

Abstract
Mosaic Variegated Aneuploidy (MVA) syndrome is a rare autosomal recessive disorder characterized by inaccurate chromosome segregation and high rates of near-diploid aneuploidy. Children with MVA syndrome die at an early age, are cancer prone, and have progeroid features like facial dysmorphisms, short stature, and cataracts. The majority of MVA cases are linked to mutations in BUBR1, a mitotic checkpoint gene required for proper chromosome segregation. Affected patients either have bi-allelic BUBR1 mutations, with one allele harboring a missense mutation and the other a nonsense mutation, or mono-allelic BUBR1 mutations combined with allelic variants that yield low amounts of wild-type BubR1 protein. Parents of MVA patients that carry single allele mutations have mild mitotic defects, but whether they are at risk for any of the pathologies associated with MVA syndrome is unknown. To address this, we engineered a mouse model for the nonsense mutation 2211insGTTA (referred to as GTTA) found in MVA patients with bi-allelic BUBR1 mutations. Here we report that both the median and maximum lifespans of the resulting BubR1 +/GTTA mice are significantly reduced. Furthermore, BubR1 +/GTTA mice develop several aging-related phenotypes at an accelerated rate, including cataract formation, lordokyphosis, skeletal muscle wasting, impaired exercise ability, and fat loss. BubR1 +/GTTA mice develop mild aneuploidies and show enhanced growth of carcinogen-induced tumors. Collectively, these data demonstrate that the BUBR1 GTTA mutation compromises longevity and healthspan, raising the interesting possibility that mono-allelic changes in BUBR1 might contribute to differences in aging rates in the general population.
Author Summary
Aging is the main risk factor for the majority of chronic diseases and the leading cause of death and disability in humans. Humans age at different rates, but the molecular genetic basis underlying this phenomenon remains largely unknown. Efforts to understand how we age have focused on genetic changes that extend lifespan or underlie progeroid disorders. One potential progeroid disorder, MVA syndrome, has been associated with mutations in the mitotic regulator BUBR1. Although MVA syndrome is rare due to its recessive nature, individuals carrying heterozygous BUBR1 mutations associated with MVA would be much more prevalent. However, whether such carriers are asymptomatic or at risk of developing aspects of MVA syndrome later in life is unknown. To investigate this, we engineered mice to carry an analogous mutation to the human MVA BUBR1 nonsense mutation 2211insGTTA. We find that these mice have a reduced lifespan and develop several age-related phenotypes at an accelerated rate. These findings suggest that bi-allelic integrity of BUBR1 is a key determinant of healthspan and longevity, and provide a conceptual framework for elucidating differences in aging rates among humans.
Introduction
Separation of duplicated chromosomes during mitosis is an intricate biological process whose molecular basis is incompletely understood. Inaccurate segregation of whole chromosomes results in numerical chromosome aberrations, referred to as aneuploidy [1]. Human aneuploidy is intimately associated with developmental defects and disease pathology [2]. For example, aneuploidy in gametes is a known cause of infertility, miscarriages and congenital birth defects [3], while somatic cell aneuploidy is a hallmark of cancer, with evidence mounting that aneuploidy can promote neoplastic transformation [4]–[6]. To safeguard against chromosome missegregation, eukaryotic organisms have developed cellular surveillance systems including the mitotic checkpoint (or spindle assembly checkpoint) and the attachment error correction machinery [7]. The mitotic checkpoint is a multi-protein network that inhibits sister chromatid separation until all chromosomes are properly attached to the mitotic spindle [8]. One of the core components of this checkpoint is BubR1, a modular protein that acts to inhibit the activity of the large multi-protein E3 ubiquitin ligase known as the anaphase promoting complex/cyclosome (APC/C), by binding to the co-activating subunit Cdc20 [9]. Once all chromosomes have achieved bi-orientation, BubR1 dissociates from Cdc20 leading to the polyubiquitination of securin and cyclin B1, two inhibitors of separase, a protease that initiates anaphase by cleaving cohesin rings that physically join duplicated sister chromosomes together. BubR1 not only promotes accurate chromosome segregation in its role as a Cdc20 inhibitor, but also acts to stabilize microtubule-kinetochore attachments [10].
BUBR1 mutations have been identified in various human malignancies, including gastrointestinal cancers [11]–[14], and in a rare human hereditary condition called mosaic variegated aneuploidy (MVA) syndrome, in which high rates of chromosome missegregation lead to systemic aneuploidy, typically involving more than 25% of cells [15], [16]. MVA syndrome has clinically heterogeneic features, including growth deficiency (with prenatal onset), mental retardation, microcephaly, facial dysmorphisms, cataracts and other eye abnormalities, short lifespan, and increased risk for childhood cancers such as rhabdomyosarcoma, Wilms' tumor and leukemia [17]. MVA patients with BUBR1 mutations fall into two groups: bi-allelic and mono-allelic mutations. Patients with bi-allelic mutations carry an allele that results in premature truncation of BubR1 protein or an absent transcript, and an allele with a missense mutation often located within the kinase domain [15]. Patients with mono-allelic mutations have either a nonsense or missense mutation combined with a non-mutated allelic BUBR1 variant that expresses low amounts of wildtype BubR1 protein [16], [17]. BubR1 protein levels are usually very low in patients with BUBR1 mutations, even in those with a missense mutation, largely because mutant BubR1 proteins produced by these alleles tend to be quite unstable [18].
Gene knockout studies in mice support the idea of a causal relationship between BubR1 insufficiency and tumorigenesis. Although homozygous BubR1 knockouts die as pre-implantation stage embryos, heterozygous knockouts are viable and show increased tumor formation when challenged with a carcinogen [19]–[21]. BubR1 depletion beyond the level of heterozygous knockout mice was achieved by the use of knockout (BubR1 −) and hypomorphic (BubR1 H) alleles [21]. Mice with one hypomorphic and one knockout allele (BubR1 −/H mice) express about 4% of normal BubR1 protein levels and die at birth from respiratory failure [21]. However, mice with two BubR1 hypomorphic alleles (BubR1 H/H mice) that produce around 10% of normal BubR1 protein levels are viable. Like MVA patients, these mice show systemic near-diploid aneuploidy, premature chromosome separation (PCS) and are susceptible to tumorigenesis [21], [22]. However, unlike other mouse models for aneuploidy that have similar features [1], BubR1 H/H mice have a very short lifespan and develop several progeroid phenotypes at a very young age (within 3 to 5 months), including growth retardation (dwarfism), facial dysmorphisms, cataracts, muscle wasting, lordokyphosis (rearward curvature of the spine), fat loss, and cardiac arrhythmias [21], [23]. This, together with observations that BubR1 protein levels decrease during natural aging in various mouse tissues, raises the possibility that BubR1 is an important regulator of aging [21], [24].
MVA syndrome has been documented as a hereditary cancer syndrome [15]–[18], [25]–[27], but could potentially be classified also as a progeroid syndrome based on its phenotypic resemblance to BubR1 progeroid mice, which includes short lifespan, dwarfism, facial dysmorphisms, and cataract formation. However, whether MVA patients have additional age-related phenotypes observed in BubR1 hypomorphic mice such as fat loss, muscle wasting and cardiac arrhythmias is a key open question that has been difficult to address, largely because MVA syndrome is very rare and because most patient die very young. While patients with the MVA disorder are rare, heterozygous carriers of BUBR1 mutant alleles are expected to be much more prevalent in the general population. If these mutations were to affect healthspan or longevity, or both, this might provide a genetic basis for why certain people develop particular age-related traits at faster rates. Little is known about the health status of parents of MVA patients. They seem to have a minor mitotic phenotype as evidenced by their predisposition to PCS, but whether they are at risk for any of the pathologies associated with MVA syndrome or progeria is unknown [28]–[30]. To address this question and to better understand the relationship between MVA syndrome and progeria, we engineered mice to carry the human MVA BUBR1 nonsense mutation 2211insGTTA [15]. We demonstrate that mice harboring this heterozygous BubR1 MVA mutation have a reduced lifespan and exhibit acceleration of various early age-related features. In addition, reduced BubR1 protein levels in BubR1 +/GTTA mice results in mild aneuploidy and increases carcinogen-induced tumor growth. These findings suggest that mono-allelic BUBR1 mutations might contribute to accelerated aging and reduced longevity, further supporting the idea that MVA syndrome is a progeria-like syndrome. Additionally, we provide important experimental evidence for the longstanding concept that variations in select genes accelerate the rate of age-related deterioration of certain tissues and organs.
