubMed] [Google Scholar]
- 91.Chen C, Chen YH, Lin WW. Involvement of p38 mitogen-activated protein kinase in lipopolysaccharide-induced iNOS and COX-2 expression in J774 macrophages. Immunology. 1999;97:124–129. doi: 10.1046/j.1365-2567.1999.00747.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Chen CC, Wang JK. p38 but not p44/42 mitogen-activated protein kinase is required for nitric oxide synthase induction mediated by lipopolysaccharide in RAW 264.7 macrophages. Mol Pharmacol. 1999;55:481–488. [PubMed] [Google Scholar]
- 93.Bhat NR, Feinstein DL, Shen Q, Bhat AN. p38 MAPK-mediated transcriptional activation of inducible nitric-oxide synthase in glial cells. Roles of nuclear factors, nuclear factor kappa B, cAMP response element-binding protein, CCAAT/enhancer-binding protein-beta, and activating transcription factor-2. J Biol Chem. 2002;277:29584–29592. doi: 10.1074/jbc.M204994200. [DOI] [PubMed] [Google Scholar]
- 94.Guan Z, Baier LD, Morrison AR. p38 mitogen-activated protein kinase down-regulates nitric oxide and up-regulates prostaglandin E2 biosynthesis stimulated by interleukin-1beta. J Biol Chem. 1997;272:8083–8089. doi: 10.1074/jbc.272.12.8083. [DOI] [PubMed] [Google Scholar]
- 95.Chan ED, Riches DW. IFN-gamma + LPS induction of iNOS is modulated by ERK, JNK/SAPK, and p38(mapk) in a mouse macrophage cell line. Am J Physiol Cell Physiol. 2001;280:C441–450. doi: 10.1152/ajpcell.2001.280.3.C441. [DOI] [PubMed] [Google Scholar]
- 96.Chan ED, Morris KR, Belisle JT, Hill P, Remigio LK, Brennan PJ, Riches DW. Induction of inducible nitric oxide synthase-NO* by lipoarabinomannan of Mycobacterium tuberculosis is mediated by MEK1-ERK, MKK7-JNK, and NF-kappaB signaling pathways. Infect Immun. 2001;69:2001–2010. doi: 10.1128/IAI.69.4.2001-2010.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Chan ED, Winston BW, Uh ST, Wynes MW, Rose DM, Riches DW. Evaluation of the role of mitogen-activated protein kinases in the expression of inducible nitric oxide synthase by IFN-gamma and TNF-alpha in mouse macrophages. J Immunol. 1999;162:415–422. [PubMed] [Google Scholar]
- 98.Cho MK, Suh SH, Kim SG. JunB/AP-1 and NF-kappa B-mediated induction of nitric oxide synthase by bovine type I collagen in serum-stimulated murine macrophages. Nitric Oxide. 2002;6:319–332. doi: 10.1006/niox.2001.0415. [DOI] [PubMed] [Google Scholar]
- 99.Yadav V, Zhang X, Liu J, Estrem S, Li S, Gong XQ, Buchanan S, Henry JR, Starling JJ, Peng SB. Reactivation of Mitogen-Activated Protein Kinase (MAPK) Pathway by FGF Receptor 3 (FGFR3)/Ras Mediates Resistance to Vemurafenib in Human B-RAF V600E Mutant Melanoma. J Biol Chem. 2012 doi: 10.1074/jbc.M112.377218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Su N, Peng L, Xia B, Zhao Y, Xu A, Wang J, Wang X, Jiang B. Lyn is involved in CD24-induced ERK1/2 activation in colorectal cancer. Mol Cancer. 2012;11:43. doi: 10.1186/1476-4598-11-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Massaoka MH, Matsuo AL, Figueiredo CR, Farias CF, Girola N, Arruda DC, Scutti JA, Romoff P, Favero OA, Ferreira MJ, Lago JH, Travassos LR. Jacaranone Induces Apoptosis in Melanoma Cells via ROS-Mediated Downregulation of Akt and p38 MAPK Activation and Displays Antitumor Activity In Vivo. PLoS One. 2012;7:e38698. doi: 10.1371/journal.pone.0038698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Huang W, Hu K, Luo S, Zhang M, Li C, Jin W, Liu Y, Griffin GE, Shattock RJ, Hu Q. Herpes Simplex Virus Type 2 Infection of Human Epithelial Cells Induces CXCL9 Expression and CD4+ T Cell Migration via Activation of p38-CCAAT/Enhancer-Binding Protein-beta Pathway. J Immunol. 2012;188:6247–6257. doi: 10.4049/jimmunol.1103706. [DOI] [PubMed] [Google Scholar]
