

Abstract
Thermal adaptation is essential in all organisms. In yeasts, the heat shock response is commanded by the heat shock transcription factor Hsf1. Here we have integrated unbiased genetic screens with directed molecular dissection to demonstrate that multiple signalling cascades contribute to thermal adaptation in the pathogenic yeast Candida albicans. We show that the molecular chaperone heat shock protein 90 (Hsp90) interacts with and down-regulates Hsf1 thereby modulating short term thermal adaptation. In the longer term, thermal adaptation depends on key MAP kinase signalling pathways that are associated with cell wall remodelling: the Hog1, Mkc1 and Cek1 pathways. We demonstrate that these pathways are differentially activated and display cross talk during heat shock. As a result ambient temperature significantly affects the resistance of C. albicans cells to cell wall stresses (Calcofluor White and Congo Red), but not osmotic stress (NaCl). We also show that the inactivation of MAP kinase signalling disrupts this cross talk between thermal and cell wall adaptation. Critically, Hsp90 coordinates this cross talk. Genetic and pharmacological inhibition of Hsp90 disrupts the Hsf1-Hsp90 regulatory circuit thereby disturbing HSP gene regulation and reducing the resistance of C. albicans to proteotoxic stresses. Hsp90 depletion also affects cell wall biogenesis by impairing the activation of its client proteins Mkc1 and Hog1, as well as Cek1, which we implicate as a new Hsp90 client in this study. Therefore Hsp90 modulates the short term Hsf1-mediated activation of the classic heat shock response, coordinating this response with long term thermal adaptation via Mkc1- Hog1- and Cek1-mediated cell wall remodelling.
Author Summary
Candida albicans is one of the most persistent yeast pathogens known to man, causing frequent mucosal infections (thrush) in otherwise healthy individuals, and potentially fatal bloodstream infections in immunocompromised patients. C. albicans colonises warm-blooded animals and occupies thermally buffered niches. Yet during its evolution this pathogen has retained the classic heat shock response whilst other stress responses have diverged significantly. We have established that the essential, evolutionarily conserved molecular chaperone, Hsp90, coordinates thermal adaptation. Hsp90 interacts with and modulates the activity of the heat shock transcription factor, Hsf1, thereby controlling the expression of heat shock proteins required for the clearance of proteins damaged by proteotoxic stresses. In addition, Hsp90 modulates the activities of key MAP kinase signalling pathways that mediate cell wall remodelling and long term adaptation to heat shock. Loss of any of these factors results in a significant reduction in thermotolerance.
Introduction
Microorganisms inhabit dynamic environments and are continually challenged with environmental stimuli and stresses. Microbial survival depends upon effective environmental response strategies that have been elaborated over evolutionary time. These cellular strategies have been intensively studied in various contemporary model organisms [1], [2], [3], [4]. The emergent paradigm is that cells react to environmental changes via a sense and respond logic: they continuously monitor their environment, and upon encountering a stimulus, mount a cellular response [5]. This is achieved through diverse signalling pathways that drive physiological adaptation to a myriad of environmental stresses that include temperature fluctuations, osmotic, oxidative and weak acid stresses, as well as nutrient limitation [6], [7].
Fungal pathogens have evolved robust stress responses that enable them to counteract the antimicrobial defences of their host, thereby promoting the colonisation of specific niches. The major fungal pathogen of humans, Candida albicans, is an opportunistic pathogen that has evolved as a relatively harmless commensal of the mucous membranes and digestive tracts of healthy individuals [8], [9]. C. albicans is a common cause of mucosal infections (thrush) and when antimicrobial defences become compromised this yeast can cause life-threatening systemic infections [8], [10]. Stress responses are critical for survival of C. albicans inside the human body, and genetic inactivation of these responses attenuates virulence of this pathogen [11], [12], [13]. However, the regulation of these stress signalling mechanisms has diverged significantly in C. albicans compared with other yeasts [14]. For example, unlike Saccharomyces cerevisiae, Schizosaccharomyces pombe or Candida glabrata [2], [15], [16], C. albicans does not activate a large core transcriptional response [3]. The core transcriptional responses of S. cerevisiae, S. pombe and C. glabrata involve the activation of common sets of stress genes by one particular stress that promote cross-protection to diverse stresses [2], [15], [16]. In S. cerevisiae and C. glabrata, this core transcriptional response and hence stress cross-protection are dependent on the transcription factors Msn2 and Msn4, which activate target genes via stress response elements (STRE) in their promoters [17]. In S. pombe, the core stress response is driven largely by the Sty1 stress activated protein kinase (SAPK: the orthologue of Hog1 in other yeasts) [15]. In contrast, C. albicans does not mount a broad core transcriptional response to stress, there is limited stress cross-protection in this yeast, and the roles of Hog1 and Msn2/Msn4-like transcription factors have diverged in this pathogen [2], [3], [18], [19], [20]. Whilst, C. albicans does appear to activate a relatively specialised core transcriptional response to osmotic, oxidative and heavy metal stresses [19], the consensus view is that this pathogen mounts relatively specific responses to particular environmental challenges. This involves activation of the corresponding signal transduction pathway, subsequent activation of the relevant set of stress genes and requisite changes in cell physiology, morphology and adherence [21], [22], [23].
A number of stress regulatory modules have been conserved between C. albicans and other yeasts. For example, the AP1-like transcription factors S. cerevisiae Yap1 [24], S. pombe Pap1 [25], C. glabrata CgYap1 [26], [27] and C. albicans Cap1 [28] play analogous roles in the activation of transcriptional responses to oxidative stress. Also, the Hog1/Sty1 SAPK is conserved in these yeasts, although the orthologues have diverged with respect to the stress responses they regulate. S. cerevisiae Hog1 is primarily involved in responses to osmotic stress, whereas C. albicans Hog1 and S. pombe Sty1 contribute to a diverse range of stress responses [29], [30], [31]. Additional mitogen activated protein kinase (MAPK) cascades have been conserved between S. cerevisiae and C. albicans. These include the cell wall integrity Mpk1/Slt2 pathway [32] and the cell wall, morphogenesis and pheromone signalling pathways involving the Cek1 and Cek2 MAPKs [33], [34]. These MAP kinase pathways contribute to thermotolerance in S. cerevisiae [35], [36], [37], although the mechanisms by which they do so remain obscure. These MAP kinase pathways are also important for virulence, as MAP kinase defective C. albicans mutants display attenuated virulence in infection models [11], [13], [33], [38].
The heat shock response is among the most fundamentally important and ubiquitous stress responses in nature. The heat shock transcription factor (Hsf1) which drives this response, is conserved from yeasts to humans [39], [40]. Indeed, the Hsf1 module and the heat shock response are even conserved in C. albicans, an organism that is obligately associated with warm blooded mammals and hence occupies thermally buffered niches [41]. Furthermore Hsf1 is essential for viability in C. albicans [42] and other yeasts [43]. These observations reflect the fundamental importance of heat shock adaptation in all organisms. Even in the absence of stress, Hsf1 binds as a trimer to canonical heat shock elements (HSEs) in the promoters of target heat shock protein (HSP) genes [44], [45], [46]. When S. cerevisiae or C. albicans cells are exposed to an acute heat shock, Hsf1 becomes hyper-phosphorylated and activated, leading to the transcriptional induction of these target HSP genes, thereby promoting cellular adaptation to the thermal insult [39], [47].
Many HSPs are molecular chaperones that promote the folding, assembly, or cellular localisation of client proteins [48]. They also minimise the aggregation of unfolded or damaged proteins and often target such proteins for degradation [48]. HSPs are critical for the survival of eukaryotic cells under normal conditions as well as following exposure to an acute heat shock. Indeed our recent exploration of the dynamic regulation of Hsf1 during thermal adaptation has suggested that the Hsf1-HSE regulon is activated even during slow thermal transitions such as the increases in temperature suffered by febrile patients [49]. This explains why the Hsf1-HSE regulon is active in C. albicans cells infecting the mammalian kidney, and why activation of this regulon is essential for virulence of C. albicans [42]. Clearly the Hsf1-HSE regulon is critical for the maintenance of thermal homeostasis, not merely for adaptation to acute heat shocks.
Hsp90 has been suggested to play a critical role in regulation of the Hsf1-HSE regulon, contributing to an autoregulatory circuit involving Hsp90 and Hsf1 [49]. In the absence of stress, Hsp90 is generally expressed at relatively high levels [50], and is thought to repress Hsf1 [49]. However, thermal and other proteotoxic stresses can induce global problems in protein folding that overwhelm the functional capacity of Hsp90 [50]. Under these conditions the repression of Hsf1 by Hsp90 was proposed to be released, allowing Hsf1 to become activated leading to increased HSP90 expression [49].
The repression of Hsf1 by Hsp90 was suggested by the observation that pharmacological inhibition of Hsp90 correlates with HSF1 activation in mammalian cells [51], [52]. Zou and colleagues demonstrated that HSF1 can be cross-linked to Hsp90 in unstressed HeLa cells, suggesting that HSF1 might interact directly with Hsp90 [52]. Additionally, the trimeric form of human HSF1 has been shown to associate with an Hsp90-immunophilin-p23 complex, and this is thought to repress HSF1 transcriptional activity [53]. Furthermore, HSP90 modulates HSF1 regulation in Xenopus oocytes [54]. Hence HSF1 is known to be a client protein of Hsp90 in metazoan cells. However, although mutations that interfere with Hsp90 function have been shown to derepress the expression of Hsf1-dependent reporter genes in S. cerevisiae [55], no physical interaction between Hsf1 and Hsp90 has been demonstrated in the fungal kingdom.
In this study, we explore the regulatory control of cellular circuitry governing the response to temperature stress through an integrated approach involving specific hypotheses and unbiased screens in C. albicans. We determined that Hsf1 is a client protein of Hsp90, establishing for the first time in the fungal kingdom that the Hsf1-Hsp90 interaction is critical for the regulation of short term adaptive responses to heat shock. We questioned how cells adapt to heat stress in the longer term by investigating which other signalling pathways contribute to thermotolerance and executing genetic screens for protein kinase mutations that confer temperature sensitivity. This revealed that the Hog1, Mkc1 and Cek1 MAP kinase pathways contribute to thermotolerance, as does casein kinase signalling. We show that these MAP kinase pathways are not essential for Hsf1 activation. Rather, they contribute to thermal adaptation in the longer term via cell wall remodelling. Hog1, Mkc1 and Cek1 are client proteins of Hsp90, and genetic depletion of Hsp90 affects this cell wall remodelling. Therefore, Hsp90 integrates the short term and long term molecular responses that underpin thermotolerance.
Materials and Methods
Strains and growth conditions
All strains are listed in Table 1, with the exception of the library of C. albicans transposon mutants [56], [57], [58]. Strains were grown in YPD (1% yeast extract, 2% bactopeptone, 2% glucose) [59]. To impose an instant heat shock of 30°C–42°C, cells were grown in YPD at 30°C to exponential phase, and mixed with an equal volume of medium that has been pre-warmed at 54°C in flasks which had been pre-warmed at 42°C. Cells were grown at 42°C for the times indicated. Doxycycline was added to YPD medium at a concentration of 20 µg/ml. Geldanamycin was added at 10 µM (A.G. Scientific, Inc., San Diego, USA), and radicicol at 20 µM (A.G. Scientific, Inc.).
Table 1. C. albicans strains.
| Strain | Genotype | Source |
| ML250 | ade2::hisG/ade2::hisG, ura3::λimm434/ura3::λimm434, ENO1/eno1::ENO1-tetR-ScHAP4AD-3XHA-ADE2, HSF1/HSF1, pACT1-FLAG-HSF1 | [49] |
| SN95 | arg4Δ/arg4Δ his1Δ/his1Δ URA3/ura3:: λimm434 IRO1/iro1::λimm434 | [102] |
| CaLC1411 (CaLC436) | arg4Δ/arg4Δ his1Δ/his1Δ URA3/ura3:: λimm434 IRO1/iro1:: λimm434 HIS1/his1::tetR-FRT FRT-tetO-HSP90/hsp90::CdHIS1 | [73] |
| CaLC501 | arg4Δ/arg4Δ his1Δ/his1Δ URA3/ura3:: λimm434 IRO1/iro1:: λimm434 HSP90/HSP90-TAP-FRT HSF1/HSF1 | [73] |
| CaLC1819 | arg4Δ/arg4Δ his1Δ/his1Δ URA3/ura3:: λimm434 IRO1/iro1:: λimm434 HSP90/HSP90 ACT1p-3xFLAG-HSF1/HSF1 | this study |
| CaLC1875 | arg4Δ/arg4Δ his1Δ/his1Δ URA3/ura3:: λimm434 IRO1/iro1::λimm434 HSP90/HSP90-TAP-FRT ACT1p-3xFLAG-HSF1/HSF1 | this study |
| CaLC1855 | arg4/arg4 his1/his1 URA3/ura3::imm434 IRO1/iro1::imm434 HSP90/HSP90-GFP-NAT | this study |
| BWP17 | ura3::λimm434/ura3::λimm434, his1::hisG/his1::hisG, arg4::hisG/arg4::hisG | [103] |
| CLM60-1 | ade2::hisG/ade2::hisG, ura3::λimm434/ura3::λimm434, ENO1/eno1::ENO1-tetR-ScHAP4AD-3XHA-ADE2, hsf1::hisG-URA3-hisG/HSF1 | [41] |
| mkc1Δ (CM1613C) | mkc1Δ::hisG-CaURA3-hisG/mkc1Δ::hisG ura3Δ::λimm434/ura3Δ::λimm434 | [32] |
| CaLC648 | As SN95, MKC1/MKC1-6xHIS-FLAG-FRT, HIS1/his1::TAR-FRT hsp90::CdHIS1/tetO-HSP90 | [86] |
| CaLC681 | As SN95, MKC1/MKC1-6xHIS-FLAG-FRT | [86] |
| hog1Δ (JC50) | ura3::λimm434/ura3::λimm434, his1::hisG/his1::hisG, hog1::loxP-ura3-loxP/hog1::loxP-HIS1-loxP CIp20 | [30] |
| cek1Δ (CK43B16) | ura3/ura3 cek1Δ::hisG-URA3-hisG/cek1Δ::hisG | [33] |
| cst20Δ (CDH22) | ura3/ura3 cst20Δ::hisG-URA3-hisG/cst20Δ::hisG | [104] |
| cka1Δ (VIC108) | cka1Δ::ARG4/cka1Δ::URA3 | [61] |
| cka2Δ (VIC84) | cka2Δ::ARG4/cka2Δ::URA3 | [61] |
| ckb1Δ (VIC138) | ckb1Δ::ARG4/ckb1Δ::URA3 | [61] |
| ckb2Δ (VIC150) | ckb2Δ::ARG4/ckb2Δ::URA3 | [61] |
| CAI4 | ura3::λimm434/ura3::λimm434 | [105] |
| CAI4+CIp10 (NGY152) | ura3::λimm434/ura3::λimm434 (RPS10-CIp10-URA3) | [106] |
| MLC67 | ura3Δ::λimm434/ura3Δ::λimm434 RPS1::pACT1-3xFLAG-HSF1/HSF1 | This study |
| MLC15 | ura3:: λimm434/ura3::λimm434, his1::hisG/his1::hisG, hog1::loxP-ura3-loxP/hog1::loxP-HIS1-loxP, RPS1::pACT1-FLAG-HSF1(NAT1) | This study |
| MLC21 | ura3/ura3 cek1Δ::hisG-URA3-hisG/cek1Δ::hisG, RPS1::pACT1-FLAG-HSF1(NAT1) | This study |
| MLC24 | cka1Δ::ARG4/cka1Δ::URA3, RPS1::pACT1-FLAG-HSF1(NAT1) | This study |
| MLC27 | cka2Δ::ARG4/cka2Δ::URA3, RPS1::pACT1-FLAG-HSF1(NAT1) | This study |
| MLC30 | mkc1Δ:: hisG/mkc1Δ::hisG ura3Δ::λimm434/ura3Δ::λimm434, RPS1::pACT1-FLAG-HSF1(NAT1) | This study |
| CaLC2259 | ckb21::ARG4/ckb21::URA3, RPS1::pACT1-FLAG-HSF1(NAT1) | This study |
| CaLC2261 | ckb22::ARG4/ckb22::URA3, RPS1::pACT1-FLAG-HSF1(NAT1) | This study |
| hog1/hst7Δ (CHH13) | ura3Δ::imm434/ura3Δ::imm434 hst7Δ::hisG/hst7Δ::hisG hog1::hisG-URA3-hisG/hog1::hisG | [81] |
| cap1Δ (JC128) | ura3:: λimm434/ura3::λimm434, his1::hisG/his1::hisG, arg4::hisG/arg4::hisG, cap1::hisG/cap1::hisG-URA3-hisG | [19] |
| CaLC2287 | arg4/arg4 his1/his1 URA3/ura3::imm434 IRO1/iro1::imm434, CEK1/CEK1-TAP-ARG4 | This study |
| CaLC2288 | arg4/arg4 his1/his1 URA3/ura3::λimm434 IRO1/iro1::λimm434, HIS1/his1::TAR-FRT, hsp90::CdHIS1/FRT-tetO-HSP90, CEK1/CEK1-TAP-ARG4 | This study |
Strain construction
For co-immunoprecipitation of Hsf1 and Hsp90, Hsf1 was tagged with FLAG and Hsp90 was tagged with the tandem affinity purification (TAP) tag. The plasmid pACT1pHSF1, containing the ACT1p-3xFLAG-HSF1 construct [41] was linearized with StuI and transformed into the strains SN95 (wild type) or CaLC501 (HSP90-TAP) (Table 1) using published procedures [60]. Nourseothricin (NAT) resistant transformants were selected on YPD containing 150 µg/mL NAT, and insertion of the ACT1p-3xFLAG-HSF1 cassette confirmed by diagnostic PCR using the primers oLC1117/oLC1628 (Table S1).