Results
Generation of a mouse model for MVA BubR1 mutation 2211insGTTA
Human BUBR1 encodes a modular 1052 amino acid serine/threonine protein kinase that is highly conserved among mammalian species (Figure 1A). Two of the four MVA patients with bi-allelic BUBR1 mutations reported by Hanks and colleagues [15] carry a nonsense mutation referred to as 2211insGTTA that results in a truncated protein that lacks the kinase domain (Figure 1A). Because of its prevalence, we mimicked this mutation in mice to understand the potential physiological consequences of MVA mutations in a heterozygous state. Using homologous recombination in embryonic stem (ES) cells, we inserted a GTTA sequence in the murine BubR1 gene at position 2178. This position corresponds to nucleotide 2211 of the human BUBR1 transcript (Figure 1A and 1B). We injected correctly targeted ES clones into blastocysts and obtained chimeric mice. These chimeras successfully transmitted the mutated BubR1 allele (referred to as BubR1 NEO;GTTA) to their offspring (Figure 1B and 1C). BubR1 +/NEO;GTTA mice were crossed to protamine-Cre transgenic mice to remove the NEO gene cassette (Figure 1B and 1D). BubR1 +/GTTA mice were obtained at the expected frequency and were overtly indistinguishable from control littermates (data not shown).
Figure 1. BubR1 +/GTTA mice are viable and mimic their human counterpart.

(A) Schematic representation of BubR1 protein. Functional domains are indicated: KEN, destruction box-motifs implicated in APC/CCdc20 inhibition (also referred to as Cdc20 BD1); TPR, tetratricopeptide motif for binding to Knl1; GLEBS, motif for Bub3 binding and kinetochore localization; Cdc20 BD2, C-terminal Cdc20 binding domain, Kinase domain, protein kinase domain homology domain. Red arrowheads indicate human and mouse GTTA insertion sites: the first amino acid of BubR1 impacted by the insertion is indicated in red font. (B) Gene targeting strategy to generate BubR1 +/GTTA mice. Shown are: the genomic mouse BubR1 locus spanning exon 12 to 21 (top); the targeting vector (TV) with the GTTA insertion in exon 17 (asterisk), loxP sites (blue triangles); the BubR1 locus after targeted recombination; and the final BubR1 +/GTTA locus after protamine-Cre mediated recombination mice to excise the NEO cassette and produce BubR1 +/GTTA mice. The Xmn1 (Xm) and Xho1 (Xh) restriction sites, the Southern blot probe, wildtype and BubR1 NEO;GTTA Southern blot fragments, and the 4 PCR primer sites (a–d) used for genotyping are indicated. (C) Southern blot containing Xmn1/Xho1 digested genomic DNA from two ES cell clones and probed with DNA fragment indicated in (B) showing the 14 kb and 9 kb fragments representing the wildtype and BubR1 NEO;GTTA alleles, respectively. (D) PCR genotyping of two pups from a BubR1 +/NEO:GTTA female crossed to a protamine-Cre male. One pup had the NEO cassette successfully removed to create a BubR1 +/GTTA allele. (E) Western blots of mitotic and asynchronous MEF extracts probed for BubR1, the mitotic marker p-H3, and actin (loading control). (F) Western blot of testis extracts probed for BubR1. Ponceau S staining was used as loading control. (G) Karyotype analysis performed on wildtype and BubR1 +/GTTA P5 MEFs. Fifty mitotic figures were counted per sample. PCS, premature chromatid separation. An unpaired t-test was used for statistical analysis. **P<0.01, ***P<0.001.
Western blot analysis demonstrated that mouse embryonic fibroblasts (MEFs) derived from BubR1 +/GTTA mice had reduced amounts of wildtype BubR1 protein (Figure 1E). The level of reduction was similar to that observed in BubR1 +/− MEFs (Figure 1E and Figure S1). The predicted 730 amino acid truncated protein encoded by the BubR1 GTTA allele was undetectable, even after long exposure times (Figure 1E), which is consistent with recent observations in cultured skin fibroblast of an MVA patient carrying the BUBR1 GTTA allele [18]. Western blot analysis of testis extracts from BubR1 +/GTTA and wildtype mice confirmed the reduction of BubR1 in cultured MEFs (Figure 1F). One plausible explanation for the absence of BubR1 GTTA encoded protein on western blots is that nonsense mutations tend to produce transcripts that are subject to non-sense mediated mRNA decay [13], [16], [18]. Alternatively, the truncated protein may be unstable and subject to rapid proteosomal degradation.
Patients with MVA syndrome show systemic near-diploid aneuploidies resulting from an inability to separate duplicated chromosomes with high accuracy during mitosis [15], [16], [29]. Although metaphase spreads from heterozygous carriers of MVA BUBR1 mutations exhibit increased rates of PCS, it is unclear whether parents of MVA patients are subject to increased aneuploidization [15], [16], [26], [31]–[34]. To examine the impact of the BubR1 GTTA allele on chromosome number integrity, we prepared metaphase spreads of passage 5 BubR1 +/GTTA and wildtype MEFs and performed chromosome counts. Aneuploidy rates of wildtype MEFs were on average 12% (Figure 1G), which is consistent with rates reported in previous studies [35]–[37]. Aneuploidy rates in BubR1 +/GTTA MEFs were significantly increased by 6%, although it should be noted that this escalation is relatively small compared to that previously observed in BubR1 H/H MEFs [21] (Table S1). Consistent with this, BubR1 H/H MEFs exhibited a more profound reduction in wildtype BubR1 protein levels than BubR1 +/GTTA MEFs (Figure 1E and Table S1). The incidence of PCS was also significantly higher in BubR1 +/GTTA MEFs than in wildtype MEFs (Figure 1G), but again not as high as in BubR1 H/H MEFs [21]. Collectively, these data indicate that the BubR1 GTTA allele that we created in mice faithfully mimics its human counterpart, and demonstrate that BubR1 +/GTTA mice have relatively mild, but significant, mitotic phenotypes.
Median and maximum lifespans of BubR1 +/GTTA mice are reduced
BubR1 H/H mice exhibit various aging-related phenotypes by 3 to 4 months of age [21] (Table S1). BubR1 +/GTTA mice, however, remained overtly indistinguishable from control littermates during this time period. To examine potential late life phenotypes associated with the GTTA mutation, we established large cohorts of BubR1 +/GTTA and wildtype mice, which we monitored for signs of ill health and the development of overt age-related phenotypes. Kaplan-Meier overall survival curves of these cohorts showed that the GTTA mutation significantly reduces median and maximum lifespan, with BubR1 +/GTTA mice having a median lifespan of 93 weeks compared to 102 weeks for wildtype mice (Figure 2). By comparison, BubR1 H/H mice had a significantly shortened median survival of only 30 weeks (Figure 2).
Figure 2. Lifespan of BubR1 +/GTTA is reduced.
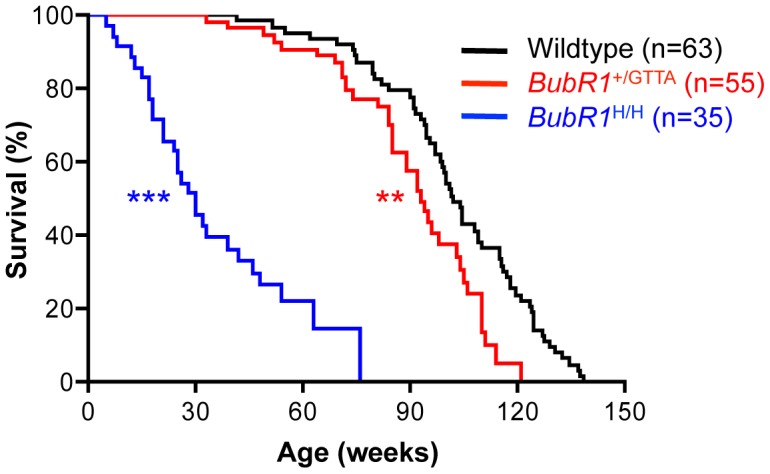
Kaplan-Meier overall survival curves. Asterisks denote significance compared to wildtype mice using a log-rank test: **P<0.01 and ***P<0.001. The maximum lifespan was significantly decreased in BubR1 +/GTTA compared to wildtype (P = 0.0008; two-sided Wang/Allison test referring to the ratio of mice alive per genotype at the 90th percentile survival point) [70].