- 103.Mansouri S, Kutky M, Hudak KA. Pokeweed antiviral protein increases HIV-1 particle infectivity by activating the cellular mitogen activated protein kinase pathway. PLoS One. 2012;7:e36369. doi: 10.1371/journal.pone.0036369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zheng Q, Tao R, Gao H, Xu J, Shang S, Zhao N. HCMV-encoded UL128 enhances TNF-alpha and IL-6 expression and promotes PBMC proliferation through the MAPK/ERK pathway in vitro. Viral Immunol. 2012;25:98–105. doi: 10.1089/vim.2011.0064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Chihara T, Hashimoto M, Osman A, Hiyoshi-Yoshidomi Y, Suzu I, Chutiwitoonchai N, Hiyoshi M, Okada S, Suzu S. HIV-1 proteins preferentially activate anti-inflammatory M2-type macrophages. J Immunol. 2012;188:3620–3627. doi: 10.4049/jimmunol.1101593. [DOI] [PubMed] [Google Scholar]
- 106.Chew T, Noyce R, Collins SE, Hancock MH, Mossman KL. Characterization of the interferon regulatory factor 3-mediated antiviral response in a cell line deficient for IFN production. Mol Immunol. 2009;46:393–399. doi: 10.1016/j.molimm.2008.10.010. [DOI] [PubMed] [Google Scholar]
- 107.Pine SO, McElrath MJ, Bochud PY. Polymorphisms in toll-like receptor 4 and toll-like receptor 9 influence viral load in a seroincident cohort of HIV-1-infected individuals. AIDS. 2009;23:2387–2395. doi: 10.1097/QAD.0b013e328330b489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Siren J, Pirhonen J, Julkunen I, Matikainen S. IFN-alpha regulates TLR-dependent gene expression of IFN-alpha, IFN-beta, IL-28, and IL-29. J Immunol. 2005;174:1932–1937. doi: 10.4049/jimmunol.174.4.1932. [DOI] [PubMed] [Google Scholar]
- 109.Rhee SH, Jones BW, Toshchakov V, Vogel SN, Fenton MJ. Toll-like receptors 2 and 4 activate STAT1 serine phosphorylation by distinct mechanisms in macrophages. J Biol Chem. 2003;278:22506–22512. doi: 10.1074/jbc.M208633200. [DOI] [PubMed] [Google Scholar]
- 110.Bhatt KH, Pandey RK, Dahiya Y, Sodhi A. Protein kinase Cdelta and protein tyrosine kinase regulate peptidoglycan-induced nuclear factor-kappaB activation and inducible nitric oxide synthase expression in mouse peritoneal macrophages in vitro. Mol Immunol. 2010;47:861–870. doi: 10.1016/j.molimm.2009.10.029. [DOI] [PubMed] [Google Scholar]
- 111.Kim DC, Kim SH, Jeong MW, Baek NI, Kim KT. Effect of rottlerin, a PKC-delta inhibitor, on TLR-4-dependent activation of murine microglia. Biochem Biophys Res Commun. 2005;337:110–115. doi: 10.1016/j.bbrc.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 112.Kurt-Jones EA, Popova L, Kwinn L, Haynes LM, Jones LP, Tripp RA, Walsh EE, Freeman MW, Golenbock DT, Anderson LJ, Finberg RW. Pattern recognition receptors TLR4 and CD14 mediate response to respiratory syncytial virus. Nat Immunol. 2000;1:398–401. doi: 10.1038/80833. [DOI] [PubMed] [Google Scholar]
- 113.Haynes LM, Moore DD, Kurt-Jones EA, Finberg RW, Anderson LJ, Tripp RA. Involvement of toll-like receptor 4 in innate immunity to respiratory syncytial virus. J Virol. 2001;75:10730–10737. doi: 10.1128/JVI.75.22.10730-10737.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Rassa JC, Meyers JL, Zhang Y, Kudaravalli R, Ross SR. Murine retroviruses activate B cells via interaction with toll-like receptor 4. Proc Natl Acad Sci U S A. 2002;99:2281–2286. doi: 10.1073/pnas.042355399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Burzyn D, Rassa JC, Kim D, Nepomnaschy I, Ross SR, Piazzon I. Toll-like receptor 4-dependent activation of dendritic cells by a retrovirus. J Virol. 2004;78:576–584. doi: 10.1128/JVI.78.2.576-584.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Otten LA, Finke D, Acha-Orbea H. Can MMTV exploit TLR4? Trends Microbiol. 2002;10:303–305. doi: 10.1016/s0966-842x(02)02385-5. discussion 305–306. [DOI] [PubMed] [Google Scholar]
- 117.Triantafilou K, Triantafilou M. Coxsackievirus B4-induced cytokine production in pancreatic cells is mediated through toll-like receptor 4. J Virol. 2004;78:11313–11320. doi: 10.1128/JVI.78.20.11313-11320.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Medzhitov R, Littman D. HIV immunology needs a new direction. Nature. 2008;455:591. doi: 10.1038/455591a. [DOI] [PubMed] [Google Scholar]
- 119.Doehle BP, Hladik F, McNevin JP, McElrath MJ, Gale M., Jr Human immunodeficiency virus type 1 mediates global disruption of innate antiviral signaling and immune defenses within infected cells. J Virol. 2009;83:10395–10405. doi: 10.1128/JVI.00849-09. [DOI] [PMC free article] [PubMed] [Google Scholar]