Localisation of Hsp90 was achieved by 3′-tagging of one HSP90 allele with GFP in the wild type strain SN95 (creating CaLC1855, Table 1). The GFP-NAT cassette was amplified using primers oLC1616/1617 (Table S1) and transformed into SN95. Proper integration of the cassette in NAT resistant transformants was confirmed by PCR using primer pairs oLC600/756 (Table S1).
To determine Hsf1 phosphorylation status, the pACT1pHSF1, containing the ACT1p-3xFLAG-HSF1 construct [41] was linearized with StuI and transformed into mkc1, hog1, cek1, cka1, cka1, ckb1, ckb2 mutants (Table 1) [30], [32], [33], [61]. NAT resistant transformants were selected and confirmed by diagnostic PCR and expression of FLAG-Hsf1 in western blots.
Cek1 was tagged with the TAP tag at its C-terminus in the wild type strain SN95 (creating CaLC2287, Table 1) and its derivative CaLC1411 (creating CaLC2288, Table 1) using a PCR based strategy as described previously [62]. Briefly, the tag and a selectable marker (ARG4) were PCR amplified from pLC573 (pFA-TAP-ARG4 [63]) using oligos oLC2292/2251 (Table S1). 200 µl of PCR product was run through a PCR clean-up and dissolved in 50 µl sterile water and transformed into C. albicans. Correct genomic integration was verified using appropriate primer pairs that anneal ∼500 bp up (oLC2252) or downstream (oLC2253) from both insertion junctions together with oLC1593 (TAP-R) and oLC1594 (ARG4-F) that target the TAP and the selectable marker (Table S1).
Western blotting
Total soluble protein was extracted and subjected to western blotting using published protocols [64], [65]. Briefly, mid-log cells were pelleted by centrifugation, washed with sterile water, and resuspended in lysis buffer (50 mM HEPES, pH 7.5, 150 mM NaCl, 5 mM EDTA, 1% Triton X-100). An equal volume of 0.5-mm acid-washed beads was added to each tube. Cells were mechanically disrupted on a BioSpec (Bartlesville, OK) mini-bead-beater for six 30 second intervals, with 1 minute on ice between each cycle. The beads and cell debris were pelleted by high-speed centrifugation and the supernatant removed for analysis. Protein concentration was determined using a Bradford reagent (Sigma-Aldrich) assay. Protein samples were mixed with one-sixth volume of 6× sample buffer containing 0.35 M Tris-HCl, 10% (w/w) SDS, 36% glycerol, 5% β-mercaptoethanol, and 0.012% bromophenol blue. Between 2 µg and 30 µg of protein was loaded in wells of an 8% SDS–PAGE gel. Separated proteins were transferred to a PVDF membrane for 1 hour at 100 V at 4°C. Membranes were blocked in 5% milk or 5% bovine serum albumin (BSA) in TBS or PBS containing 0.1% Tween-20 at room temperature for 1 hour and subsequently incubated in primary antibody as follows. All primary antibodies (except those against p38 and p44/42 MAPK) were left on the membrane for one hour at room temperature. The p38 MAPK and p44/42 MAPK antibodies were incubated overnight at 4°C. Membranes were washed with 1×PBS-T or TBS-T and probed for one hour with secondary antibody dissolved in 1×PBS-T and 5% milk or BSA. Membranes were washed in PBS-T and signals detected using an ECL western blotting kit as per the manufacturer's instructions (Pierce).
To detect FLAG-Hsf1, a 1∶25000 dilution of anti-FLAG HRP conjugated antibody (Sigma, A8592) was used in PBS-T+5% milk [PBS 0.1% Tween-20, 5% (w/v) milk]. To detect Act1, an anti-Act1 antibody was used (Santa Cruz Biotechnology, sc47778) at a 1∶1000 dilution in PBS-T+5% milk. To detect Hsp90, a 1∶10000 dilution of anti-Hsp90 antibody was used (courtesy of Bryan Larson) in PBS-T+5% milk. To detect Hsp70, a 1∶1000 dilution of anti-Hsp70 antibody (Enzo Life Sciences, ADI-SPA-822) was used in PBS-T+5% milk.
To detect Mkc1 and Cek1 phosphorylation [22], [66], a 1∶2000 dilution of phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) Rabbit mAb was used (New England Biolabs, Hitchin, Hertfordshire, UK, #4370) in TBS-T+5% BSA [TBS 0.1% Tween-20, 5% (w/v) BSA]. For Hog1 phosphorylation [30], a 1∶2000 dilution of phospho-p38 MAPK (Thr180/Tyr182) rabbit mAB was used in TBS-T+5% BSA (New England Biolabs, Hitchin, Hertfordshire, UK, #9211).
To detect Mkc1-6XHis-FLAG, the anti-FLAG-HRP antibody was used as above. To detect total Hog1 an anti-Hog1 antibody (Santa Cruz Biotechnology, y-215) was diluted 1∶1000 in PBS-T+5% milk. TAP-tagged Cek1 was detected using a 1∶5000 dilution of anti-TAP tag rabbit polyclonal antibody (Thermoscientific, CAB1001) in PBS-T+5% milk.
Co-immunoprecipitations
C. albicans cultures were grown to mid-log phase (OD600 = 0.5), cells harvested, washed with sterile H20 and resuspended in 1 ml of lysis buffer (20 mM Tris pH 7.5, 100 mM KCl, 5 mM MgCl and 20% glycerol, with one protease inhibitor cocktail per 50 ml (complete, EDTA-free tablet, Roche Diagnostics, Indianapolis, IN, USA), 1 mM PMSF (EMD Chemicals, Gibbstown, NJ, USA) and 20 mM sodium molybdate (Sigma Aldrich Co., St Louis, MO, USA)). Cells were then disrupted by bead beating twice for 4 minutes with a 7 minute break on ice between cycles. Lysates were centrifuged at 1300×g for three 5-minute cycles, recovering the supernatants at each stage. The combined lysate was then cleared by centrifugation at 21,000×g for 10 minutes at 4°C and protein concentrations determined using the Bradford assay [67].
Anti-FLAG immunoprecipitations were performed by diluting protein samples to 2 mg/ml in lysis buffer containing 20 mM sodium molybdate and 0.2% Tween, and incubating with anti-FLAG M2 affinity agarose (Sigma Aldrich) at 4°C overnight as per the manufacturer's specifications. Unbound material was discarded, the beads washed five times with 1 ml lysis buffer containing 0.1% Tween, and the bound proteins eluted by boiling in one volume of 2× sample buffer (125 mM Tris-HCl, pH 6.8, 5% glycerol, 2.5% SDS, 2.5% beta-mecarptoethanol, dH2O, bromophenol blue). Anti-IgG immunoprecipitations were performed using the same approach, but using rabbit IgG agarose (Sigma Aldrich) as per the manufacturer's specifications.
Protein samples were then electrophoresed on 8% SDS-PAGE gels. Proteins were then electrotransferred to PVDF membranes (Bio-Rad Laboratories, Inc., Hercules, CA, USA) and blocked with PBS-T+5% milk. Blots were incubated with antibodies against CaHsp90 (courtesy of Bryan Larsen) (1∶10000 dilution, [68]), or FLAG (1∶10000, Sigma Aldrich Co.).
qRT-PCR
To monitor gene expression changes in response to tetO-HSP90 depletion, strains SN95 and CaLC1411 were grown overnight at 30°C in YPD while shaking at 200 rpm. Stationary phase cultures were split, adjusted to an OD600 of 0.04 where one culture was treated with doxycycline (BD Biosciences), while the other was left untreated. Cells were grown for 7 hours at 30°C. To monitor gene expression changes in response to heat shock, wild type and SN95 cells were grown to mid-log phase, subjected to a 30°C–42°C heat shock and 50 ml was harvested from each culture at the specified time, centrifuged at 3000 rpm for 2 minutes at 4°C, washed once with dH2O before being frozen at −80°C. RNA was then isolated using the QIAGEN RNeasy kit and cDNA synthesis was performed using the AffinityScript cDNA synthesis kit (Stratagene). PCR was carried out using the SYBR Green JumpStart Taq ReadyMix (Sigma-Aldrich) with the following cycle conditions: 94°C for 2 minutes, and 94°C for 15 seconds, 60°C for 1 minute, 72°C for 1 minute, for 40 cycles. All reactions were done in triplicate using the following primer pairs: HSP104 (oLC1620/1621), HSP90 (oLC754/755), PGA13 (oLC2256/2257), PMT4 (oLC2262/2263), RHR2 (oLC2266/2267). Transcript levels were normalised to ACT1 (oLC2285/2286) (Table S1). Data were analysed in the StepOne analysis software (Applied Biosystems).
Temperature sensitivity screen
The C. albicans transposon insertion mutant library was generously provided by Aaron Mitchell (Carnegie Mellon University) [56], [58]. Strains were inoculated in 100 µl YPD in 96 well plates and incubated overnight at 30°C with shaking at 200 rpm. Cells were then diluted 1∶10 in YPD and incubated at 30°C with shaking at 200 rpm for 4 hours. After 4 hours of exponential growth, cells were then exposed to a one hour 30°C–42°C heat shock by addition of 100 µl pre-warmed YPD at 54°C and incubating at 42°C, and the growth of each culture monitored continuously for 6 hours by measuring the OD600. Non-heat shocked control cells received 100 µl YPD at 30°C and were incubated at 30°C. This screen was repeated independently three times, and only those mutants that displayed consistent phenotypes were taken for further analysis. The validity of key transposon mutants highlighted by this screen was then confirmed by subjecting corresponding homozygous null mutants to the same screen.
Minimum inhibitory concentration assays and stress phenotypes
Unless otherwise stated, the susceptibilities of strains were determined with the following stressors: heat stress (42°C heat shock), Calcofluor White (CFW: 100 µg/ml), Congo Red (CR: 100 µg/ml), H2O2 (5 mM) and NaCl (1 M). All stress assays were performed in YPD.
MIC assays were performed in flat bottom, 96-well microtiter plates (Corning Costar). Briefly, assays were set up in 0.2 ml/well, with 2× concentrations of NaCl, CFW and CR prepared in 100 µl YPD. Final concentrations of NaCl were 0, 0.25, 0.5, 1.0, 1.5 and 2 M; and for CFW and CR were 0, 25, 50, 100, 150 and 200 µg/ml. Cell densities of overnight cultures were determined and dilutions were prepared in YPD such that ∼103 cells were inoculated into each well. Plates were placed statically at either 25°C, 30°C, 37°C or 42°C for 48 hours, after which plates were sealed and cells resuspended by agitation. Absorbance was determined at 600 nm using a spectrophotometer (VERSA max, Molecular Devices), and was corrected for background from the corresponding medium. Every strain was tested in duplicate on three separate occasions. MIC data were quantitatively displayed with colour using the program Java TreeView 1.1.6 (http://jtreeview.sourceforge.net).
For stress cross-protection assays, cells were grown overnight in YPD at 30°C with shaking at 200 rpm. These were diluted to an OD600 = 0.2 in fresh YPD, and grown for a further 4 hours at 30°C. Cells were then subjected to a 30 minute heat shock at 42°C, incubated at 30°C and stressed for one hour with CFW, CR, H2O2 or NaCl. Cells were diluted, plated onto YPD and viability determined (CFUs). Control cells were not subjected to the prior heat shock. In other experiments cells were exposed to a prior CFW, CR, H2O2 or NaCl stress for one hour at 30°C before being heat shocked at 42°C for 30 min, and then cell viability determined.
To test stress sensitivity following Hsp90 depletion, C. albicans tetO-HSP90 cells (CaLC1411: Table 1) were incubated for 7 hours in YPD containing 20 µg/ml doxycycline at a starting OD600 of 0.04. These cells were left untreated or stressed for one hour with CFW, CR, a 30°C–42°C heat shock, H2O2 or NaCl at the concentrations specified above, and CFUs determined. Controls included CaLC1411 cells that were not treated with doxycycline, and parental SN95 cells that were subjected to the same treatments. Growth curves of CaLC1411 (tetO-HSP90/hsp90) were determined in the absence and presence of doxycycline to ensure adequate viability of this strain (Supplementary Figure S1C).