BubR1 H/H mice develop quite severe cardiac arrhythmias that are thought to be the primary cause of premature death of the animals [38] (Table S1). This prompted us to test whether reduced lifespan of BubR1 +/GTTA mice might be due to cardiac problems. However, the frequency of cardiac arrhythmias in BubR1 +/GTTA mice was not elevated (Figure S2A). Subsequent cardiac stress tolerance tests, in which a lethal dose of the β-adrenergic agonist isoproterenol was injected and the time to death measured [39], further indicated that the cardiac performance of BubR1 +/GTTA mice is not compromised (Figure S2B).
Muscle wasting and cataract formation are accelerated in BubR1 +/GTTA mice
A prominent phenotype of BubR1 hypomorphic mice is lordokyphosis [21]. The underlying condition here is skeletal muscle deterioration rather than osteoporosis [23]. BubR1 +/GTTA and wildtype mice in our cohorts were biweekly monitored for this phenotype. Wildtype mice are known to develop lordokyphosis as part of their normal aging process [40], a finding commonly attributed to a combination of muscle wasting and osteoporosis [41], [42]. While mice in both our cohorts indeed developed lordokyphosis with aging, the median onset of this age-related phenotype was markedly accelerated in BubR1 +/GTTA mice (89 weeks versus 116 weeks; Figure 3A).
Figure 3. Accelerated deterioration of skeletal muscle in BubR1 +/GTTA mice.

(A) Incidence and latency of lordokyphosis. A log rank test was used for statistical analysis: ***P<0.001. (B) Mean fiber diameter of gastrocnemius (Gastro), abdominal (ABD), and paraspinal muscles of 15-month-old mice. (C) Expression of the senescence markers p16 Ink4a and p19 Arf in gastrocnemius muscles of 15 month-old mice analyzed by qRT-PCR. (D–F) Treadmill exercise ability of 3- and 15-month-old mice: presented are running time (D), distance travelled (E), and workload (F) performed. Error bars represent SEM. Males mice were used for all analysis in D–F: wildtype 3 months, n = 3 males; BubR1 +/GTTA 3 months, n = 5 males, wildtype and BubR1 +/GTTA 15 months, n = 4 males each. An unpaired t-test was used for statistical analyses in (B–F): *P<0.05.
To determine whether this acceleration might be due to early muscle degeneration, we sacrificed 15-month old BubR1 +/GTTA and wildtype mice and performed muscle fiber diameter measurements on cross sections of the gastrocnemius, paraspinal and abdominal muscles. As shown in Figure 3B, average muscle fiber diameters of BubR1 +/GTTA mice were significantly reduced in all three muscle groups. No such reductions were observed in 3-month-old BubR1 +/GTTA mice (Figure S3). Using qRT-PCR we analyzed the gastrocnemius of aged BubR1 +/GTTA and wildtype mice for expression of p16 Ink4a and p19 Arf and found that both senescence markers were expressed at elevated levels [23], [38], suggesting that senescent cells might contribute to accelerated muscle degeneration in BubR1 +/GTTA mice (Figure 3C). Furthermore, 15-month old BubR1 +/GTTA mice had similar bone mineral densities and contents as wildtype mice as measured by dual energy x-ray absorptiometry (DEXA; Figure S4), demonstrating that the early kyphosis is not due to accelerated osteoporosis.
To assess whether early muscle wasting resulted in decreased muscle function, we performed treadmill exercise tests on BubR1 +/GTTA and wildtype mice at various ages [38], [43]. As shown in Figure 3D–3F, wildtype mice showed similar exercise ability at 3 and 15 months of age. However, while BubR1 +/GTTA mice showed normal exercise ability at 3 months, the duration of exercise, distance travelled and overall amount of work performed were all significantly decreased at 15 months.
A second early aging-associated phenotype of BubR1 hypomorphic mice is cataract formation [21]. MVA patients are also prone to cataracts, as well as other eye anomalies [16], [29]. Our biweekly inspections of BubR1 +/GTTA and wildtype mice revealed that cataract formation was significantly accelerated in BubR1 +/GTTA mice, with 50% of BubR1 +/GTTA mice having cataracts at 101 weeks versus 116 weeks for wildtype mice (Figure 4A). Affected lenses of both BubR1 +/GTTA and wildtype mice had nuclear cataracts, as revealed by histological evaluation (Figure 4B). Similar to BubR1 H/H lenses [21], BubR1 +/GTTA lenses exhibited posteriorly located epithelial cells (Figure 4B and 4C), although Morgagnian globules, which are a distinguishing feature of hypomorphic lenses, were not detected. In contrast, posterior epithelial cells were rarely observed in wildtype lenses. Taken together, the above data demonstrate that skeletal muscle degeneration and cataract formation, two hallmarks of chronological aging in humans [44], are accelerated in mice carrying the BubR1 GTTA mutation found in human MVA syndrome.
Figure 4. Cataract formation is accelerated in BubR1 +/GTTA mice.

(A) Incidence and onset of cataracts in BubR1 +/GTTA and wildtype mice. Eyes were dilated and screened for cataracts using a slit light. A log rank test was used for statistical analysis: *P<0.05. (B) Representative cross sections of cataractous lenses stained with hematoxylin and eosin. Note that the BubR1 +/GTTA lens section contains more posteriorly located (arrowheads). Scale bars represent 2 mm and 100 µm. (C) Quantitation of posteriorly (P) located epithelial cells in cross sections of cataractous lenses. Error bars represent SEM. In C we used: n = 10 wildtype and n = 9 BubR1 +/GTTA 24-month-old animals.
BubR1 GTTA carriers exhibit early age-related fat loss
In humans, the amount of fat tissue increases during middle age but then decreases at advanced age [45]. Furthermore, during and after middle age, fat redistributes from subcutaneous to intra-abdominal visceral depots. In turn, these deposits shrink in the elderly as a result of fat redistribution to bone marrow, muscle, and liver [45]. Previous studies have demonstrated that BubR1 H/H progeroid mice prematurely lose fat from various depots and the subdermal adipose layer [38], [46], [47]. To probe for premature changes in fat mass and redistribution in BubR1 +/GTTA mice, we measured the overall amount of fat (using DEXA scanning on live animals), the mass of various fat deposits, the subdermal adipose thickness, and the fat cell size of 15- and 24-month-old BubR1 +/GTTA and wildtype males. Body weight, total fat mass, and weights of major fat depots, including inguinal adipose tissue (IAT), subscapular adipose tissue (SSAT) and mesenteric adipose tissue (MES), were all normal in 15-month-old BubR1 +/GTTA mice (Figure 5A–5C). All these values remained unchanged in 24-month-old wildtype mice. In contrast, however, body weight, percentage of body fat, and total fat mass of BubR1 +/GTTA were all significantly reduced at 24 months. In addition, several fat depots shrank significantly, including IAT and brown fat, while SSAT and MES were trending downward.
Figure 5. Age-related fat loss in BubR1 +/GTTA mice.

(A) Body fat percentage, lean mass and fat mass of wildtype and BubR1 +/GTTA mice measured by DEXA scanning. (B) Fat depot sizes in wildtype and BubR1 +/GTTA mice at the indicated ages. (C) Body weight of wildtype and BubR1 +/GTTA mice at the indicated ages. (D) Average fat cell diameters in IAT of mice at the indicated ages. (E) Average subcutaneous adipose layer thickness of lateral skin of the indicated mice. (F) Expression of p16 Ink4a and p19 Arf in gastrocnemius muscles of 24 month-old mice analyzed by qRT-PCR. For all analyses error bars represent SEM. For C-F we used: n = 4 15-month-old wildtype and BubR1 +/GTTA males; n = 7 24-month-old wildtype males; and n = 8 BubR1 +/GTTA 24-month-old males. An unpaired t-test was used for statistical analysis in A–F: *P<0.05 and **P<0.01.
Histological analysis showed that the average diameter of fat cells in IAT of BubR1 +/GTTA mice declined significantly between 15 and 24 months (Figure 5D). Fat cell size also decreased in wildtype mice, but to a lesser extent than in BubR1 +/GTTA mice. Cross sections prepared from lateral skin from 15- and 24-month-old BubR1 +/GTTA and wildtype males revealed a dramatic decline in subdermal adipose layer thickness with aging selectively in BubR1 +/GTTA males (Figure 5E). In contrast, dermal layer thickness did not decline (Figure S5).