To determine kinase activation in response to Hsp90 depletion, wild type SN95 and tetO-HSP90 (CaLC1411) were grown overnight in YPD at 30°C. Cells were diluted in YPD with or without doxycycline at a starting OD600 of 0.04 and incubated for 7 hours. Cells were then left untreated or stressed with H2O2 for 10 minutes and NaCl for 12 minutes to determine Hog1 activation. For Mkc1 and Cek1 activation, tagged versions of these proteins in SN95 and tetO-HSP90 (CaLC1411) were grown as above and stressed with CFW for 30 minutes or a 30°C–42°C heat shock for 30 minutes. Mkc1 total levels were assayed using Mkc1-6XHis-FLAG tagged in SN95 and tetO-HSP90 (CaLC681 and CaLC648 respectively, Table 1). Total Cek1 levels were assayed using Cek1-TAP tagged in SN95 and tetO-HSP90 (CaLC2287 and CaLC2288 respectively, Table 1).
Chitin content
The chitin content of cells was measured as described previously [69]. Briefly, formalin fixed cells were stained with 25 µg/ml Calcofluor White and fluorescence was preserved with Vectashield mounting medium (Vector Laboratories, Peterborough, United Kingdom). All samples were examined by differential interference contrast (DIC) and fluorescence microscopy (456 nm) with a Zeiss Axioplan 2 microscope. Images were captured using a C4742-95 digital camera (Hamamatsu Photonics, Hamamatsu, Japan) and analysed using Openlab software (version 4.04: Improvision, Coventry, United Kingdom). CFW fluorescence was quantified for 50 individual yeast cells from each sample, using region-of-interest measurements. Mean fluorescence intensities were then calculated and expressed as arbitrary units.
Fluorescence microscopy
Exponentially growing Hsp90-GFP (CaLC1855, Table 1) cells were heat shocked as described above, and 1 ml of cells were removed at 0, 10, 60 and 120 minutes post-heat shock. Cells were harvested and washed in 1 ml of 42°C pre-warmed 1×PBS. Supernatant was removed and cells were resuspended in the remaining 50 µl. Of this, 5 µl was placed on a slide, and cells were heat fixed by placing the slide on a 70°C hot plate for 1 minute. Slides were cooled for a few seconds before 4 µl of 1 µg/ml DAPI (Fluke, Sigma-Aldrich) were added. Imaging was performed on a Zeiss Imager M1 upright microscope and AxioCam MRm with AxioVision 4.7 software. An X-Cite series120 light source with ET green fluorescent protein (GFP) and 4′,6-diamidino-2-phenylindole (DAPI) hybrid filter sets from ChromaTechnology (Bellows Falls, VT) were used for fluorescence microscopy. DAPI fluorescence was viewed under the DAPI hybrid filter and GFP-tagged proteins under the GFP filter.
Transmission electron microscopy
For high pressure freezing transmission electron microscopy (HPF-TEM), cells were prepared by high-pressure freezing with a Leica EM PACT2 (Leica Microsystems (UK) Ltd, Milton Keynes). After freezing, cells were freeze-substituted in substitution reagent (1% OsO4/0.1% uranyl acetate in acetone) with a Leica EM AFS2. Samples were encapsulated in 3% (w/v) low melting point agarose prior to processing to Spurr resin. Additional infiltration was provided under vacuum at 60°C before embedding in TAAB capsules and polymerizing at 60°C for 48 h. Semi-thin survey sections of 0.5 µM thickness were stained with 1% toluidine blue to identify areas of best cell density. Ultrathin sections (60 nm) were prepared with a Diatome diamond knife on a Leica UC6 ultramicrotome, and stained with uranyl acetate and lead citrate for examination with a Philips CM10 transmission microscope (FEI UK Ltd, Cambridge, UK) and imaging with a Gatan Bioscan 792 (Gatan UK, Abingdon, UK).
Results
Hsp90 negatively regulates Hsf1
An autoregulatory loop, whereby Hsf1 activates HSP90 expression and Hsp90 interacts with and down-regulates Hsf1, is thought to lie at the heart of heat shock adaptation in fungi. This presumption provided the basis for mathematical modelling of thermal adaptation in C. albicans [49], but had not been confirmed experimentally. If this presumption is true, one would expect that inhibition of Hsp90, or Hsp90 depletion would lead to Hsf1 activation [49]. To test this, we examined the impact of the Hsp90 inhibitors, radicicol and geldanamycin [70], [71], upon Hsf1 phosphorylation. C. albicans cells (ML250: Table 1) were incubated with radicicol or geldanamycin for up to one hour, and Hsf1 phosphorylation was monitored via the resultant band shift revealed by western blot analysis, as described previously [41] (Figure 1A). These Hsp90 inhibitors induced Hsf1 phosphorylation after one hour, as confirmed by the band shift observed after controlled dephosphorylation with lambda phosphatase. To exclude the possibility that this Hsf1 phosphorylation was induced by a general effect of the drugs upon protein folding, we examined the impact of dithiothreitol and tunicamycin, known inducers of the unfolded protein response in C. albicans [72]. Hsf1 was not activated following treatment with 5 mM dithiothreitol or 4.73 µM tunicamycin for 1 hour (data not shown), suggesting that the induction of Hsf1 phosphorylation by radicicol and geldanamycin related to Hsp90 function rather than some general effect upon protein folding. Therefore, Hsp90 inhibits Hsf1, as predicted.
Figure 1. Hsf1 down-regulates Hsp90.

(A) Inhibiting Hsp90 with geldanamycin leads to Hsf1 activation. C. albicans cells were incubated with 20 µM geldanamycin for one hour, and Hsf1 phosphorylation examined by western blotting. (B) Depleting Hsp90 leads to Hsf1 activation. Doxycycline treatment of C. albicans tetO-HSP90 cells was used to ectopically down-regulate Hsp90 levels, and Hsf1 phosphorylation was then examined by western blotting: black arrow, phosphorylated Hsf1; white arrow, non-phosphorylated Hsf1. Hsf1 phosphorylation was confirmed using lambda phosphatase controls. (C) Hsp90 depletion causes protracted Hsf1 phosphorylation during a 30°C–42°C heat shock. Exponentially growing C. albicans tetO-HSP90 cells were transferred from 30°C to 42°C, and 20 µg/ml doxycycline was added. Hsf1 phosphorylation was examined every hour up to 4 hours. Data reflect the outcomes for at least two independent replicate experiments. (D) HSP104 transcript levels increase following Hsp90 depletion in tetO-HSP90 cells. WT (wild type, SN95: Table 1) and tetO-HSP90 cells (CaLC1411) were treated with 0 or 20 µg/ml doxycycline for 7 hours, and HSP104 transcript levels were measured and normalised to the ACT1 loading control. Wild-type cells were also subjected to a 30 minute 30°C–42°C heat shock for HSP104 comparison.
Next, we validated our pharmacological findings using a genetic approach. A doxycycline conditional C. albicans HSP90 mutant (tetO-HSP90) [73], in which HSP90 expression is independent of Hsf1, was used to ectopically down-regulate Hsp90 levels (Figure 1B and S1A). Doxycycline treatment led to Hsf1 phosphorylation after approximately 6 hours, at which point Hsp90 levels were reduced by 50% (Figure 1B and S1A). Therefore, ectopic down-regulation of HSP90 caused Hsf1 phosphorylation even in the absence of a heat shock, further reinforcing the hypothesis that Hsp90 inhibits Hsf1.
A key finding from our modelling of thermal adaptation was that this system displays perfect adaptation: i.e. Hsf1 activation returns to basal levels within two hours once cells have adapted to their new ambient temperature [49]. If Hsp90 down-regulates the heat shock response, then one would expect this perfect adaptation to be dependent on Hsp90. Also, if new Hsp90 synthesis is inhibited after a heat shock, Hsf1 would remain phosphorylated and the system would not adapt to this stress. We tested this by examining Hsf1 phosphorylation levels in doxycycline-treated C. albicans tetO-HSP90 cells after a 30°C–42°C heat shock. As predicted, these Hsp90-depleted cells were unable to recover, as revealed by the maintenance of Hsf1 phosphorylation four hours after the heat shock (Figure 1C). Therefore, perfect thermal adaptation is dependent upon Hsp90.
To further test the impact of depleting Hsp90 on the heat shock response, we looked at induction of HSP104, a known target of Hsf1 [49]. In wild type cells (SN95: Table 1) HSP104 was up-regulated approximately 10-fold in response to a 30 minute 30°C–42°C heat shock (Figure 1D), which was consistent with previous findings [49], and HSP104 expression was not affected by doxycycline treatment. HSP104 mRNA levels were elevated in tetO-HSP90 cells even in the absence of doxycycline, presumably because Hsp90 levels are significantly reduced under these conditions (Figure S1B). An additional increase in HSP104 expression was observed after doxycycline treatment when Hsp90 levels were reduced further (Figure S1B). Taken together, these data strongly imply that Hsp90 is a master regulator of the heat shock response.
The proposed Hsf1-Hsp90 autoregulatory loop [49] suggests a physical interaction between Hsf1 and Hsp90. There is some evidence for this in mammalian cells [52], [53], but none in fungal systems. Therefore we tested this experimentally by co-immunoprecipitation. First, proteins were extracted from C. albicans cells expressing a FLAG-tagged Hsf1 protein (CaLC1819), and from control cells in which Hsf1 was not FLAG-tagged (WT, SN95). Protein extracts were incubated with anti-FLAG beads, and the resulting immunoprecipitates probed for Hsp90 on western blots. Hsp90 was observed reproducibly in immunoprecipitates from the FLAG-Hsf1 expressing strain, but not in those from the untagged control strain (Figure 2A). Reciprocal immunoprecipitations were then performed to test the validity of this apparent Hsf1-Hsp90 interaction. C. albicans cells expressing both TAP-tagged Hsp90 and FLAG-tagged Hsf1 (CaLC1875) were used in these experiments, and cells lacking the FLAG-tagged Hsf1 (CaLC501: Table 1) were used as a control. Hsp90 was immunoprecipitated using IgG beads, which bind the Protein A in the TAP tag. These immunoprecipitates were then probed for the FLAG-tagged Hsf1, revealing a band of the appropriate mass from cells expressing TAP-tagged Hsp90, but not from the controls (Figure 2B). This confirmed that Hsp90 interacts physically with Hsf1. This is the first demonstration of a physical interaction between Hsf1 and Hsp90 in any yeast.
Figure 2. Hsf1 is an Hsp90 client in C. albicans.

Hsf1 and Hsp90 physically interact as revealed by co-immunoprecipitation. (A) Hsp90 co-purifies with FLAG-Hsf1 after immunoprecipitation of extracts from unstressed cells with anti-FLAG M2 affinity agarose. Hsp90 was not co-immunoprecipitated from control cells lacking tagged Hsf1. (B) Reciprocal co-immunoprecipitation was performed using Hsp90-TAP showing that FLAG-Hsf1 copurifies with Hsp90-TAP upon immunoprecipitation with IgG agarose. FLAG-Hsf1 was not co-immunoprecipiated with IgG agarose in control cells lacking FLAG-Hsf1. Data reflect the outcomes for three independent replicate experiments. (C) Hsp90 co-immunoprecipitates with FLAG-Hsf1 0, 10 and 60 minutes after a 30°C to 42°C heat shock, but does not co-immunoprecipitate in control cells lacking FLAG-Hsf1. The membrane was re-probed for Hsp70, which co-purifies with FLAG-Hsf1 at 60 minutes post-heat shock. The reciprocal co-immunopreciptation validates these results: FLAG-Hsf1 co-immunoprecipitates with Hsp90-TAP at 0, 10 and 60 minutes post-heat shock. Re-probing the membranes for Hsp70, reveals that Hsp70 interacts with Hsp90-TAP at 60 minutes post-heat shock. (D) Localisation of Hsp90-GFP in response to elevated temperature. C. albicans CaLC1855 cells were treated with a 42°C heat shock and fixed at 0, 10 and 60 minutes post-heat shock. Hsp90 is localised in the cytosol at 0 and 10 minutes post-heat shock. Significant accumulation of Hsp90 in the nucleus is observed 60 minutes post-heat shock, as determined by co-staining with DAPI. Scale bars, 5 µm.
An Hsf1-Hsp90 autoregulatory loop, as inferred by the model [49], [74], predicts dynamic changes in the Hsf1-Hsp90 interaction during a heat shock. The model predicts that Hsp90 releases Hsf1 following a heat shock, rebinding Hsf1 as the response is down-regulated. We tested this hypothesis by co-immunoprecipitation, examining Hsp90-Hsf1 interactions over a 60 minute period following heat shock. Rather than decreasing when Hsf1 becomes activated (as was predicted), the Hsp90-Hsf1 interaction increased over 60 minutes (Figure 2C) during the period when Hsf1 activation is maximal [49]. Therefore, Hsf1 is not released from Hsp90 after heat shock, and this mechanism cannot account for Hsf1 activation. We examined the Hsf1-Hsp90 interaction two hours after heat shock, when the response is down-regulated [49], and found that the interaction was still increased when compared to untreated samples or a 10 minute heat shock (Figure S2B, bottom panel). This increase in the Hsf1-Hsp90 interaction after a prolonged heat shock (Figure 2B), accompanies the decline in Hsf1 activity and the down-regulation of the heat shock response [49].
To determine whether other components of the Hsp90 chaperone machinery might also interact with Hsf1, we focused on Hsp70, which operates within the Hsp90 chaperone system [74], and has been implicated in Hsf1 regulation. Indeed, the deletion of SSA1 and SSA2, which encode cytosolic isoforms of Hsp70 derepresses Hsf1 transcriptional activity in S. cerevisiae [75]. However, a physical interaction between Hsp70 and Hsf1 has not been reported in any yeast. Therefore, we re-probed our FLAG-Hsf1 immunoprecipitations with an antibody against Hsp70. We found that Hsp70 interacts with Hsf1, but only in response to a prolonged heat shock (Figure 2C, bottom left panel, and Figure S2A). We also re-probed our Hsp90-TAP immunoprecipitations for Hsp70. Our data suggest that Hsp90 and Hsp70 interact in response to prolonged heat shock (Figure 2C, bottom right panel and Figure S2B, top panel).
Given our findings that the Hsf1-Hsp90 interaction strengthens upon heat shock, and that Hsf1 is thought to bind DNA constitutively [44], one might predict that Hsp90 localises to the nucleus upon heat shock. Indeed, a recent study by Lamoth and colleagues shows Hsp90 nuclear localisation upon a 55°C heat shock in Aspergillus fumigatus [76]. Therefore we followed the localisation of Hsp90-GFP in C. albicans in response to a heat shock (CaLC1855, Table 1). Under steady state conditions, Hsp90-GFP was distributed throughout the cell, with no obvious localisation (Figure 2D), and this was also the case 10 minutes after a 42°C heat shock. However, 60 minutes post-heat shock, nuclear accumulation of Hsp90-GFP was clearly evident (Figure 2D), and Hsp90-GFP remained in the nucleus 120 minutes post heat shock (Figure S2C). Therefore the dynamics of the nuclear accumulation of Hsp90-GFP correlated with the dynamics of the Hsf1-Hsp90 interaction. These data reinforce the view that Hsp90 and the Hsp90 chaperone machine plays an important role in the down-regulation of Hsf1 and the heat shock response.
How is Hsf1 activated?