Loss of fat tissue in BubR1 H/H mice is, at least in part, due to accumulation of senescent cells [38], [47]. To determine whether accelerated fat loss in BubR1 +/GTTA mice might involve cellular senescence, we measured p16 Ink4a and p19 Arf transcript levels in IAT of 24-month-old BubR1 +/GTTA and wildtype mice by qRT-PCR. Levels of both senescence markers were markedly increased in BubR1 +/GTTA mice (Figure 5F), indicating that early accumulation of senescent cells contributes to age-related fat loss in heterozygous carriers of the BubR1 GTTA mutation found in MVA patients.
BubR1 +/GTTA mice develop large carcinogen-induced lung tumors
MVA patients are prone to tumor formation, including patients with mono-or bi-allelic BUBR1 mutations [15], [16]. Consistent with this, BubR1 H/H mice are highly susceptible to carcinogen-induced tumors, although it should be noted that spontaneous tumor rates are not increased [21], [23] (Table S1). Similar results have been reported for mice in which one BubR1 allele has been inactivated [19]. As a first step to evaluate whether BubR1 +/GTTA mice might be tumor prone we sacrificed BubR1 +/GTTA and wildtype mice at 24 months of age and screened internal organs for overt tumors. This analysis revealed that the incidence of spontaneous tumors was similar for both genotypes (Figure S6A). Analysis of the tumor spectra revealed no significant increases in incidence of individual tumor types (Figure S6B), although there was a trend for increased tumor multiplicity in BubR1 +/GTTA mice.
To complement these studies, we treated BubR1 +/GTTA and wildtype mice with the carcinogen 7,12-dimethylbenz(a)anthracene (DMBA) at postnatal day 5. The mice were then sacrificed at 4 months and analyzed for lung tumor formation. We found that the tumor incidence and the tumor multiplicity were both very similar in BubR1 +/GTTA and wildtype mice (Figure 6A and 6B). Interestingly, however, tumor size was dramatically increased in BubR1 +/GTTA mice (Figure 6C). These findings suggest that the BubR1 GTTA mutation has no obvious impact on tumor initiation but can promote growth of established tumors.
Figure 6. Carcinogen-induced lung tumor size is increased in BubR1 +/GTTA mice.

(A) Incidence of DMBA-induced lung tumors. (B) Lung tumor multiplicity. (C) Average lung tumor size. An unpaired t-test was utilized to determine statistical significance: **P<0.01. Color codes in B and C are as in A. For all analysis error bars represent SEM; n = 19 wildtype and n = 20 BubR1 +/GTTA mice.
Mono-allelic loss of BubR1 reduces lifespan
The finding that BubR1 +/GTTA and BubR1 +/− MEFs have similar wildtype BubR1 protein levels and the truncated protein encoded by the GTTA allele is expressed at non-detectable levels raised the question whether BubR1 +/− mice might be phenotypically similar to BubR1 +/GTTA mice. BubR1 +/− MEFs show karyotypic similarity to BubR1 +/GTTA MEFs in that their aneuploidy rates are also modestly increased [21]. On the other hand, PCS, a hallmark of MVA patients [15]–[18], [25]–[27], is elevated in BubR1 +/GTTA MEFs but not in BubR1 +/− MEFs. In an earlier study, in which survival of BubR1 H/H, BubR1 +/−, BubR1 +/H, and BubR1 +/+ mice was analyzed for up to 15 month of age, yielded no difference in survival between BubR1 +/− and BubR1 +/+ mice [21]. However, a retrospective analysis of survival records of BubR1 +/− and BubR1 +/+ animals that were maintained until natural death revealed that mono-allelic loss of BubR1 significantly reduces the median lifespan (90 weeks compared to 102 weeks for the corresponding BubR1 +/+ mice, Figure 7). There was no statistically significant decrease in maximum lifespan. We note that these earlier BubR1 +/− and BubR1 +/+ cohorts of mice were not analyzed for any age-related phenotypes (see Table S1).
Figure 7. Lifespan of BubR1 +/− mice is reduced.
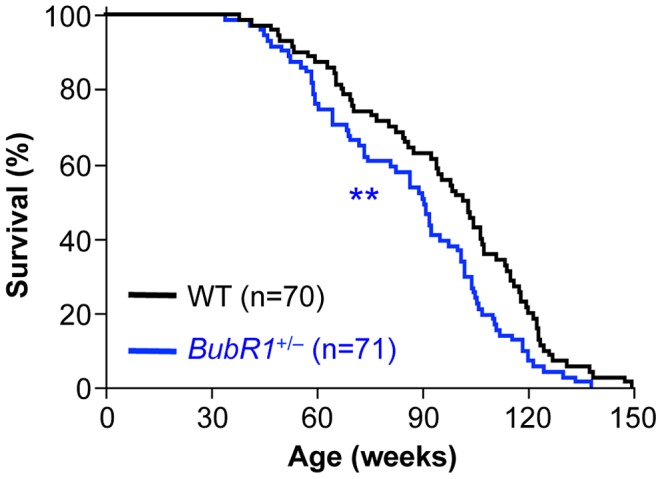
Kaplan-Meier overall survival curves. Asterisks denote significance compared to wildtype mice using a log-rank test. **P<0.01.
Discussion
Biallelic mutations in WRN, CSA and CSB, and DNA repair genes such as XPB, XPD and TTD are associated with human diseases that have features of premature aging [48]–[50]. MVA syndrome has some progeroid traits, but unlike the above syndromes has not been widely recognized as a progeroid disorder [17]. Whether the spectrum of age-related phenotypes of MVA patients is broader than reported has been difficult to assess, mainly because MVA patients are very rare and die early [15], [16]. It is also unknown whether parents of MVA patients are susceptible to any of the pathologies associated with MVA syndrome. We engineered a mouse model to mimic the BubR1 nonsense mutation 2211insGTTA found in MVA patients with bi-allelic BUBR1 mutations and show that these mice have a significantly shorter lifespan and develop several age-related disorders at accelerated rates, including sarcopenia, cataracts, and loss of fat tissue. These findings strengthen the notion that MVA syndrome is a progeroid syndrome, and provide important experimental evidence for the longstanding concept that variations in select genes may affect the rate of age-related deterioration in certain tissues and organs.
To our knowledge, accelerated age-related pathologies have not been reported in parents of affected individuals with any of the classical recessive human progeroid syndromes [48]–[51]. Furthermore, while homozygous knockout or mutant mice have been established for most of the implicated genes, including WRN, CSA and CSB, XPB, XPD, and TTD, whether heterozygotes of these models have reduced longevity or a faster than normal onset of age-related functional decline in particular tissues has not been studied in detail [50], [52]–[56]. Thus, based on the findings presented here, it will not only be important to determine whether heterozygous MVA BubR1 mutations other than 2211insGTTA cause age-related phenotypes in mice, but also to perform similar studies on mice heterozygous for other progeria-associated genes. Three observations suggest that the phenotypes of BubR1 +/GTTA mice are likely to be caused by reduced expression of wildtype BubR1 protein without a contribution of truncated BubR1 protein. First, the truncated protein encoded by the GTTA allele is non-detectable by western blotting, indicating that its level of expression is very low. Furthermore, residual levels of wildtype BubR1 protein are similar in BubR1 +/GTTA and BubR1 +/− MEFs. Second, BubR1 +/GTTA and BubR1 +/− MEFs have similar aneuploidy rates, suggesting that the extent to which BubR1 is dysfunctional is the same. Third, BubR1 +/GTTA and BubR1 +/− mice show very similar reductions in median lifespan. It will be important to complement the survival data of BubR1 +/− mice with a comprehensive analysis of age-related phenotypes, and new cohorts of BubR1 +/− and BubR1 +/+ mice are currently being established for this purpose.
Previously, we have shown that clearance of p16Ink4a-positive senescent cells from BubR1 H/H mice results in attenuation of sarcopenia, fat loss, and cataracts, indicating that accumulation of senescent cells in skeletal muscle, adipose tissue, and eye drives functional decline in these tissues [38]. This, combined with the observation that p16 Ink4a and p19 Arf transcript levels are elevated in skeletal muscle and fat of BubR1 +/GTTA mice suggests that senescence contributes to the accelerated functional decline in these animals. On the other hand, accelerated cataractogenesis in BubR1 +/GTTA mice seems to be senescent cell independent (data not shown). Perhaps, the mere accumulation of epithelial cells in the posterior portion of the lens is sufficient to accelerate cataract formation. The main difference between BubR1 +/GTTA and BubR1 H/H lenses is that the latter have posteriorly located Morgagnian globules [21], which may be associated with senescence and explain why cataractogenesis is much more accelerated in BubR1 H/H than in BubR1 +/GTTA mice.