The above observations indicate that while Hsp90 down-regulates Hsf1, Hsf1 activation is mediated by Hsp90-independent mechanisms. How then is Hsf1 activated? C. albicans Hsf1 is activated by phosphorylation [41], but the protein kinase responsible for this in yeast remains unknown [52], [53]. To determine which kinase is responsible for Hsf1 phosphorylation in C. albicans, we exploited the recent availability of the C. albicans transposon insertion kinase mutant collection which was kindly provided by Aaron Mitchell [58]. This collection of mutants which comprises homozygous insertion or deletion mutations in 67 protein kinase genes and 13 protein kinase-related genes, were screened for temperature sensitivity by monitoring their growth following a one hour 30°C–42°C heat shock. Mutants that consistently displayed a >10% growth defect relative to the wild-type controls in three independent screens were considered temperature sensitive, and those that consistently displayed >10% growth acceleration were considered temperature resistant (Figure 3). The tetO-HSF1 mutant (CLM60-1: Table 1) was always temperature sensitive, and the known temperature sensitivity of mkc1 mutants [32] was consistently recapitulated. (Mkc1 is the orthologue of S. cerevisiae Slt2 [32].) These observations leant weight to the validity of the output from this screen. In this study we focussed on those kinases whose inactivation confers temperature sensitivity.
Figure 3. Key MAP kinase signalling pathways contribute to thermal adaptation in C. albicans.
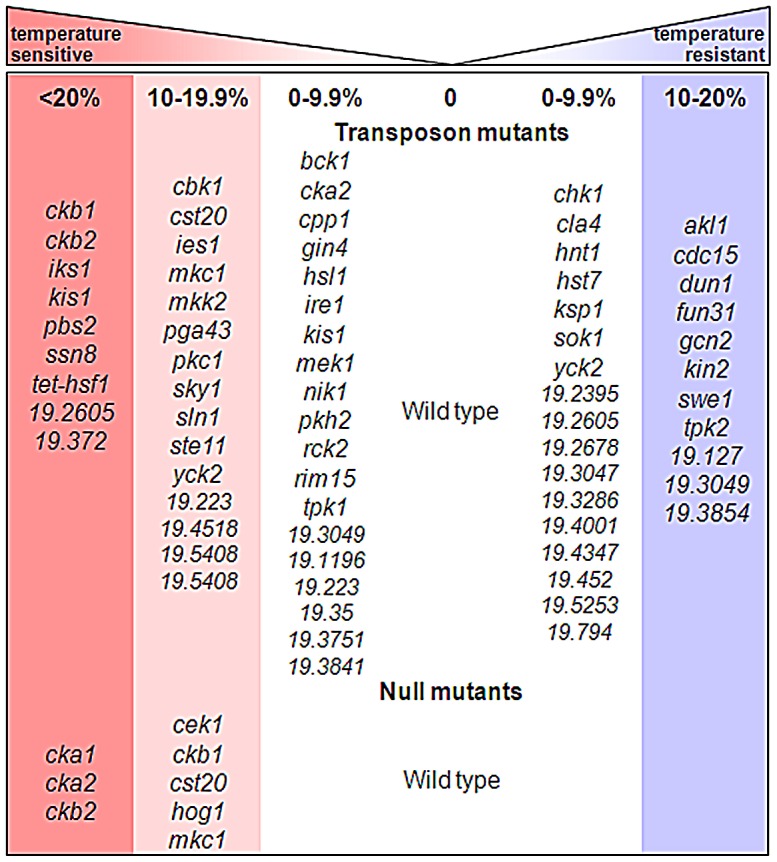
Figure depicting protein kinase mutants screened for temperature phenotypes. Temperature sensitivity was assayed by monitoring the growth of C. albicans mutants following a 30°C–42°C heat shock. Triplicate primary screens examined transposon mutants [57], and triplicate secondary screens tested C. albicans null mutants (Table 1). Temperature resistant kinases are displayed in the blue column, kinases with no apparent role in thermal adaptation are displayed in the pale columns, and temperature sensitive kinases are present in the pink (10–19%) to red (>20%) columns.
Numerous kinase mutants displayed significant and reproducible temperature sensitivity in our screen, notably the casein kinase subunits as well as components of key MAP kinase pathways such as the cell wall integrity pathway (Mkc1, Pkc1), the osmotic stress pathway (Pbs2) and the starvation and cell wall stress pathway (Ste11) (Figure 3). Several downstream kinases of these pathways were absent from the mutant collection, prompting us to perform a secondary screen of null mutants. This secondary screen confirmed the temperature sensitivity of all of the transposon mutants tested from the primary screen (Figure 3), including the casein kinase subunits (Cka1/2 and Ckb1/2) and Mkc1. Furthermore, the secondary screen tested the MAP kinases Cek1 and Hog1 (which were missing from the collection of transposon mutants), thereby confirming the involvement of these pathways in C. albicans thermotolerance (Figure 3).
Our next aim was to test whether any of the kinases identified in the above screen are responsible for Hsf1 phosphorylation in response to heat shock. Hsf1 was FLAG3-tagged at its N-terminus in CAI4 (MLC67) and in each of the following temperature sensitive null mutants: mkc1 (MLC30), hog1 (MLC15), cek1 (MLC21), cka1 (MLC24), cka2 (MLC27), ckb1 (CaLC2259) and ckb2 (CaLC2261) (Table 1). Each of these mutants was then subjected to a 30°C–42°C heat shock, and Hsf1 phosphorylation assayed at 0, 10, 30 and 60 minutes post-heat shock (Figure S3). All mutants displayed similar Hsf1 phosphorylation dynamics to wild-type cells, indicating that none of the kinases alone is essential for Hsf1 phosphorylation. Therefore, there might be functional redundancy with respect to Hsf1 phosphorylation during heat shock. Alternatively, Hsf1 might be phosphorylated by an essential kinase that was not represented in the kinase mutant collection. These observations also suggest that the MAP kinase pathways might contribute to thermal adaptation in C. albicans by mechanisms other than via Hsf1 phosphorylation and the activation of the heat shock regulon.
MAP kinase pathways display differential activation profiles during thermal adaptation
Next we explored how the signalling pathways contribute to thermotolerance. A recent and elegant study by Diezmann and colleagues [62] demonstrated that casein kinase 2 (CK2) regulates Hsp90 phosphorylation and activity. Therefore, the mutations in CK2 subunits (cka1, cka2, ckb1 and ckb2) probably reduce thermotolerance by interfering with Hsp90 function. In addition to regulating Hsf1 (Figures 1 and 2), which is critical for thermal adaptation [49], Hsp90 has numerous other client proteins, some of which may contribute to thermotolerance [62].
We therefore focussed on the roles of the three MAP kinase pathways that were highlighted by our screen: the cell wall integrity Mkc1 pathway, the osmolarity/oxidative stress Hog1 pathway and the starvation/cell wall response Cek1 pathway. First we monitored the activation of each MAP kinase during heat shock. Wild-type CAI4 cells were subjected to a 30°C–42°C heat shock, proteins extracted at 0, 10, 30 and 60 minutes, and MAP kinase phosphorylation probed by western blotting. Each MAP kinase responded differently to the thermal upshift (Figure 4A). Mkc1 was rapidly dephosphorylated before being re-phosphorylated in the longer term. Cek1 phosphorylation levels remained relatively stable. Hog1 was rapidly dephosphorylated, and was not reactivated during the one hour period examined (Figure 4A). These data were entirely consistent with previous studies that have reported effects of temperature on Hog1, Cek1 and Mkc1 phosphorylation [22], [30], [77], [78]. Significantly, total kinase levels remain unchanged during the 60 minute heat shock.
Figure 4. Differential activation profiles of MAP kinases in response to heat shock.

(A) Phosphorylation of C. albicans Mkc1, Cek1 and Hog1 during a 30°C–42°C heat shock revealed by western analysis using phospho-specific antibodies. Total kinase levels were monitored using antibodies against total Hog1, FLAG-tagged Mkc1 and TAP-tagged Cek1. Actin served as an internal loading control. (B) Expression of target genes for Mkc1 (PGA13), Cek1 (PMT4) and Hog1 (RHR2) during a 30°C–42°C heat shock, as determined by qRT-PCR of the corresponding transcripts relative to the internal ACT1 mRNA control.
To further validate the effects of heat shock upon these signalling pathways, we examined the expression of key MAP kinase targets: PGA13 is an Mkc1 target [78], PMT4 is a Cek1 target [79] and RHR2 is a Hog1 target [30]. RNA was extracted from cells 0, 10, 30 and 60 minutes after a 30°C–42°C heat shock, and transcript levels assayed by qRT-PCR relative to the internal ACT1 mRNA control. The levels of these targets reflected the activation profiles of the corresponding MAP kinase (Figure 4B).
Cross-talk between the Mkc1, Hog1 and Cek1 pathways during heat shock
MAP kinase signalling is often represented in terms of linear pathways. However, in C. albicans, as in other organisms there is emerging evidence for cross-talk between these pathways [22], [80], [81], [82]. Therefore we tested whether they interact during thermal adaptation by comparing the phosphorylation status of the terminal kinases in wild type and MAP kinase mutants following a 30°C–42°C heat shock (Figure 5). First, we examined Cek1 and Hog1 activation in an mkc1 mutant (Figure 5A). Mkc1 inactivation did not affect the responses of either Hog1 or Cek1. Second, we monitored Hog1 and Mkc1 phosphorylation in a cek1 mutant (Figure 5B). After Cek1 inactivation, the transient dephosphorylation of Mkc1 was inhibited. Hog1 phosphorylation levels still declined over the duration of the heat shock. Third, we tested the effects of Hog1 inactivation upon Mkc1 and Cek1 (Figure 5C). Mkc1 phosphorylation was minimal and remained low when hog1 cells were exposed to the heat shock. In contrast, we observed that Cek1 was hyperphosphorylated in hog1 cells, as reported previously [22], [81], [83]. Our data reveal that there are significant interactions between the MAP kinase signalling pathways during thermal adaptation.
Figure 5. Cross-talk between MAP kinase pathways during heat shock.
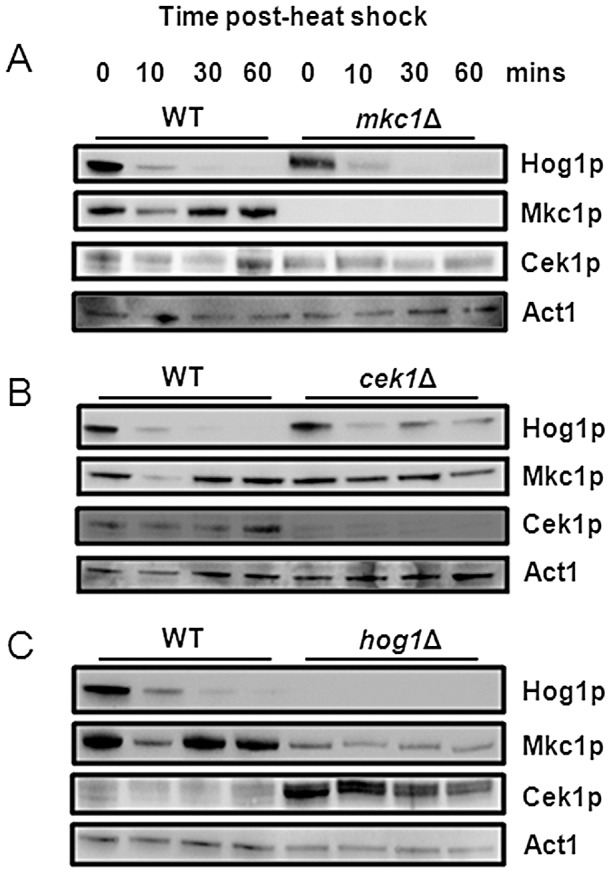
Differential MAP kinase activation in response to a 30°C–42°C heat shock in MAP kinase mutants. (A) Activation of Hog1, Mkc1 and Cek1 was determined in an mkc1Δ mutant relative to the internal Act1 control. (B) Activation of Hog1, Mkc1 and Cek1 was assayed in a cek1Δ mutant relative to the internal Act1 control. (C) Activation of Hog1, Mkc1 and Cek1 was determined in a hog1Δ mutant relative to the internal Act1 control.
Thermal adaptation affects Mkc1, Hog1 and Cek1-mediated resistance to other stresses
Given this cross-talk and the modulation of MAP kinase activities after thermal up-shifts, we reasoned that ambient temperature is likely to influence the resistance of C. albicans cells to those stresses normally associated with these signalling pathways. For example, Hog1 signalling mediates osmotic stress adaptation [84], and the Mkc1 and Cek1 pathways promote resistance to the cell wall stresses Calcofluor White and Congo Red [22]. Therefore we performed MICs to test the effects of ambient temperature upon the resistance of wild-type (NGY152: Table 1), hog1, cek1 and mkc1 cells to these stresses (Figure 6). The mkc1 cells were temperature sensitive, as reported [32], [84]. Also, as reported previously, hog1 cells were sensitive to NaCl [84]. Furthermore, mkc1 and cek1 cells displayed sensitivity to Calcofluor White [22], [81], [83]. These controls faithfully replicated previous observations.
Figure 6. Impact of MAP kinase cross-talk upon stress resistance during thermal adaptation.

Cek1 signalling is required for the Calcofluor White resistance of hog1 cells at high temperatures. MIC assays were performed in YPD medium supplemented with different concentrations of NaCl, Calcofluor White or Congo Red. Plates were incubated statically at 25°C, 30°C, 37°C and 42°C for 48 hours. For each strain, optical densities were averaged for duplicate measurements and growth is quantitatively displayed with colour as indicated with the colour bar. Data are representative of three biological replicates. WT, wild type (NGY152: Table 1).
We observed that ambient temperature significantly influences the sensitivity of C. albicans cells to cell wall, but not osmotic stresses (Figure 6). Firstly, wild-type cells were more sensitive to Calcofluor White at lower temperatures (25°C and 30°C). Secondly, hog1 cells were relatively resistant to Calcofluor White at all temperatures tested, and phenocopied wild type cells on Congo Red. Thirdly, cek1 cells were resistant to Congo Red at most temperatures, but sensitive at 42°C. Clearly ambient temperature significantly affects cell wall stress resistance. These data reinforce the notion of cross-talk between thermal and cell wall stress signalling pathways.
We reasoned that the Calcofluor resistance of hog1 cells at low temperatures (Figure 6) might be Cek1 dependent. This is because inactivation of Hog1 led to elevated Cek1 phosphorylation levels (Figure 5C), and Cek1 promotes Calcofluor White resistance (Figure 6). Hog1 and cek1 mutations are synthetically lethal [81], and hence we could not examine a hog1 cek1 double mutant. Therefore, instead we tested the phenotype of a hog1/hog1 hst7/hst7 double mutant, in which Cek1 signalling is blocked [81]. As predicted, the inactivation of Cek1 signalling attenuated the Calcofluor White resistance of hog1 cells at low temperatures (Figure 6). This indicates that Hog1 inactivation promotes cell wall stress resistance at low temperatures via Cek1 signalling. These data reinforce the importance of cross-talk between the MAP kinase signalling pathways and highlight the relevance of this cross-talk for thermal adaptation.