The mechanism by which BubR1 insufficiency induces senescence appears to be more complicated than anticipated [21], [23], [57]. It is unlikely that aneuploidy represents the primary lesion that drives senescence, mainly because other aneuploidy models with substantially higher aneuploidy rates do not undergo premature senescence and aging [1], [4], [6], [58]. BubR1 is expressed in interphase where it apparently continues to serve as an inhibitor of APC/CCdc20 activity [37]. Consistent with this, recent reports indicate that APC/CCdc20 E3 ubiquitin ligase activity orchestrates key developmental processes in post-mitotic neurons, including dendrite growth and presynaptic differentiation [59], [60]. These findings raise the interesting possibility that BubR1 insufficiency might lead to unscheduled degradation of APC/CCdc20 substrates in interphase cells, which, in turn, could lead to cellular stresses that engage p16Ink4a and induce senescence. Key progeroid phenotypes of BubR1 H/H mice are also observed in BubR1 +/GTTA, but are considerably milder, which correlates with less profound BubR1 protein insufficiency (Table S1). Various phenotypes seem unique to BubR1 H/H mice including cardiac dysfunction, dwarfism, facial dysmorphisms, and thinning of the dermis, suggesting that a more extreme level of BubR1 insufficiency is required for their induction.
BubR1 +/GTTA mice are not prone to spontaneous tumors and show normal tumor incidence and multiplicity when challenged with the carcinogen DMBA. The most straightforward explanation would be that the level of aneuploidization is insufficient to promote neoplastic transformation. Consistent with this, Bub3 +/− mice have similarly mild aneuploidy rates as BubR1 +/GTTA mice and are also not prone to spontaneous or DMBA-induced tumors [61]. However, a significant feature of DBMA-induced lung tumors of BubR1 +/GTTA mice is their large size, indicating that the mutation promotes tumor aggressiveness without impacting tumor initiation. It will be interesting to determine whether individuals carrying the 2211insGTTA mutation are prone to lethal malignancies.
Our current study and previous data support the notion that BubR1 protein levels tightly correlate with aneuploidy rates, cancer susceptibility, lifespan and aging-related phenotypes (Table S1), indicating that BubR1 is a key determinant of healthspan and lifespan and warrants a comprehensive analysis of the health status of parents of MVA patients and relatives that are also heterozygous carriers of the same MVA BUBR1 mutations. In addition, it would be interesting to screen for BUBR1 mutations in the general population, either in an unbiased manner or more selectively in cohorts prone to conditions associated with BubR1 insufficiency in mice, including sarcopenia, cataracts and fat tissue dysfunction. Subsequent characterization of these mutations, for instance for impact on BubR1 protein stability, might lead to the identification of additional BUBR1 variants that influence rates of age-related deterioration in certain tissues and organs.
Materials and Methods
Generation of BubR1 +/GTTA mice
The BubR1 GTTA allele was produced by a recombineering based approach [62]. Briefly, a genomic BubR1 gene fragment of 10 kb spanning exons 14–19 was retrieved from BAC #bMQ_294E2 (129S7/SvEv ES Cell, Source BioScience) and transferred into pDTA. Insertion of the GTTA sequence into the exon 17 (c.2178_2179) was done as follows: a tetracyclin-resistance gene cassette was made by PCR using pKOEZ-40 plasmid as a template (gift from Dr. Pumin Zhang), the tetra-partite forward primer (50 bp homology to the target region, GTTA, ClaI, 24 bp homology to the Tet) 5′-CCTGG TGTTCACAGTATCGCCTACAACTGTTAAAATCCCTACTAGAATTAGTTAATCGATGGTCGA CGGTATCGATAAGCTTGA-3′ and the tri-partite reverse primer (50 bp homology to the target region, ClaI, 24 bp homology to the Tet) 5′-TCCAGCACAGGCATCGGTC GGTCTTCCACAGAAAACTCCGCAAAAGCACTATCGATTTGGATGGTGAATCC GTTAGCGA-3′. The GTTA-ClaI-Tet-ClaI cassette was inserted into the pDTA-BubR1(E14-E19) by recombineering. The resulting construct was digested with ClaI and re-ligated for removal of the Tet gene. Next, a loxP-neomycin phosphotransferase II (neo) gene-loxP cassette was inserted into the pDTA-BubR1 GTTA (E14-E19) construct, 141 bp upstream of exon 17, using recombineering [62]. The final targeting vector was linearized with MluI and electroporated into TL1 129Sv/E ES cells. Transfectants were selected in 350 µg/ml G418 and 0.2 µM FIAU, and expanded for Southern blot analysis using a 1155 bp 5′ external probe on XmnI/XhoI -digested genomic ES cell DNA. The probe was amplified by PCR from 129Sv/E genomic DNA using the following primers: 5′-GCAGAGTATCCTGACAGGTTAAGGCAC-3′ and 5′-CATAATAATTATCCAACCATGAATGATC-3′. Chimeric mice were produced by microinjection of three independent ES cell targeted clones with 40 chromosomes into C57BL/6 blastocysts. Chimeric males were mated with C57BL/6 females and germline transmission of the BubR1 NEO;GTTA allele was verified by PCR analysis of tail DNA from pups with a agouti coat color. By crossing BubR1 +/NEO;GTTA mice to protamine-Cre transgenics [63] the NEO cassette was excised. The following primer combinations were used for PCR genotyping of mice used in our studies: primers a (5′-TCAGATCTCCTAGAGCTGGGGTTA-3′) and b (5′-AATTCTAGTAGGGATTTTAA CAGTTG-3′) for BubR1 GTTA; primers c (5′-GTCTTGTCGATCAGGATGATCTG-3′) and d (5′-GAAGTAGTATTGTTCCTGTGG CAT-3′) for BubR1 NEO;GTTA. The targeting vector, targeted ES cells as well as BubR1 NEO;GTTA mice were sequenced for the presence of GTTA insertion by PCR amplifying the 1.1 kb fragment using primers c and d. Note: in humans, 2211insGTTA results into S738fsX753 (see Mutation 1, shared by Family 2 and Family 3 [15]). In mice, 2178–9insGTTA results in S727fsX750. Insertion of GTTA followed by ClaI coding sequence, as in our targeting strategy, leads to S727fsX730. All mice, including BubR1 +/− and BubR1 +/+ mice used for survival analysis, were on a mixed 129 X C57BL/6 genetic background and housed in a pathogen-free barrier and maintained on a 12 hours dark/light cycle throughout the study. Mice had ad libitum access to food containing 10% fat and were inspected daily. Animals used for survival analysis were mice found dead or sacrificed when moribund. Mice sacrificed at 15 and 24-months of age were screened for lymphomas, carcinomas and sarcomas to assess spontaneous tumorigenesis. Animals sacrificed at 3, 15 and 24-months for analyses were omitted from survival, lordokyphosis and cataract incidence curves. All animal protocols were reviewed and approved by the Mayo Clinic institutional animal care and use committee.
Generation and culture of MEFs
Wildtype and BubR1 +/GTTA MEFs were generated and cultured as previously described [46]. MEFs were frozen at passage 2 or 3 and used for experimentation at the indicated passages. At least three wildtype and BubR1 +/GTTA lines were used for all experiments. Mitotic MEFs were generated as previously described [36].
Western blotting
Western blot analysis was performed as previously described [64]. Tissue lysates were prepared by first snap freezing the tissue in liquid nitrogen upon sacrifice. Frozen tissue was ground into fine powder with pestle and mortar. 20 mg of tissue powder was suspended in 200 µl lysis buffer (0.1% NP-40, 10% glycerol in PBS, plus protease inhibitors) and vortexed for 10 min at 4°C. After centrifugation at 14000 rpm at 4°C, 150 µl supernatant was transferred to a 0.5 ml PCR tube and 150 µl Laemmli lysis buffer was added. The lysate was boiled for 10 min before loading on Tris-HCl Polyacrylamide gel. Blots were probed with antibodies for BubR1 (BD), pH3Ser10 (Millipore) and ß-actin (Sigma). Ponceau S stain was used as a loading control. Quantification of BubR1 protein levels in BubR1 +/GTTA and BubR1 +/− MEF lysates was done as described [65].
Karyotype analysis
Karyotype analysis on P5 wildtype and BubR1 +/GTTA MEFs was performed as described [66].
Analysis of age-related phenotypes
Bi-weekly monitoring for lordokyphosis and cataract incidence was performed as described [23]. Fiber diameter measurements were performed on cross sections of the gastrocnemius and abdominal muscle from 3 and 15-month-old mice according to previously described methods [23]. Dissection of the paraspinal muscle was performed halfway between the front and hind limb, and processed and measured as the other skeletal muscles. The mean was calculated from a total of fifty measurements obtained with a calibrated program (Olympus MicroSuite Five). Measurements of fat cell diameters were performed according to the same method. Measurements of the total thickness of dermis and subcutaneous adipose layer of lateral skin were performed as described [46]. For histological evaluation of cataracts, whole eyes were paraffin embedded, sagittally sectioned through the middle of the lens and stained with hematoxylin and eosin. The number of cells that had migrated past the epithelial bow of the lens was counted as posterior localized epithelial cells. DEXA scanning was used to measure bone mineral density, bone mineral content, percentage of total body fat, lean mass and fat mass. These measurements were done as described [67]. Treadmill exercise tests were performed as described [43]. For isoproterenol stress tests, a lethal dose of isoproterenol (680 mg/kg) was injected in the chest cavity and time to death was recorded. Mice were monitored for cardiac arrhythmias using a Vevo2100 ultrasound system (Visualsonics) as described [68].
Quantitative RT–PCR
qRT-PCR analysis on cDNA derived from RNA isolated from various mouse tissues was as described [23].
Tumor susceptibility
Mice were tested for DMBA induced tumor formation as previously described [69].
Supporting Information
Quantification of BubR1 protein levels in BubR1 +/GTTA and BubR1 +/−MEFs. (A) Western blot analysis of serially diluted wildtype and representative BubR1 +/GTTA and BubR1 +/− MEF lysates probed for BubR1. Actin was used as a loading control. (B) The average BubR1 signal intensity of three independent wildtype MEF lines plotted against percentage of lysate volume loaded using the equation (Top left). (C) Relative BubR1 protein amount in 3 independent BubR1 +/GTTA and BubR1 +/− MEF lines presented as average ± SD.
(TIF)
Heart function appears normal in BubR1 +/GTTA mice. (A) Cardiac arrhythmia measurements of wildtype and BubR1 +/GTTA 15-month-old male mice shown as percentage of sinus pause disturbances per heartbeat. BPM, beats per minute. (B) Cardiac stress tolerance was determined by injection of a lethal dose of isoproterenol (680 mg/kg) and time to death was recorded. Error bars represent SEM. For isoproterenol experiments 4 wildtype and 4 BubR1 +/GTTA 15-month-old males were used.
(TIF)
BubR1 +/GTTA mice show no evidence of sarcopenia at a young age. Mean fiber diameter measurements on cross sections of the gastrocnemius (Gastro), abdominal (ABD) and paraspinal muscle in wildtype and BubR1 +/GTTA mice at 3 months of age. Error bars represent SEM. For all analysis n = 3 wildtype and n = 5 BubR1 +/GTTA males.
(TIF)
Bone composition is similar in aged BubR1 +/GTTA mice. Bone composition of 15-month-old wildtype and BubR1 +/GTTA mice as determined by DEXA scanning. BMD, bone mineral density; BMC, bone mineral content.
(TIF)
Normal age-related decline of dermal thickness in BubR1 +/GTTA mice. Measurements of the average dermal thickness on cross sections from the lateral skin at the indicated ages. Error bars represent SEM. n = 4 15-month-old wildtype and BubR1 +/GTTA males; n = 7 24-month-old wildtype mice; and n = 8 BubR1 +/GTTA 24-month-old mice.
(TIF)
BubR1 +/GTTA mice are not prone to spontaneous tumors. (A) Spontaneous tumor incidence in 24-month-old wildtype and BubR1 +/GTTA animals. Mice were sacrificed and screened for lymphomas, carcinomas and sarcomas.(B) Tumor spectrum of 24-month-old wildtype and BubR1 +/GTTA mice. We note that values in A and B were not statistically different (Fishers' exact test).
(TIF)
Summary of aging-related phenotypes in wildtype, BubR1 +/GTTA, BubR1 +/− and BubR1 H/H mice. Aneuploidy rates in MEFs are indicated as percentage increase over wildtype controls (wildtype controls for BubR1+/GTTA MEFs had 12% aneuploidy, whereas those of BubR1 +/− and BubR1 H/H MEFs had 9% aneuploidy). ND, not determined. 1 denotes previous published results [21]; 2 [38]; and 3 [42].
(TIF)
Acknowledgments
We thank Wei Zhou and Ming Li of Mayo Clinic's Gene Knockout Mouse Core Facility for ES cell microinjection and chimera breeding, Paul Galardy for helpful discussions and feedback on the manuscript.
Funding Statement
This work was supported by NIH grant R01-CA96985 and the Noaber Foundation. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Ricke RM, van Ree JH, van Deursen JM (2008) Whole chromosome instability and cancer: a complex relationship. Trends Genet 24: 457–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Siegel JJ, Amon A (2012) New Insights into the Troubles of Aneuploidy. Annu Rev Cell Dev Biol 28: 189–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Nagaoka SI, Hassold TJ, Hunt PA (2012) Human aneuploidy: mechanisms and new insights into an age-old problem. Nat Rev Genet 13: 493–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Holland AJ, Cleveland DW (2012) Losing balance: the origin and impact of aneuploidy in cancer. EMBO Rep 13: 501–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bakhoum SF, Compton DA (2012) Chromosomal instability and cancer: a complex relationship with therapeutic potential. J Clin Invest 122: 1138–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pfau SJ, Amon A (2012) Chromosomal instability and aneuploidy in cancer: from yeast to man. EMBO Rep 13: 515–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Baker DJ, Dawlaty MM, Galardy P, van Deursen JM (2007) Mitotic regulation of the anaphase-promoting complex. Cellular and molecular life sciences: CMLS 64: 589–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Musacchio A, Salmon ED (2007) The spindle-assembly checkpoint in space and time. Nature Reviews Molecular Cell Biology 8: 379–393. [DOI] [PubMed] [Google Scholar]
- 9. Peters JM (2006) The anaphase promoting complex/cyclosome: a machine designed to destroy. Nat Rev Mol Cell Biol 7: 644–656. [DOI] [PubMed] [Google Scholar]
- 10. Lampson MA, Kapoor TM (2005) The human mitotic checkpoint protein BubR1 regulates chromosome-spindle attachments. Nature Cell Biology 7: 93–+. [DOI] [PubMed] [Google Scholar]
- 11. Cahill DP, Lengauer C, Yu J, Riggins GJ, Willson JK, et al. (1998) Mutations of mitotic checkpoint genes in human cancers. Nature 392: 300–303. [DOI] [PubMed] [Google Scholar]
- 12. Lengauer C, Kinzler KW, Vogelstein B (1997) Genetic instability in colorectal cancers. Nature 386: 623–627. [DOI] [PubMed] [Google Scholar]
- 13. Rio Frio T, Lavoie J, Hamel N, Geyer FC, Kushner YB, et al. (2010) Homozygous BUB1B mutation and susceptibility to gastrointestinal neoplasia. N Engl J Med 363: 2628–2637. [DOI] [PubMed] [Google Scholar]
- 14. Weaver BA, Cleveland DW (2006) Does aneuploidy cause cancer? Curr Opin Cell Biol 18: 658–667. [DOI] [PubMed] [Google Scholar]
- 15. Hanks S, Coleman K, Reid S, Plaja A, Firth H, et al. (2004) Constitutional aneuploidy and cancer predisposition caused by biallelic mutations in BUB1B. Nat Genet 36: 1159–1161. [DOI] [PubMed] [Google Scholar]