MAP kinase cross-talk affects acquired tolerance to heat shock and other stresses
In some yeasts, the core transcriptional response to stress underpins the phenomenon of stress cross-protection, whereby exposure to one stress protects the cell against subsequent exposure to an alternative type of stress via the up-regulation of key stress response genes [2], [4], [15], [16]. The core stress response is limited in C. albicans [19]. Nevertheless, it remained conceivable that the effects of ambient temperature upon the resistance of C. albicans to certain stresses might be mediated through stress cross-protection. To test this, mid-exponential C. albicans cells (NGY152: Table 1) were subjected to a 30 minute 30°C–42°C heat shock and subsequently exposed to a cell wall stress (Congo Red or Calcofluor White), osmotic stress (NaCl), or oxidative stress (hydrogen peroxide). Cell wall or osmotic stress resistance was not enhanced by prior exposure to heat shock (Figure 7A). Furthermore, prior exposure to cell wall or osmotic stress resistance did not enhance resistance to a subsequent heat shock (Figure 7B). This was consistent with our observation that ambient temperature does not significantly affect osmotic stress resistance (Figure 6), and indicated that the influence of ambient temperature upon Calcofluor White resistance is not mediated by stress cross-protection. This was consistent with the divergent core stress response in C. albicans [3], [19].
Figure 7. The cross-talk between thermal adaptation and cell wall stress resistance is not mediated via stress cross-protection.

(A) Stress cross-protection in C. albicans wild-type cells (NGY152: Table 1) was not observed for cells pre-treated with a heat stress and then subjected to cell wall stress, but was observed for cells exposed to a secondary oxidative stress. The data represent cell survival after exposure to a 30°C–42°C heat shock followed by a subsequent cell wall (CFW, CR), osmotic (NaCl) or peroxide (H2O2) stress (see Materials and Methods) and the data are expressed relative to unstressed cells (dark bars). Control cells (grey bars), were not exposed to the prior 30°C–42°C heat shock. (B) The reciprocal assay was performed, whereby C. albicans wild type cells were exposed to a prior stress (cell wall: CFW, CR; osmotic: NaCl; peroxide: H2O2) followed by a 30°C–42°C heat shock (dark bars). These data are expressed relative to unstressed cells. Control cells (grey bars) correspond to cells exposed only to the 30°C–42°C heat shock. (C) Wild type, hog1Δ (JC50) or cap1Δ cells (JC128: Table 1) were pre-treated with a 30°C–42°C heat shock, followed by a H2O2 stress (dark bars). Control cells (grey bars) were not exposed to the 30°C–42°C heat shock. The data represent the level of survival compared to unstressed cells. All data are the means from three independent assays: ** paired, two-tailed t-test, p<0.01.
We included oxidative stress as a control in the above experiments because a prior heat shock has been reported to protect C. albicans against peroxide stress [3]. The mechanisms by which a heat shock protects cells against a subsequent oxidative stress have not been elucidated. We noted that two uncharacterised genes that are induced by oxidative stress are also up-regulated by heat shock: orf19.7882 and orf19.7085 [3], [41], [85]. Both genes are induced in response to oxidative stress in a Cap1-dependent fashion, and are down-regulated by Hog1. Therefore we tested whether Hog1 and Cap1 are required for the observed stress cross-protection (Figure 7C). The hog1 mutant (JC50) displayed a comparable increase in survival to wild type cells when cells were pre-treated with a 30°C–42°C heat shock and then exposed to hydrogen peroxide. In contrast, cap1 cells (JC128: Table 1) lost this stress cross-protection. Therefore, the acquired resistance to hydrogen peroxide after exposure to heat shock is dependent on Cap1. This probably occurs through the Cap1 dependent up-regulation of oxidative stress genes such as orf19.7882 and orf19.7085 in response to heat shock.
Cross-talk between thermotolerance and stress adaptation is mediated by Hsp90
What mechanisms are responsible for the cross-talk between thermotolerance and stress adaptation if this is not mediated by stress cross-protection? Hsp90 is a key regulator of thermal adaptation, regulating its own expression via Hsf1 (Figure 1). In addition, Hsp90 modulates the activities of multifarious client proteins [62]. This list of Hsp90 interactors in C. albicans includes CK2 subunits [62], Mkc1, a well-defined Hsp90 protein client in C. albicans [86], and Hog1 [62]. Mkc1 and Hog1 orthologues are also known Hsp90 client proteins in S. cerevisiae [87], [88]. As these protein kinases were identified in our screen as being important for thermotolerance, we reasoned that Hsp90 might play a significant role in coordinating thermal adaptation in C. albicans. According to this hypothesis, changes in ambient temperature are expected to influence Hsp90 availability [88] and this in turn modulates the activity of its client proteins [49]. These client proteins include Hog1 and Mkc1 which, when activated, induce expression of target genes that promote cellular adaptation to elevated temperatures and other stresses.
A clear prediction of this hypothesis is that Hsp90 depletion should attenuate the cell's ability to withstand specific stresses. To test this, tetO-HSP90 cells were treated with 20 µg/ml doxycycline for 7 hours, by which point Hsp90 levels were significantly reduced (Figure S1A and S1B) and growth was beginning to slow (Figure S1C). These cells were then stressed for one hour with Calcofluor White, Congo Red, a 30°C–42°C heat shock, hydrogen peroxide or NaCl (Materials and Methods). As predicted, Hsp90 depletion significantly attenuated cellular resistance to all of the stresses tested except NaCl when compared to wild type or tetO-HSP90 cells not treated with doxycycline (Figure 8A).
Figure 8. Hsp90 depletion decreases stress resistance.
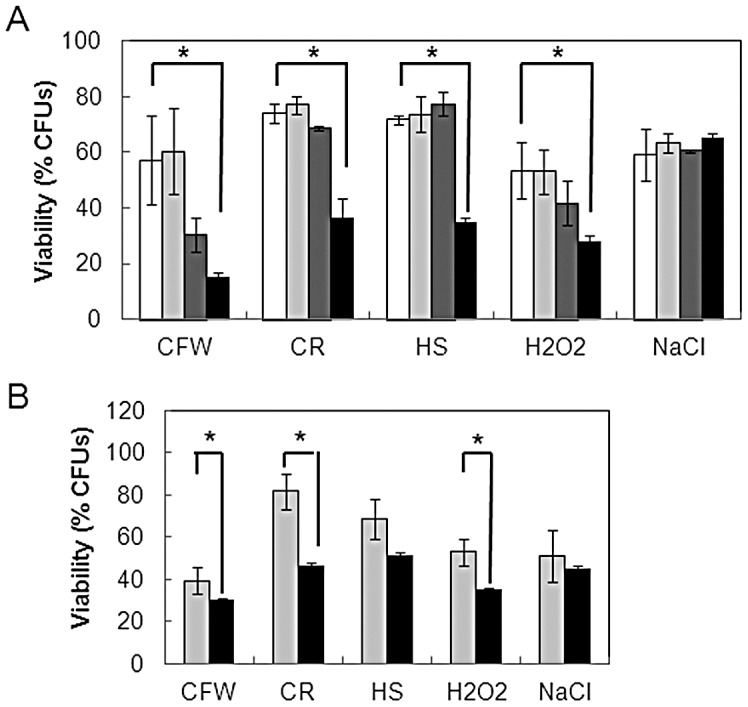
(A) Impact of Hsp90 depletion upon stress resistance (CFUs) was measured following doxycycline treatment of C. albicans wild-type and tetO-HSP90 cells (SN95 and CaLC1411: Table 1): white bars, wild type, no doxycycline; light grey bars, wild type plus 20 µg/ml doxycycline; dark grey bars, tetO-HSP90 no doxycycline; black bars, tetO-HSP90 plus 20 µg/ml doxycycline; CFW, 100 µg/ml Calcofluor White; CR, 100 µg/ml Congo Red; HS, 30°C–42°C heat shock; 5 mM H2O2; 1 M NaCl. (B) Impact of Hsp90 inhibition with the pharmacological inhibitor geldanamycin. Cells were grown for 7 hours in the absence or presence of 10 µM geldanamycin and subjected to the same stresses as in (A). CFUs determined from untreated cells. All data are the means from three independent assays: ** paired, two-tailed t-test, p<0.01.
To test this further, we examined the effects of the Hsp90 inhibitor geldanamycin upon stress resistance. Although the differences were not as dramatic as for genetic depletion of Hsp90 (Figure 8A), similar effects were observed following geldanamycin treatment (Figure 8B). The fact that Hsp90 depletion did not affect osmotic stress resistance was entirely consistent with our previous findings that ambient temperature did not influence osmotic stress resistance (Figure 6), and that there was no stress cross-protection for thermal and osmotic stresses (Figure 7).
Clearly Hsp90 influences cellular responses to a range of stresses, not only to heat shock. To determine whether these effects are mediated through its client proteins we assessed the impact of Hsp90 depletion upon the activation of known client proteins, Mkc1 and Hog1, and the potential client protein, Cek1 (Figure 9). The basal activation of each MAP kinase was examined in wild type and tetO-HSP90 (SN95 and CaLC1411: Table 1) in the absence of stress, and following the imposition of stress. Mkc1 activation levels were attenuated following Hsp90 depletion. This was the case in the absence of stress and following heat shock or Calcofluor White treatment (Figures 9A and 9B). This corresponds with a decrease in total Mkc1 kinase levels following Hsp90 depletion. In contrast, Hsp90 depletion had no effect on the levels of Cek1 activation in the absence of stress, but led to an increase in Cek1 phosphorylation following Calcofluor White treatment (Figures 9A and 9B). With regard to Hog1, the basal levels of phosphorylation were maintained following Hsp90 depletion in the absence of stress (Figure 9C), and Hog1 activation was not attenuated in response to osmotic stress although total Hog1 levels decreased (Figure 9D). This was entirely consistent with our other findings, whereby ambient temperature did not affect osmotic tress resistance (Figure 6) and a prior heat shock did not protect cells against a subsequent osmotic stress (Figure 7). However, Hsp90 depletion blocked Hog1 activation following exposure to hydrogen peroxide (Figure 9C). We conclude that Hsp90 depletion exerts differential effects upon Hog1, Mkc1 and Cek1. Furthermore the data are consistent with our prediction that Hsp90 modulates the activities of these MAP kinases.
Figure 9. Hsp90 depletion affects MAP kinase signalling in the presence and absence of stress.
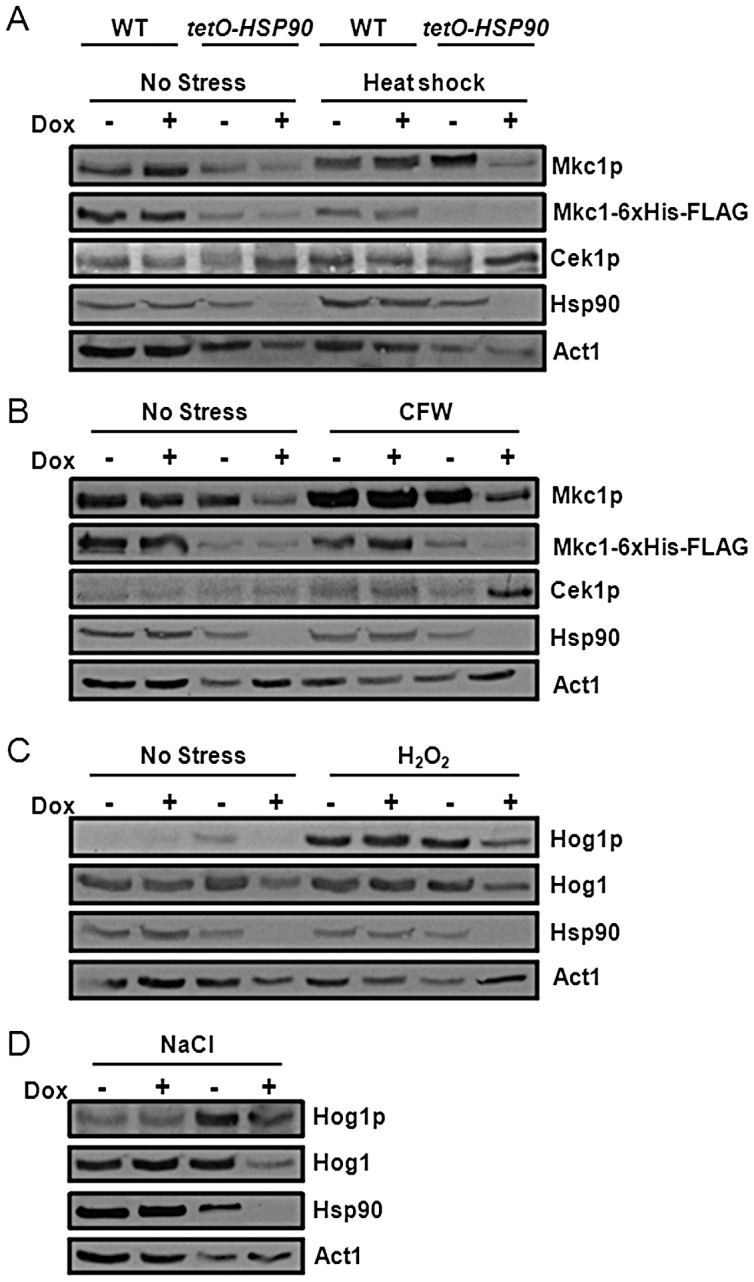
C. albicans wild-type and tetO-HSP90 cells (SN95 and CaLC1411: Table 1) were treated with 0 or 20 µg/ml doxycycline for seven hours. Mkc1, Cek1 and Hog1 phosphorylation levels were then assayed by western analysis in unstressed cells or cells treated as follows: (A) 30 minute 30°C–42°C heat shock; (B) 30 minutes with 100 µg/ml Calcofluor White (CFW); (C) 10 minutes with 5 mM H2O2; or (D) 12 minutes with 1 M NaCl. Total Mkc1 levels were assayed using Mkc1-6xHis-FLAG tagged cells in SN95 (CaLC681) and tetO-HSP90 (caLC648) cells (Table 1). Hsp90 levels and total kinase levels for Hog1 were also examined by western blotting relative to the internal Act1 loading control.
Cek1 is an Hsp90 client
It was conceivable that Cek1 is an Hsp90 client protein. To test this we determined the impact of Hsp90 depletion on Cek1 stability as Hsp90 client proteins are generally destabilised in the absence of Hsp90 [62], [86], [89]. Cek1 was TAP-tagged at its C-terminus and the specificity of this tagging was verified by western blotting alongside untagged SN95 and CaLC1411 controls (Figure S4). The levels of this Cek1-TAP protein were then monitored in the tetO-HSP90 strain CaLC1411 (CaLC2288, Table 1) and in wild-type SN95 (CaLC2287, Table 1) cells (Figure 10). Cek1-TAP protein levels decreased in response to Hsp90 depletion in the absence of stress, as well as in response to heat shock and Calcofluor White treatment (Figure 10). This suggested that Cek1 is destabilised by Hsp90 depletion and that Cek1 is a client protein of Hsp90. Therefore the contribution of Cek1 to thermal adaptation appears to be modulated by Hsp90.