- 16. Matsuura S, Matsumoto Y, Morishima K, Izumi H, Matsumoto H, et al. (2006) Monoallelic BUB1B mutations and defective mitotic-spindle checkpoint in seven families with premature chromatid separation (PCS) syndrome. Am J Med Genet A 140: 358–367. [DOI] [PubMed] [Google Scholar]
- 17. Garcia-Castillo H, Vasquez-Velasquez AI, Rivera H, Barros-Nunez P (2008) Clinical and genetic heterogeneity in patients with mosaic variegated aneuploidy: Delineation of clinical subtypes. Am J Med Genet A 146A: 1687–1695. [DOI] [PubMed] [Google Scholar]
- 18. Suijkerbuijk SJ, van Osch MH, Bos FL, Hanks S, Rahman N, et al. (2010) Molecular causes for BUBR1 dysfunction in the human cancer predisposition syndrome mosaic variegated aneuploidy. Cancer Res 70: 4891–4900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dai W, Wang Q, Liu T, Swamy M, Fang Y, et al. (2004) Slippage of mitotic arrest and enhanced tumor development in mice with BubR1 haploinsufficiency. Cancer Res 64: 440–445. [DOI] [PubMed] [Google Scholar]
- 20. Wang Q, Liu T, Fang Y, Xie S, Huang X, et al. (2004) BUBR1 deficiency results in abnormal megakaryopoiesis. Blood 103: 1278–1285. [DOI] [PubMed] [Google Scholar]
- 21. Baker DJ, Jeganathan KB, Cameron JD, Thompson M, Juneja S, et al. (2004) BubR1 insufficiency causes early onset of aging-associated phenotypes and infertility in mice. Nature genetics 36: 744–749. [DOI] [PubMed] [Google Scholar]
- 22. Baker DJ, Jeganathan KB, Malureanu L, Perez-Terzic C, Terzic A, et al. (2006) Early aging-associated phenotypes in Bub3/Rae1 haploinsufficient mice. J Cell Biol 172: 529–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Baker DJ, Perez-Terzic C, Jin F, Pitel K, Niederlander NJ, et al. (2008) Opposing roles for p16Ink4a and p19Arf in senescence and ageing caused by BubR1 insufficiency. Nature cell biology 10: 825–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Matsumoto T, Baker DJ, d'Uscio LV, Mozammel G, Katusic ZS, et al. (2007) Aging-associated vascular phenotype in mutant mice with low levels of BubR1. Stroke 38: 1050–1056. [DOI] [PubMed] [Google Scholar]
- 25. Hanks S, Coleman K, Summersgill B, Messahel B, Williamson D, et al. (2006) Comparative genomic hybridization and BUB1B mutation analyses in childhood cancers associated with mosaic variegated aneuploidy syndrome. Cancer letters 239: 234–238. [DOI] [PubMed] [Google Scholar]
- 26. Limwongse C, Schwartz S, Bocian M, Robin NH (1999) Child with mosaic variegated aneuploidy and embryonal rhabdomyosarcoma. Am J Med Genet 82: 20–24. [DOI] [PubMed] [Google Scholar]
- 27. Hanks S, Rahman N (2005) Aneuploidy-cancer predisposition syndromes: a new link between the mitotic spindle checkpoint and cancer. Cell Cycle 4: 225–227. [PubMed] [Google Scholar]
- 28. Kajii T, Ikeuchi T, Yang ZQ, Nakamura Y, Tsuji Y, et al. (2001) Cancer-prone syndrome of mosaic variegated aneuploidy and total premature chromatid separation: Report of five infants. American Journal of Medical Genetics 104: 57–64. [DOI] [PubMed] [Google Scholar]
- 29. Garcia-Castillo H, Vasquez-Velasquez AI, Rivera H, Barros-Nunez P (2008) Clinical and genetic heterogeneity in patients with mosaic variegated aneuploidy: Delineation of clinical subtypes. American Journal of Medical Genetics Part A 146A: 1687–1695. [DOI] [PubMed] [Google Scholar]
- 30. Kajii T, Kawai T, Takumi T, Misu H, Mabuchi O, et al. (1998) Mosaic variegated aneuploidy with multiple congenital abnormalities: Homozygosity for total premature chromatid separation trait. American Journal of Medical Genetics 78: 245–249. [DOI] [PubMed] [Google Scholar]
- 31. Plaja A, Vendrell T, Smeets D, Sarret E, Gili T, et al. (2001) Variegated aneuploidy related to premature centromere division (PCD) is expressed in vivo and is a cancer-prone disease. Am J Med Genet 98: 216–223. [DOI] [PubMed] [Google Scholar]
- 32. Ganmore I, Smooha G, Izraeli S (2009) Constitutional aneuploidy and cancer predisposition. Hum Mol Genet 18: R84–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Matsuura S, Ito E, Tauchi H, Komatsu K, Ikeuchi T, et al. (2000) Chromosomal instability syndrome of total premature chromatid separation with mosaic variegated aneuploidy is defective in mitotic-spindle checkpoint. Am J Hum Genet 67: 483–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kawame H, Sugio Y, Fuyama Y, Hayashi Y, Suzuki H, et al. (1999) Syndrome of microcephaly, Dandy-Walker malformation, and Wilms tumor caused by mosaic variegated aneuploidy with premature centromere division (PCD): report of a new case and review of the literature. J Hum Genet 44: 219–224. [DOI] [PubMed] [Google Scholar]
- 35. Ricke RM, Jeganathan KB, van Deursen JM (2011) Bub1 overexpression induces aneuploidy and tumor formation through Aurora B kinase hyperactivation. J Cell Biol 193: 1049–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. van Ree JH, Jeganathan KB, Malureanu L, van Deursen JM (2010) Overexpression of the E2 ubiquitin-conjugating enzyme UbcH10 causes chromosome missegregation and tumor formation. J Cell Biol 188: 83–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Malureanu L, Jeganathan KB, Jin F, Baker DJ, van Ree JH, et al. (2010) Cdc20 hypomorphic mice fail to counteract de novo synthesis of cyclin B1 in mitosis. J Cell Biol 191: 313–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Baker DJ, Wijshake T, Tchkonia T, LeBrasseur NK, Childs BG, et al. (2011) Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 479: 232–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Knollmann BC, Casimiro MC, Katchman AN, Sirenko SG, Schober T, et al. (2004) Isoproterenol exacerbates a long QT phenotype in Kcnq1-deficient neonatal mice: Possible roles for human-like Kcnq1 isoform 1 and slow delayed rectifier K+ current. Journal of Pharmacology and Experimental Therapeutics 310: 311–318. [DOI] [PubMed] [Google Scholar]
- 40. Wang L, Yang L, Debidda M, Witte D, Zheng Y (2007) Cdc42 GTPase-activating protein deficiency promotes genomic instability and premature aging-like phenotypes. Proc Natl Acad Sci U S A 104: 1248–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tyner SD, Venkatachalam S, Choi J, Jones S, Ghebranious N, et al. (2002) p53 mutant mice that display early ageing-associated phenotypes. Nature 415: 45–53. [DOI] [PubMed] [Google Scholar]
- 42. Baker DJ, Jeganathan KB, Malureanu L, Perez-Terzic C, Terzic A, et al. (2006) Early aging-associated phenotypes in Bub3/Rae1 haploinsufficient mice. The Journal of cell biology 172: 529–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. LeBrasseur NK, Schelhorn TM, Bernardo BL, Cosgrove PG, Loria PM, et al. (2009) Myostatin Inhibition Enhances the Effects of Exercise on Performance and Metabolic Outcomes in Aged Mice. Journals of Gerontology Series a-Biological Sciences and Medical Sciences 64: 940–948. [DOI] [PubMed] [Google Scholar]
- 44. Roubenoff R (2000) Sarcopenia and its implications for the elderly. Eur J Clin Nutr 54 Suppl 3: S40–47. [DOI] [PubMed] [Google Scholar]
- 45. Tchkonia T, Morbeck DE, Von Zglinicki T, Van Deursen J, Lustgarten J, et al. (2010) Fat tissue, aging, and cellular senescence. Aging cell 9: 667–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Baker DJ, Jeganathan KB, Cameron JD, Thompson M, Juneja S, et al. (2004) BubR1 insufficiency causes early onset of aging-associated phenotypes and infertility in mice. Nat Genet 36: 744–749. [DOI] [PubMed] [Google Scholar]
- 47. Baker DJ, Perez-Terzic C, Jin F, Pitel K, Niederlander NJ, et al. (2008) Opposing roles for p16Ink4a and p19Arf in senescence and ageing caused by BubR1 insufficiency. Nat Cell Biol 10: 825–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Burtner CR, Kennedy BK (2010) Progeria syndromes and ageing: what is the connection? Nat Rev Mol Cell Biol 11: 567–578. [DOI] [PubMed] [Google Scholar]