Figure 10. Hsp90 depletion destabilises Cek1.
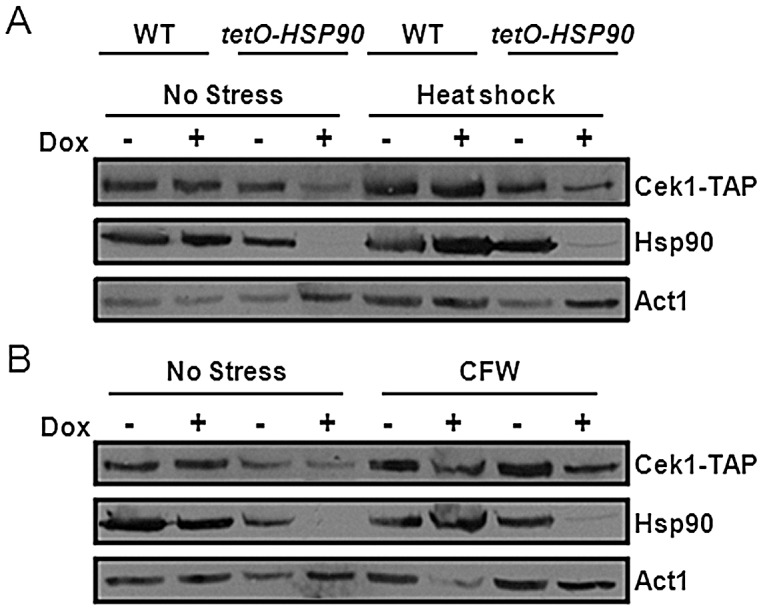
C. albicans CEK1-TAP (CaLC2287) and tetO-HSP90 CEK1-TAP (CaLC2288: Table 1) cells were treated with 0 or 20 µg/ml doxycycline for seven hours and subjected to western analysis. Decreased Cek1-TAP levels were observed following Hsp90 depletion in unstressed cells and in cells treated as follows: (A) 30 minute 30°C–42°C heat shock; or (B) 30 minutes with 100 µg/ml Calcofluor White (CFW). Hsp90 protein levels were examined confirming significant depletion following doxycycline treatment of tetO-HSP90 cells. Actin served as the internal loading control.
Hsp90 influences cell wall remodelling
Several observations infer a link between stress adaptation and cell wall architecture in C. albicans. For example, the Pkc1/Mkc1 cell wall salvage pathway is activated by certain stresses [58], [66], [90]. Also, the Hog1 stress pathway has been implicated in cell wall biosynthesis [81], partly by regulating chitin synthesis [66], [69]. If Hsp90 depletion modulates Hog1, Mkc1 and Cek1 signalling (Figure 9), and these pathways contribute to cell wall biogenesis, we reasoned that Hsp90 could regulate cell wall architecture. We took two approaches to test this hypothesis. First, we tested the effects of Hsp90 depletion on chitin levels by Calcofluor White staining. Chitin content increased more than two-fold following doxycycline treatment of tetO-HSP90 cells compared to control cells (Figures 11A and 11B). Second, we examined the impact of Hsp90 depletion upon cell wall architecture by transmission electron microscopy (Figure 11C). Cell wall thickness increased two-fold after Hsp90 depletion compared to the controls. Therefore, Hsp90 is essential for normal cell wall structure, providing the first ever link between Hsp90 and cell wall architecture.
Figure 11. Hsp90 depletion affects cell wall architecture.

C. albicans wild-type cells (SN95), and tetO-HSP90 (CaLC1411: Table 1) were treated with 0 or 20 µg/ml doxycycline for seven hours. Chitin levels were assayed by Calcofluor White staining, scale bars are 10 µm (A) and quantification of fluorescence levels (B). Data represent means from fifty cells: ** paired, two-tailed t-test, p<0.01. The architecture of the cell wall was examined by transmission electron microscopy where scale bars are 200 µm (C), and the thickness of the cell wall quantified in n = 30 cells (D): ** paired, two-tailed t-test, p<0.01.
Discussion
Our results illuminate novel functional connections between key cellular regulators required for thermotolerance, and establish distinct roles for Hsp90 in orchestrating short term versus long term mechanisms of thermal adaptation. We provide evidence that Hsf1 is a client protein of Hsp90 for the first time in any fungus, demonstrating that Hsp90 contributes to the short term regulation of thermal adaptation. Furthermore, we identified several key signalling pathways that contribute to thermotolerance in C. albicans. These include the Mkc1, Hog1 and Cek1 pathways, each of which plays a role in cell wall integrity. These pathways contribute to thermal adaptation in the longer term via cell wall remodelling, and Hsp90 links many of these pathways, as critical components of these pathways are Hsp90 client proteins. Finally, we establish a role for Hsp90 in cell wall biogenesis for the first time in any organism. Overall, we see that Hsp90 drives short term thermal adaptation via down-regulation of Hsf1, and longer term adaptation through modulation of other client proteins, leading to a more robust cell wall.
Hsf1 is known to activate HSP90 expression [41] and Hsp90 has been predicted to regulate Hsf1 [49]. Here we demonstrate that inhibiting Hsp90 in C. albicans, either pharmacologically or genetically, derepresses Hsf1 (Figure 1), indicating that Hsp90 down-regulates Hsf1. Furthermore, Hsf1 physically interacts with Hsp90 under steady state conditions (Figure 2A), confirming for the first time in any fungus that Hsf1 is an Hsp90 client. What's more, we have shown that this interaction increases during heat shock, and also leads to the recruitment of Hsp70, which binds Hsf1 and Hsp90 during a prolonged heat shock (Figures 2B and S1). This increased interaction correlates with an increase in the levels of both Hsp90 and Hsf1 that occurs during heat shock. Consistent with these findings, we found that Hsp90 accumulates in the nucleus upon a prolonged heat shock (Figure 2C and Figure S1). Similarly, the S. cerevisiae Hsp70 chaperone Ssa1 has been shown to enter the nucleus one hour after a 42°C heat shock [91]. This is consistent with our observation that Hsp70 binds Hsf1 and Hsp90 one hour after a 37°C–42°C heat shock. Therefore short-term thermal adaptation involves the activation of Hsf1 by Hsp90-independent mechanisms. Hsf1 induces the expression of Hsp90 and other chaperones that promote protein folding and repair proteotoxic damage [39], [41]. Hsp90 then down-regulates Hsf1 thereby dampening the heat shock response once adaptation is achieved. This regulatory circuit is central to thermal adaptation in C. albicans [41], [49] and may be conserved across the eukaryotic kingdom as pharmacological inhibition of Hsp90 derepresses Hsf1 orthologues in S. cerevisiae and mammalian systems [52], [55].
With a view to identifying the kinase responsible for phosphorylating Hsf1 in C. albicans we performed a screen for protein kinase mutants that are temperature sensitive (Figure 3). This highlighted several critical regulators on key MAP kinase pathways, including the Mkc1, Hog1 and Cek1 pathways. None of these pathways are essential for Hsf1 phosphorylation (Figure S3), suggesting that there is functional redundancy with respect to Hsf1 activation during heat shock, or that these pathways act independently of Hsf1 in promoting thermotolerance. These pathways are differentially activated during heat shock (Figure 4), and there is cross-talk between these pathways under these conditions (Figure 5). Furthermore, ambient temperature significantly affects the resistance of C. albicans cells to cell wall stresses, and these effects are influenced by Cek1, Hog1 and Mkc1 (Figure 6). Each of these MAP kinase pathways is known to contribute to cell wall remodelling [32], [81], [83], [90], and mutations that interfere with cell wall synthesis are known to confer temperature sensitivity upon C. albicans. For example, the inactivation of certain protein mannosyltransferases of the PMT family, or the deletion of OCH1 can confer temperature sensitivity [92], [93]. Additionally, deletion of SSR1, a GPI-anchored cell wall protein causes sensitivity to elevated temperatures [94]. These data strongly suggest that Mkc1, Hog1 and Cek1 signalling promotes longer term thermotolerance via the maintenance of a robust cell wall (Figure 12).
Figure 12. Hsp90 coordinates the activities of multiple signalling pathways that contribute to thermotolerance in C. albicans - a model.
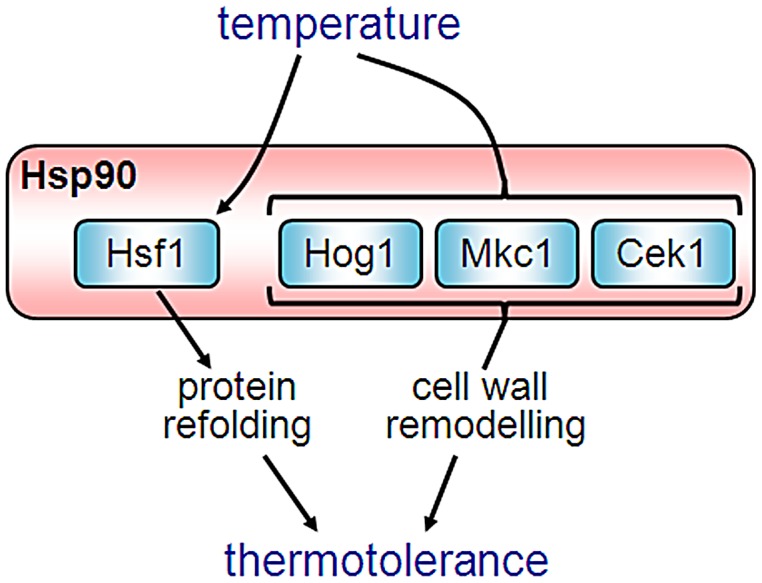
Hsf1 activation is required for thermotolerance [42]. Hog1, Mkc1 and Cek1 signalling are also required for thermotolerance (Figure 3), but these MAP kinases are not essential for Hsf1 phosphorylation (Figure 4). Instead, these pathways promote thermotolerance in part via cell wall remodelling [81], [90]. Hsp90 coordinates much of this activity. Hsf1 (Figures 1 and 2), Hog1, Mkc1 and Cek1 (Figure 10) are all Hsp90 client proteins [62], [86]. Changes in ambient temperature affect interactions between Hsp90 and Hsf1 (Figure 2), and probably affect Hsp90 interactions with Hog1, Mkc1 and Cek1 [49] thereby modulating the activities of these signalling pathways and their inputs to thermal adaptation. Increases in ambient temperature activate Hsf1, thereby inducing the expression of protein chaperones (HSPs) including Hsp90, which promotes thermal adaptation in the shorter term. Hsp90 then down-regulates Hsf1 and modulates Mkc1, Hog1 and Cek1 signalling, which in the longer term influences cell wall architecture (Figure 11), leading to the thermotolerance of C. albicans.
As an environmentally contingent hub of protein homeostasis and regulatory circuitry, Hsp90 has profound effects on biology, disease, and evolution. Hsp90 modulates the phenotypic effects of genetic variation in an environmentally responsive manner [95], [96], [97], [98], influencing approximately 20% of observed natural genetic variation and serving to maintain phenotypic robustness and promote diversification [96]. Mkc1 was defined as a protein client of Hsp90 after Mkc1 signalling was shown to contribute to antifungal drug tolerance in C. albicans [86]. Hog1 was subsequently shown to be an Hsp90 client protein after Diezmann and co-workers identified this MAPK in a chemogenetic screen of the C. albicans Hsp90 interactome [62]. However, Cek1 was not highlighted in this screen. We demonstrate here that, like Mkc1 and Hog1, Cek1 is an Hsp90 client protein (Figure 10). This is in keeping with a recent study by Taipale and colleagues which demonstrates that Hsp90 binds about 60% of mammalian kinases [99]. Therefore we tested whether Mkc1, Hog1 and Cek1 signalling is influenced by Hsp90 during thermal upshifts. We found that Hsp90 does influence the activation of these kinases during heat shock and in response to their respective stresses (Figure 9). Furthermore, Hsp90 depletion influenced the sensitivity of C. albicans cells to specific stresses such as cell wall and oxidative stress, as well as to heat shock (Figure 8). Clearly Hsp90 modulates Mkc1, Hog1 and Cek1 signalling and their outputs, and these pathways are known to contribute to cell wall architecture. Therefore, we reasoned that Hsp90 might, in part, influence cell wall remodelling (Figure 12). We confirmed this hypothesis by demonstrating that Hsp90 depletion significantly increases the chitin content and thickness of C. albicans cell walls (Figure 11). We note that cell wall robustness does not correlate with cell wall thickness. Indeed Ene and co-workers have recently shown that cell wall architecture is altered by growth on different carbon sources, yielding thinner cell walls that are more robust, leading to increased stress resistance [100]. Therefore, Hsp90 coordinates both short term thermal adaptation (via Hsf1 down-regulation), and long term thermal adaptation (via its client proteins Mkc1, Hog1 and Cek1) (Figure 12).
Additional pathways that were not highlighted in our screen might contribute to thermotolerance. For example some protein kinases that encode essential functions were missing from the transposon library. Indeed, the protein kinase responsible for Hsf1 phosphorylation remains obscure. It is worth noting that as Hsf1 is activated in response to Hsp90 depletion, and as such it is unlikely that the Hsf1 kinase requires stabilisation by Hsp90. One must also note that Hsf1 might be phosphorylated by multiple protein kinases, and hence that there may be functional redundancy with respect to Hsf1 phosphorylation. Indeed, NetPhos2.0 analysis of Hsf1 suggests multiple phosphorylation sites for multiple kinases.
Clearly ambient temperature plays a major role in fungal pathogenicity [42]. Furthermore our data indicate that ambient temperature strongly influences physiological attributes that contribute to fungal pathogenicity, such as a robust cell wall and effective stress adaptation [11], [13], [42], [49]. In addition we show that increases in ambient temperature lead to elevated oxidative stress resistance, but that the reverse is not true (Figure 7). This observation is consistent with the idea of “asymmetric adaptive prediction”, whereby microbes appear to have “learned” over evolutionary timescales that exposure to one stress is likely to be followed by exposure to a second unrelated stress [101]. As a result, exposure to the first stress results in the activation of an adaptive response that prepares the cell for exposure to the second stress [101]. With respect to C. albicans, the elevated temperatures associated with localised inflammation, appear to protect the fungal cells against the imminent exposure to oxidative stress that will follow exposure to macrophages and neutrophils.
In conclusion this study reveals new Hsp90 client proteins that play central roles in the control of cellular adaptation: Hsf1 and Cek1. Furthermore, this work provides important new insights into the mechanisms by which Hsp90 coordinates short and long term mechanisms that contribute to thermotolerance in a major fungal pathogen.
Supporting Information
Dynamics of Hsp90 depletion in C. albicans tetO-HSP90 cells. (A) Doxycycline-conditional C. albicans tetO-HSP90 (CaLC1411: Table 1) cells were treated with 20 µg/ml doxycycline, and Hsp90 levels were examined by western blotting and quantified relative to the Act1 internal control. (B) C. albicans WT (wild type SN95: Table 1) cells and tetO-HSP90 cells were treated with 0 or 20 µg/ml doxycycline for 7 hours. Proteins were extracted and probed for Hsp90. (C) Effect of 20 µg/ml doxycycline on the growth of these C. albicans tetO-HSP90 cells.