- 49. Hasty P, Campisi J, Hoeijmakers J, van Steeg H, Vijg J (2003) Aging and genome maintenance: lessons from the mouse? Science 299: 1355–1359. [DOI] [PubMed] [Google Scholar]
- 50. Garinis GA, van der Horst GT, Vijg J, Hoeijmakers JH (2008) DNA damage and ageing: new-age ideas for an age-old problem. Nat Cell Biol 10: 1241–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Moser MJ, Bigbee WL, Grant SG, Emond MJ, Langlois RG, et al. (2000) Genetic instability and hematologic disease risk in Werner syndrome patients and heterozygotes. Cancer Res 60: 2492–2496. [PubMed] [Google Scholar]
- 52. Lebel M, Leder P (1998) A deletion within the murine Werner syndrome helicase induces sensitivity to inhibitors of topoisomerase and loss of cellular proliferative capacity. Proc Natl Acad Sci U S A 95: 13097–13102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. van der Horst GT, Meira L, Gorgels TG, de Wit J, Velasco-Miguel S, et al. (2002) UVB radiation-induced cancer predisposition in Cockayne syndrome group A (Csa) mutant mice. DNA Repair (Amst) 1: 143–157. [DOI] [PubMed] [Google Scholar]
- 54. Andressoo JO, Weeda G, de Wit J, Mitchell JR, Beems RB, et al. (2009) An Xpb mouse model for combined xeroderma pigmentosum and cockayne syndrome reveals progeroid features upon further attenuation of DNA repair. Mol Cell Biol 29: 1276–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. de Boer J, Donker I, de Wit J, Hoeijmakers JH, Weeda G (1998) Disruption of the mouse xeroderma pigmentosum group D DNA repair/basal transcription gene results in preimplantation lethality. Cancer Res 58: 89–94. [PubMed] [Google Scholar]
- 56. de Boer J, Andressoo JO, de Wit J, Huijmans J, Beems RB, et al. (2002) Premature aging in mice deficient in DNA repair and transcription. Science 296: 1276–1279. [DOI] [PubMed] [Google Scholar]
- 57. Baker DJ, Jin F, van Deursen JM (2008) The yin and yang of the Cdkn2a locus in senescence and aging. Cell Cycle 7: 2795–2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Schvartzman JM, Sotillo R, Benezra R (2010) Mitotic chromosomal instability and cancer: mouse modelling of the human disease. Nat Rev Cancer 10: 102–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kim AH, Puram SV, Bilimoria PM, Ikeuchi Y, Keough S, et al. (2009) A centrosomal Cdc20-APC pathway controls dendrite morphogenesis in postmitotic neurons. Cell 136: 322–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Yang Y, Kim AH, Yamada T, Wu B, Bilimoria PM, et al. (2009) A Cdc20-APC ubiquitin signaling pathway regulates presynaptic differentiation. Science 326: 575–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Babu JR, Jeganathan KB, Baker DJ, Wu X, Kang-Decker N, et al. (2003) Rae1 is an essential mitotic checkpoint regulator that cooperates with Bub3 to prevent chromosome missegregation. The Journal of cell biology 160: 341–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Malureanu LA (2011) Targeting vector construction through recombineering. Methods Mol Biol 693: 181–203. [DOI] [PubMed] [Google Scholar]
- 63. O'Gorman S, Dagenais NA, Qian M, Marchuk Y (1997) Protamine-Cre recombinase transgenes efficiently recombine target sequences in the male germ line of mice, but not in embryonic stem cells. Proc Natl Acad Sci U S A 94: 14602–14607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kasper LH, Brindle PK, Schnabel CA, Pritchard CEJ, Cleary ML, et al. (1999) CREB binding protein interacts with nucleoporin-specific FG repeats that activate transcription and mediate NUP98-HOXA9 oncogenicity. Molecular and Cellular Biology 19: 764–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Hamada M, Malureanu LA, Wijshake T, Zhou W, van Deursen JM (2012) Reprogramming to pluripotency can conceal somatic cell chromosomal instability. PLoS Genet 8: e1002913 doi:10.1371/journal.pgen.1002913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Babu JR, Jeganathan KB, Baker DJ, Wu X, Kang-Decker N, et al. (2003) Rae1 is an essential mitotic checkpoint regulator that cooperates with Bub3 to prevent chromosome missegregation. J Cell Biol 160: 341–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Pajvani UB, Trujillo ME, Combs TP, Iyengar P, Jelicks L, et al. (2005) Fat apoptosis through targeted activation of caspase 8: a new mouse model of inducible and reversible lipoatrophy. Nature Medicine 11: 797–803. [DOI] [PubMed] [Google Scholar]
- 68. Martinez-Fernandez A, Nelson TJ, Yamada S, Reyes S, Alekseev AE, et al. (2009) iPS programmed without c-MYC yield proficient cardiogenesis for functional heart chimerism. Circulation Research 105: 648–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Serrano M, Lee HW, Chin L, CordonCardo C, Beach D, et al. (1996) Role of the INK4a locus in tumor suppression and cell mortality. Cell 85: 27–37. [DOI] [PubMed] [Google Scholar]
- 70. Wang C, Li Q, Redden DT, Weindruch R, Allison DB (2004) Statistical methods for testing effects on “maximum lifespan”. Mech Ageing Dev 125: 629–632. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Quantification of BubR1 protein levels in BubR1 +/GTTA and BubR1 +/−MEFs. (A) Western blot analysis of serially diluted wildtype and representative BubR1 +/GTTA and BubR1 +/− MEF lysates probed for BubR1. Actin was used as a loading control. (B) The average BubR1 signal intensity of three independent wildtype MEF lines plotted against percentage of lysate volume loaded using the equation (Top left). (C) Relative BubR1 protein amount in 3 independent BubR1 +/GTTA and BubR1 +/− MEF lines presented as average ± SD.
(TIF)
Heart function appears normal in BubR1 +/GTTA mice. (A) Cardiac arrhythmia measurements of wildtype and BubR1 +/GTTA 15-month-old male mice shown as percentage of sinus pause disturbances per heartbeat. BPM, beats per minute. (B) Cardiac stress tolerance was determined by injection of a lethal dose of isoproterenol (680 mg/kg) and time to death was recorded. Error bars represent SEM. For isoproterenol experiments 4 wildtype and 4 BubR1 +/GTTA 15-month-old males were used.
(TIF)
BubR1 +/GTTA mice show no evidence of sarcopenia at a young age. Mean fiber diameter measurements on cross sections of the gastrocnemius (Gastro), abdominal (ABD) and paraspinal muscle in wildtype and BubR1 +/GTTA mice at 3 months of age. Error bars represent SEM. For all analysis n = 3 wildtype and n = 5 BubR1 +/GTTA males.
(TIF)
Bone composition is similar in aged BubR1 +/GTTA mice. Bone composition of 15-month-old wildtype and BubR1 +/GTTA mice as determined by DEXA scanning. BMD, bone mineral density; BMC, bone mineral content.
(TIF)
Normal age-related decline of dermal thickness in BubR1 +/GTTA mice. Measurements of the average dermal thickness on cross sections from the lateral skin at the indicated ages. Error bars represent SEM. n = 4 15-month-old wildtype and BubR1 +/GTTA males; n = 7 24-month-old wildtype mice; and n = 8 BubR1 +/GTTA 24-month-old mice.
(TIF)
BubR1 +/GTTA mice are not prone to spontaneous tumors. (A) Spontaneous tumor incidence in 24-month-old wildtype and BubR1 +/GTTA animals. Mice were sacrificed and screened for lymphomas, carcinomas and sarcomas.(B) Tumor spectrum of 24-month-old wildtype and BubR1 +/GTTA mice. We note that values in A and B were not statistically different (Fishers' exact test).
(TIF)
Summary of aging-related phenotypes in wildtype, BubR1 +/GTTA, BubR1 +/− and BubR1 H/H mice. Aneuploidy rates in MEFs are indicated as percentage increase over wildtype controls (wildtype controls for BubR1+/GTTA MEFs had 12% aneuploidy, whereas those of BubR1 +/− and BubR1 H/H MEFs had 9% aneuploidy). ND, not determined. 1 denotes previous published results [21]; 2 [38]; and 3 [42].
(TIF)