(TIF)
Dynamics of the Hsf1-Hsp90 interaction in C. albicans . (A) Immunoprecipitation of FLAG-Hsf1 with anti-FLAG M2 affinity agarose during a heat shock shows the interaction of Hsf1 with Hsp70 120 minutes after a 42°C heat shock. (B) FLAG-Hsf1 co-immunoprecipitates with Hsp90-TAP on IgG agarose 0, 10 and 120 minutes post-heat shock. Re-probing these membranes for Hsp70, shows that Hsp70 interacts with Hsp90 120 minutes post-heat shock. (C) Localisation of Hsp90-GFP 120 minutes after heat shock. Cells were treated with a 30°C–42°C heat shock and fixed 120 minutes post-heat shock revealing significant accumulation in the nucleus (localised by DAPI staining). Scale bars, 5 µm.
(TIF)
Protein kinases that contribute to thermal adaptation in C. albicans are not required for Hsf1 phosphorylation. Effect of Mkc1, Cek1, Hog1 and CK2 inactivation upon Hsf1 phosphorylation dynamics during a 30°C–42°C heat shock. Wild-type and mutant cells were subjected to a heat shock and proteins harvested at 0, 10, 30 and 60 minutes post-heat shock. All strains exhibited full activation of Hsf1, as seen by the band shift 10 minutes post-heat shock: black arrow, phosphorylated Hsf1; white arrow, non-phosphorylated Hsf1.
(TIF)
Specific detection of Cek1-TAP. C. albicans wild-type cells (SN95), CEK1-TAP (CaLC2287) treated with 0 or 20 µg/ml doxycycline for seven hours (−/+), tetO-HSP90 (CaLC1411) and tetO-HSP90 CEK1-TAP (CaLC2288: Table 1). Western analyses were performed to confirm efficient tagging of Cek1, and that doxycycline does not affect Cek1-TAP protein levels directly. The membrane was reprobed with the internal Act1 control to confirm even loading.
(TIF)
Primers used in this study.
(PDF)
Acknowledgments
We thank Rebecca Shapiro for creating CaLC1819, CaLC1855 and CaLC1875, Gillian Milne for help with EM, Aaron Mitchell for generously providing the transposon insertion mutant library, Jesus Pla for generously providing the hog1 hst7 mutant, and Cathy Collins for technical assistance.
Funding Statement
MDL is grateful to the Caledonian Research Foundation [www.calres.co.uk] for awarding her a Carnegie/Caledonian PhD Scholarship (2008–2011), and to the Wellcome Trust [www.wellcome.ac.uk] for their support through her Sir Henry Wellcome Postdoctoral Fellowship (096072). LEC is grateful to the Burroughs Wellcome Fund [www.bwfund.org] for her Career Award in Biomedical Sciences, and to the Canadian Ministry of Research [www.mri.gov.on.ca] for an Innovation Early Researcher Award and Canada Research Chair. LEC is also supported by research grants from the Canadian Institutes of Health [www.cihr-irsc.gc.ca] (MOP-86452; MOP-119520). AJPB is supported by research grants from the Wellcome Trust [www.wellcome.ac.uk] (080088; 097377), the UK Biotechnology and Biological Research Council [www.bbsrc.ac.uk] (BB/D009308/1; BB/F00513X/1), the European Commission [http://ec.europa.eu/research/fp7] (FINSysB MC-ITN: PITN-GA-2008-214004) and by an Advanced Grant from the European Research Council [http://erc.europa.eu/] (STRIFE: C-2009-AdG-249793). These funding bodies had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Tirosh I, Weinberger A, Carmi M, Barkai N (2006) A genetic signature of interspecies variations in gene expression. Nat Genet 38: 830–834. [DOI] [PubMed] [Google Scholar]
- 2. Gasch AP, Spellman PT, Kao CM, Carmel-Harel O, Eisen MB, et al. (2000) Genomic expression programs in the response of yeast cells to environmental changes. Mol Biol Cell 11: 4241–4257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Enjalbert B, Nantel A, Whiteway M (2003) Stress-induced gene expression in Candida albicans: absence of a general stress response. Mol Biol Cell 14: 1460–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Causton HC, Ren B, Koh SS, Harbison CT, Kanin E, et al. (2001) Remodeling of yeast genome expression in response to environmental changes. Mol Biol Cell 12: 323–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kussell E, Leibler S (2005) Phenotypic diversity, population growth, and information in fluctuating environments. Science 309: 2075–2078. [DOI] [PubMed] [Google Scholar]
- 6. Estruch F (2000) Stress-controlled transcription factors, stress-induced genes and stress tolerance in budding yeast. FEMS Microbiol Rev 24: 469–486. [DOI] [PubMed] [Google Scholar]
- 7. de Dios CH, Roman E, Monge RA, Pla J (2010) The role of MAPK signal transduction pathways in the response to oxidative stress in the fungal pathogen Candida albicans: implications in virulence. Curr Protein Pept Sci 11: 693–703. [DOI] [PubMed] [Google Scholar]
- 8.Odds FC, editor (1988) Candida and Candidosis. 2nd ed. London, United Kingdom: Bailliere Tindall.
- 9. Piispanen AE, Hogan DA (2008) PEPped up: induction of Candida albicans virulence by bacterial cell wall fragments. Cell Host Microbe 4: 1–2. [DOI] [PubMed] [Google Scholar]
- 10.Calderone RA (2002) Candida and Candidiasis. Washington, DC: ASM Press.
- 11. Alonso-Monge R, Navarro-Garcia F, Molero G, Diez-Orejas R, Gustin M, et al. (1999) Role of the mitogen-activated protein kinase Hog1p in morphogenesis and virulence of Candida albicans . J Bacteriol 181: 3058–3068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wysong DR, Christin L, Sugar AM, Robbins PW, Diamond RD (1998) Cloning and sequencing of a Candida albicans catalase gene and effects of disruption of this gene. Infect Immun 66: 1953–1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Diez-Orejas R, Molero G, Navarro-Garcia F, Pla J, Nombela C, et al. (1997) Reduced virulence of Candida albicans MKC1 mutants: a role for mitogen-activated protein kinase in pathogenesis. Infect Immun 65: 833–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nikolaou E, Agrafioti I, Stumpf M, Quinn J, Stansfield I, et al. (2009) Phylogenetic diversity of stress signalling pathways in fungi. BMC Evol Biol 9: 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chen D, Toone WM, Mata J, Lyne R, Burns G, et al. (2003) Global transcriptional responses of fission yeast to environmental stress. Mol Biol Cell 14: 214–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Roetzer A, Gregori C, Jennings AM, Quintin J, Ferrandon D, et al. (2008) Candida glabrata environmental stress response involves Saccharomyces cerevisiae Msn2/4 orthologous transcription factors. Mol Microbiol 69: 603–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Garreau H, Hasan RN, Renault G, Estruch F, Boy-Marcotte E, et al. (2000) Hyperphosphorylation of Msn2p and Msn4p in response to heat shock and the diauxic shift is inhibited by cAMP in Saccharomyces cerevisiae . Microbiology 146 (Pt 9) 2113–2120. [DOI] [PubMed] [Google Scholar]
- 18. Nicholls S, Straffon M, Enjalbert B, Nantel A, Macaskill S, et al. (2004) Msn2- and Msn4-like transcription factors play no obvious roles in the stress responses of the fungal pathogen Candida albicans . Eukaryot Cell 3: 1111–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Enjalbert B, Smith DA, Cornell MJ, Alam I, Nicholls S, et al. (2006) Role of the Hog1 stress-activated protein kinase in the global transcriptional response to stress in the fungal pathogen Candida albicans . Mol Biol Cell 17: 1018–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ramsdale M, Selway L, Stead D, Walker J, Yin Z, et al. (2008) MNL1 regulates weak acid-induced stress responses of the fungal pathogen Candida albicans . Mol Biol Cell 19: 4393–4403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Alonso-Monge R, Navarro-Garcia F, Roman E, Negredo AI, Eisman B, et al. (2003) The Hog1 mitogen-activated protein kinase is essential in the oxidative stress response and chlamydospore formation in Candida albicans . Eukaryot Cell 2: 351–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Monge RA, Roman E, Nombela C, Pla J (2006) The MAP kinase signal transduction network in Candida albicans . Microbiology 152: 905–912. [DOI] [PubMed] [Google Scholar]
- 24. Kuge S, Jones N, Nomoto A (1997) Regulation of yAP-1 nuclear localization in response to oxidative stress. EMBO J 16: 1710–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Toone WM, Kuge S, Samuels M, Morgan BA, Toda T, et al. (1998) Regulation of the fission yeast transcription factor Pap1 by oxidative stress: requirement for the nuclear export factor Crm1 (Exportin) and the stress-activated MAP kinase Sty1/Spc1. Genes Dev 12: 1453–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chen K-H, Miyazaki T, Tsai H-F, Bennett JE (2007) The bZip transcription factor Cgap1p is involved in multidrug resistance and required for activation of multidrug transporter gene CgFLR1 in Candida glabrata . Gene 386: 63–72. [DOI] [PubMed] [Google Scholar]
- 27. Roetzer A, Klopf E, Gratz N, Marcet-Houben M, Hiller E, et al. (2011) Regulation of Candida glabrata oxidative stress resistance is adapted to host environment. FEBS Lett 585: 319–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhang X, De Micheli M, Coleman ST, Sanglard D, Moye-Rowley WS (2000) Analysis of the oxidative stress regulation of the Candida albicans transcription factor, Cap1p. Mol Microbiol 36: 618–629. [DOI] [PubMed] [Google Scholar]
- 29. Brewster JL, de Valoir T, Dwyer ND, Winter E, Gustin MC (1993) An osmosensing signal transduction pathway in yeast. Science 259: 1760–1763. [DOI] [PubMed] [Google Scholar]
- 30. Smith DA, Nicholls S, Morgan BA, Brown AJ, Quinn J (2004) A conserved stress-activated protein kinase regulates a core stress response in the human pathogen Candida albicans . Mol Biol Cell 15: 4179–4190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Degols G, Shiozaki K, Russell P (1996) Activation and regulation of the Spc1 stress-activated protein kinase in Schizosaccharomyces pombe . Mol Cell Biol 16: 2870–2877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Csank C, Schroppel K, Leberer E, Harcus D, Mohamed O, et al. (1998) Roles of the Candida albicans mitogen-activated protein kinase homolog, Cek1p, in hyphal development and systemic candidiasis. Infect Immun 66: 2713–2721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chen J, Chen J, Lane S, Liu H (2002) A conserved mitogen-activated protein kinase pathway is required for mating in Candida albicans . Mol Microbiol 46: 1335–1344. [DOI] [PubMed] [Google Scholar]
- 35. Kamada Y, Jung US, Piotrowski J, Levin DE (1995) The protein kinase C-activated MAP kinase pathway of Saccharomyces cerevisiae mediates a novel aspect of the heat shock response. Genes Dev 9: 1559–1571. [DOI] [PubMed] [Google Scholar]
- 36. Winkler A, Arkind C, Mattison CP, Burkholder A, Knoche K, et al. (2002) Heat stress activates the yeast high-osmolarity glycerol mitogen-activated protein kinase pathway, and protein tyrosine phosphatases are essential under heat stress. Eukaryotic Cell 1: 163–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Elion EA, Brill JA, Fink GR (1991) FUS3 represses CLN1 and CLN2 and in concert with KSS1 promotes signal transduction. Proceedings of the National Academy of Sciences 88: 9392–9396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Guhad FA, Jensen HE, Aalbaek B, Csank C, Mohamed O, et al. (1998) Mitogen-activated protein kinase-defective Candida albicans is avirulent in a novel model of localized murine candidiasis. FEMS Microbiol Lett 166: 135–139. [DOI] [PubMed] [Google Scholar]
- 39. Sorger PK, Lewis MJ, Pelham HR (1987) Heat shock factor is regulated differently in yeast and HeLa cells. Nature 329: 81–84. [DOI] [PubMed] [Google Scholar]
- 40. Wiederrecht G, Shuey DJ, Kibbe WA, Parker CS (1987) The Saccharomyces and Drosophila heat shock transcription factors are identical in size and DNA binding properties. Cell 48: 507–515. [DOI] [PubMed] [Google Scholar]
- 41. Nicholls S, Leach MD, Priest CL, Brown AJ (2009) Role of the heat shock transcription factor, Hsf1, in a major fungal pathogen that is obligately associated with warm-blooded animals. Mol Microbiol 74: 844–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Nicholls S, MacCallum DM, Kaffarnik FA, Selway L, Peck SC, et al. (2011) Activation of the heat shock transcription factor Hsf1 is essential for the full virulence of the fungal pathogen Candida albicans . Fungal Genet Biol 48: 297–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sorger PK, Pelham HRB (1988) Yeast heat shock factor is an essential DNA-binding protein that exhibits temperature-dependent phosphorylation. Cell 54: 855–864. [DOI] [PubMed] [Google Scholar]
- 44. Jakobsen BK, Pelham HR (1988) Constitutive binding of yeast heat shock factor to DNA in vivo . Mol Cell Biol 8: 5040–5042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wiederrecht G, Seto D, Parker CS (1988) Isolation of the gene encoding the S. cerevisiae heat shock transcription factor. Cell 54: 841–853. [DOI] [PubMed] [Google Scholar]
- 46. Giardina C, Lis JT (1995) Sodium salicylate and yeast heat shock gene transcription. J Biol Chem 270: 10369–10372. [DOI] [PubMed] [Google Scholar]
- 47. Gallo GJ, Prentice H, Kingston RE (1993) Heat shock factor is required for growth at normal temperatures in the fission yeast Schizosaccharomyces pombe . Mol Cell Biol 13: 749–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Feder ME, Hofmann GE (1999) Heat-shock proteins, molecular chaperones, and the stress response: evolutionary and ecological physiology. Annu Rev Physiol 61: 243–282. [DOI] [PubMed] [Google Scholar]
- 49. Leach MD, Tyc KM, Brown AJP, Klipp E (2012) Modelling the regulation of thermal adaptation in Candida albicans, a major fungal pathogen of humans. PLoS ONE 7: e32467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Jarosz DF, Taipale M, Lindquist S (2010) Protein homeostasis and the phenotypic manifestation of genetic diversity: principles and mechanisms. Annu Rev Genet 44: 189–216. [DOI] [PubMed] [Google Scholar]
- 51. Hegde RS, Zuo J, Voellmy R, Welch WJ (1995) Short circuiting stress protein expression via a tyrosine kinase inhibitor, herbimycin A. J Cell Physiol 165: 186–200. [DOI] [PubMed] [Google Scholar]
- 52. Zou J, Guo Y, Guettouche T, Smith DF, Voellmy R (1998) Repression of heat shock transcription factor HSF1 activation by HSP90 (HSP90 complex) that forms a stress-sensitive complex with HSF1. Cell 94: 471–480. [DOI] [PubMed] [Google Scholar]
- 53. Guo Y, Guettouche T, Fenna M, Boellmann F, Pratt WB, et al. (2001) Evidence for a mechanism of repression of heat shock factor 1 transcriptional activity by a multichaperone complex. J Biol Chem 276: 45791–45799. [DOI] [PubMed] [Google Scholar]
- 54. Ali A, Bharadwaj S, O'Carroll R, Ovsenek N (1998) HSP90 interacts with and regulates the activity of heat shock factor 1 in Xenopus oocytes. Mol Cell Biol 18: 4949–4960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Duina AA, Kalton HM, Gaber RF (1998) Requirement for Hsp90 and a CyP-40-type cyclophilin in negative regulation of the heat shock response. J Biol Chem 273: 18974–18978. [DOI] [PubMed] [Google Scholar]
- 56. Davis DA, Bruno VM, Loza L, Filler SG, Mitchell AP (2002) Candida albicans Mds3p, a conserved regulator of pH responses and virulence identified through insertional mutagenesis. Genetics 162: 1573–1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Nobile CJ, Bruno VM, Richard ML, Davis DA, Mitchell AP (2003) Genetic control of chlamydospore formation in Candida albicans . Microbiology 149: 3629–3637. [DOI] [PubMed] [Google Scholar]
- 58. Blankenship JR, Fanning S, Hamaker JJ, Mitchell AP (2010) An extensive circuitry for cell wall regulation in Candida albicans . PLoS Pathog 6: e1000752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Sherman F (1991) Getting started with yeast. Methods Enzymol 194: 3–21. [DOI] [PubMed] [Google Scholar]
- 60. Walther A, Wendland J (2003) An improved transformation protocol for the human fungal pathogen Candida albicans . Curr Genet 42: 339–343. [DOI] [PubMed] [Google Scholar]
- 61. Bruno VM, Mitchell AP (2005) Regulation of azole drug susceptibility by Candida albicans protein kinase CK2. Mol Microbiol 56: 559–573. [DOI] [PubMed] [Google Scholar]
- 62. Diezmann S, Michaut M, Shapiro RS, Bader GD, Cowen LE (2012) Mapping the Hsp90 genetic interaction network in Candida albicans reveals environmental contingency and rewired circuitry. PLoS Genetics 8: e1002562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lavoie H, Sellam A, Askew C, Nantel A, Whiteway M (2008) A toolbox for epitope-tagging and genome-wide location analysis in Candida albicans . BMC Genomics 9: 578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Leach MD, Stead DA, Argo E, MacCallum DM, Brown AJ (2011) Molecular and proteomic analyses highlight the importance of ubiquitination for the stress resistance, metabolic adaptation, morphogenetic regulation and virulence of Candida albicans . Mol Microbiol 79: 1574–1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Leach MD, Stead DA, Argo E, Brown AJ (2011) Identification of sumoylation targets, combined with inactivation of SMT3, reveals the impact of sumoylation upon growth, morphology, and stress resistance in the pathogen Candida albicans . Mol Biol Cell 22: 687–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Munro CA, Selvaggini S, de Bruijn I, Walker L, Lenardon MD, et al. (2007) The PKC, HOG and Ca2+ signalling pathways co-ordinately regulate chitin synthesis in Candida albicans . Mol Microbiol 63: 1399–1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72: 248–254. [DOI] [PubMed] [Google Scholar]
- 68. Burt ET, Daly R, Hoganson D, Tsirulnikov Y, Essmann M, et al. (2003) Isolation and partial characterization of Hsp90 from Candida albicans . Ann Clin Lab Sci 33: 86–93. [PubMed] [Google Scholar]
- 69. Walker LA, Munro CA, de Bruijn I, Lenardon MD, McKinnon A, et al. (2008) Stimulation of chitin synthesis rescues Candida albicans from echinocandins. PLoS Pathog 4: e1000040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Smith DF, Whitesell L, Nair SC, Chen S, Prapapanich V, et al. (1995) Progesterone receptor structure and function altered by geldanamycin, an Hsp90 binding agent. Mol Cell Biol 15: 6804–6812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Roe SM, Prodromou C, O'Brien R, Ladbury JE, Piper PW, et al. (1999) Structural basis for inhibition of the Hsp90 molecular chaperone by the antitumor antibiotics radicicol and geldanamycin. J Med Chem 42: 260–266. [DOI] [PubMed] [Google Scholar]
- 72. Wimalasena TT, Enjalbert B, Guillemette T, Plumridge A, Budge S, et al. (2008) Impact of the unfolded protein response upon genome-wide expression patterns, and the role of Hac1 in the polarized growth, of Candida albicans . Fungal Genet Biol 45: 1235–1247. [DOI] [PubMed] [Google Scholar]
- 73. Shapiro RS, Uppuluri P, Zaas AK, Collins C, Senn H, et al. (2009) Hsp90 orchestrates temperature-dependent Candida albicans morphogenesis via Ras1-PKA signaling. Curr Biol 19: 621–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Leach MD, Klipp E, Cowen LE, Brown AJ (2012) Fungal Hsp90: a biological transistor that tunes cellular outputs to thermal inputs. Nat Rev Microbiol 10: 693–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Craig EA, Jacobsen K (1984) Mutations of the heat inducible 70 kilodalton genes of yeast confer temperature sensitive growth. Cell 38: 841–849. [DOI] [PubMed] [Google Scholar]
- 76. Lamoth F, Juvvadi PR, Fortwendel JR, Steinbach WJ (2012) Heat-shock protein 90 (Hsp90) is required for conidiation and cell wall integrity in Aspergillus fumigatus . Eukaryot Cell 11: 1324–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Gónzalez-Párraga P, Alonso-Monge R, Plá J, Argüelles JC (2010) Adaptive tolerance to oxidative stress and the induction of antioxidant enzymatic activities in Candida albicans are independent of the Hog1 and Cap1-mediated pathways. FEMS Yeast Research 10: 747–756. [DOI] [PubMed] [Google Scholar]
- 78. Bruno VM, Kalachikov S, Subaran R, Nobile CJ, Kyratsous C, et al. (2006) Control of the C. albicans cell wall damage response by transcriptional regulator Cas5. PLoS Pathog 2: e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Cantero PD, Ernst JF (2011) Damage to the glycoshield activates PMT-directed O-mannosylation via the Msb2–Cek1 pathway in Candida albicans . Mol Microbiol 80: 715–725. [DOI] [PubMed] [Google Scholar]
- 80. Arana DM, Nombela C, Alonso-Monge R, Pla J (2005) The Pbs2 MAP kinase kinase is essential for the oxidative-stress response in the fungal pathogen Candida albicans . Microbiology 151: 1033–1049. [DOI] [PubMed] [Google Scholar]
- 81. Eisman B, Alonso-Monge R, Roman E, Arana D, Nombela C, et al. (2006) The Cek1 and Hog1 mitogen-activated protein kinases play complementary roles in cell wall biogenesis and chlamydospore formation in the fungal pathogen Candida albicans . Eukaryot Cell 5: 347–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. O'Rourke SM, Herskowitz I (1998) The Hog1 MAPK prevents cross talk between the HOG and pheromone response MAPK pathways in Saccharomyces cerevisiae . Genes Dev 12: 2874–2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Roman E, Nombela C, Pla J (2005) The Sho1 adaptor protein links oxidative stress to morphogenesis and cell wall biosynthesis in the fungal pathogen Candida albicans . Mol Cell Biol 25: 10611–10627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. San Jose C, Monge RA, Perez-Diaz R, Pla J, Nombela C (1996) The mitogen-activated protein kinase homolog HOG1 gene controls glycerol accumulation in the pathogenic fungus Candida albicans . J Bacteriol 178: 5850–5852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Enjalbert B, Moran GP, Vaughan C, Yeomans T, MacCallum DM, et al. (2009) Genome-wide gene expression profiling and a forward genetic screen show that differential expression of the sodium ion transporter Ena21 contributes to the differential tolerance of Candida albicans and Candida dubliniensis to osmotic stress. Mol Microbiol 72: 216–228. [DOI] [PubMed] [Google Scholar]
- 86. LaFayette SL, Collins C, Zaas AK, Schell WA, Betancourt-Quiroz M, et al. (2010) PKC signaling regulates drug resistance of the fungal pathogen Candida albicans via circuitry comprised of Mkc1, calcineurin, and Hsp90. PLoS Pathog 6: e1001069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Millson SH, Truman AW, King V, Prodromou C, Pearl LH, et al. (2005) A two-hybrid screen of the yeast proteome for Hsp90 interactors uncovers a novel Hsp90 chaperone requirement in the activity of a stress-activated mitogen-activated protein kinase, Slt2p (Mpk1p). Eukaryotic Cell 4: 849–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Hawle P, Horst D, Bebelman JP, Yang XX, Siderius M, et al. (2007) Cdc37p is required for stress-induced high-osmolarity glycerol and protein kinase C mitogen-activated protein kinase pathway functionality by interaction with Hog1p and Slt2p (Mpk1p). Eukaryotic Cell 6: 521–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Singh SD, Robbins N, Zaas AK, Schell WA, Perfect JR, et al. (2009) Hsp90 governs echinocandin resistance in the pathogenic yeast Candida albicans via calcineurin. PLoS Pathog 5: e1000532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Shulga N, James P, Craig EA, Goldfarb DS (1999) A nuclear export signal prevents Saccharomyces cerevisiae Hsp70 Ssb1p from stimulating nuclear localization signal-directed nuclear transport. J Biol Chem 274: 16501–16507. [DOI] [PubMed] [Google Scholar]
- 92. Prill SK, Klinkert B, Timpel C, Gale CA, Schroppel K, et al. (2005) PMT family of Candida albicans: five protein mannosyltransferase isoforms affect growth, morphogenesis and antifungal resistance. Mol Microbiol 55: 546–560. [DOI] [PubMed] [Google Scholar]
- 93. Bates S, Hughes HB, Munro CA, Thomas WP, MacCallum DM, et al. (2006) Outer chain N-glycans are required for cell wall integrity and virulence of Candida albicans . J Biol Chem 281: 90–98. [DOI] [PubMed] [Google Scholar]
- 94. Plaine A, Walker L, Da Costa G, Mora-Montes HM, McKinnon A, et al. (2008) Functional analysis of Candida albicans GPI-anchored proteins: Roles in cell wall integrity and caspofungin sensitivity. Fungal Genet Biol 45: 1404–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Cowen LE, Lindquist S (2005) Hsp90 potentiates the rapid evolution of new traits: drug resistance in diverse fungi. Science 309: 2185–2189. [DOI] [PubMed] [Google Scholar]
- 96. Jarosz DF, Lindquist S (2010) Hsp90 and environmental stress transform the adaptive value of natural genetic variation. Science 330: 1820–1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Queitsch C, Sangster TA, Lindquist S (2002) Hsp90 as a capacitor of phenotypic variation. Nature 417: 618–624. [DOI] [PubMed] [Google Scholar]
- 98. Rutherford SL, Lindquist S (1998) Hsp90 as a capacitor for morphological evolution. Nature 396: 336–342. [DOI] [PubMed] [Google Scholar]
- 99. Taipale M, Krykbaeva I, Koeva M, Kayatekin C, Westover Kenneth D, et al. (2012) Quantitative analysis of Hsp90-client interactions reveals principles of substrate recognition. Cell 150: 987–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Ene IV, Adya AK, Wehmeier S, Brand AC, Maccallum DM, et al. (2012) Host carbon sources modulate cell wall architecture, drug resistance and virulence in a fungal pathogen. Cell Microbiol 14: 1319–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Mitchell A, Romano GH, Groisman B, Yona A, Dekel E, et al. (2009) Adaptive prediction of environmental changes by microorganisms. Nature 460: 220–224. [DOI] [PubMed] [Google Scholar]
- 102. Noble SM, Johnson AD (2005) Strains and strategies for large-scale gene deletion studies of the diploid human fungal pathogen Candida albicans . Eukaryot Cell 4: 298–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Wilson RB, Davis D, Mitchell AP (1999) Rapid hypothesis testing with Candida albicans through gene disruption with short homology regions. J Bacteriol 181: 1868–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Leberer E, Harcus D, Broadbent ID, Clark KL, Dignard D, et al. (1996) Signal transduction through homologs of the Ste20p and Ste7p protein kinases can trigger hyphal formation in the pathogenic fungus Candida albicans . Proc Natl Acad Sci U S A 93: 13217–13222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Fonzi WA, Irwin MY (1993) Isogenic strain construction and gene mapping in Candida albicans . Genetics 134: 717–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Walker LA, MacCallum DM, Bertram G, Gow NAR, Odds FC, et al. (2009) Genome-wide analysis of Candida albicans gene expression patterns during infection of the mammalian kidney. Fungal Genet Biol 46: 210–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Dynamics of Hsp90 depletion in C. albicans tetO-HSP90 cells. (A) Doxycycline-conditional C. albicans tetO-HSP90 (CaLC1411: Table 1) cells were treated with 20 µg/ml doxycycline, and Hsp90 levels were examined by western blotting and quantified relative to the Act1 internal control. (B) C. albicans WT (wild type SN95: Table 1) cells and tetO-HSP90 cells were treated with 0 or 20 µg/ml doxycycline for 7 hours. Proteins were extracted and probed for Hsp90. (C) Effect of 20 µg/ml doxycycline on the growth of these C. albicans tetO-HSP90 cells.
(TIF)
Dynamics of the Hsf1-Hsp90 interaction in C. albicans . (A) Immunoprecipitation of FLAG-Hsf1 with anti-FLAG M2 affinity agarose during a heat shock shows the interaction of Hsf1 with Hsp70 120 minutes after a 42°C heat shock. (B) FLAG-Hsf1 co-immunoprecipitates with Hsp90-TAP on IgG agarose 0, 10 and 120 minutes post-heat shock. Re-probing these membranes for Hsp70, shows that Hsp70 interacts with Hsp90 120 minutes post-heat shock. (C) Localisation of Hsp90-GFP 120 minutes after heat shock. Cells were treated with a 30°C–42°C heat shock and fixed 120 minutes post-heat shock revealing significant accumulation in the nucleus (localised by DAPI staining). Scale bars, 5 µm.
(TIF)
Protein kinases that contribute to thermal adaptation in C. albicans are not required for Hsf1 phosphorylation. Effect of Mkc1, Cek1, Hog1 and CK2 inactivation upon Hsf1 phosphorylation dynamics during a 30°C–42°C heat shock. Wild-type and mutant cells were subjected to a heat shock and proteins harvested at 0, 10, 30 and 60 minutes post-heat shock. All strains exhibited full activation of Hsf1, as seen by the band shift 10 minutes post-heat shock: black arrow, phosphorylated Hsf1; white arrow, non-phosphorylated Hsf1.
(TIF)
Specific detection of Cek1-TAP. C. albicans wild-type cells (SN95), CEK1-TAP (CaLC2287) treated with 0 or 20 µg/ml doxycycline for seven hours (−/+), tetO-HSP90 (CaLC1411) and tetO-HSP90 CEK1-TAP (CaLC2288: Table 1). Western analyses were performed to confirm efficient tagging of Cek1, and that doxycycline does not affect Cek1-TAP protein levels directly. The membrane was reprobed with the internal Act1 control to confirm even loading.
(TIF)
Primers used in this study.
(PDF)