

Graphical abstract
Highlights
► New drugs for Chagas disease are needed with improved safety and efficacy. ► Sterol 14-demethylase (CYP51) is essential for de novo sterol biosynthesis. ► Antifungal azole drugs kill Trypanosoma cruzi by inhibiting CYP51 activity. ► Posaconazole and ravuconazole are in Phase II clinical trials for Chagas disease. ► Investigational CYP51 inhibitors are being developed as alternatives.
Keywords: Trypanosoma cruzi, Chagas disease, CYP51, Sterol 14-demethylase
Abstract
The protozoan parasite, Trypanosoma cruzi, causes the most prevalent parasitic infection in the American continent. It gives rise to life-long infection in humans and results in severe cardiomyopathy or other life-threatening manifestations (Chagas disease) in ∼30% of those infected. Animal models and clinical studies indicate that etiological treatment of the infection reduces the risk of developing the disease manifestations. Unfortunately, the existing chemotherapeutics have suboptimal antiparasitic activity and cause significant side effects in many patients, thus better anti-trypanosomal drugs are greatly needed. The sterol biosynthesis pathway has received attention as a target for the development of new drugs for Chagas disease. In particular, inhibitors of sterol 14-demethylase (CYP51) are shown to be extremely active on T. cruziin vitro and in animal models. Antifungal drugs (i.e. azoles) in clinical use or in clinical studies have been extensively tested preclinically on T. cruzi with posaconazole and ravuconazole demonstrating the most promising activity. As a result, posaconazole and a pro-drug of ravuconazole (E1224) are currently being evaluated in Phase II studies for Chagas disease. Additional CYP51 inhibitors that are specifically optimized for anti-T. cruzi activity are in development by academia. These represent an alternative to proprietary antifungal drugs if the latter fall short in clinical trials or are too expensive for widespread clinical use in disease endemic countries. The research over the next few years will help define the role of CYP51 inhibitors, alone or in combination with other drugs, for managing patients with Chagas disease.
1. Introduction to Chagas disease
The etiologic agent of Chagas disease, the protozoan Trypanosoma cruzi (T. cruzi), is primarily transmitted to humans by blood feeding Triatomine bugs. Vectorial transmission is confined to the American continent, whereas infection by blood transfusion or by mother-to-baby transmission occurs wherever individuals harboring this chronic infection reside or immigrate. Approximately 8–10 million people are infected in Latin America making it the most prevalent parasitic disease in the American continent and the first cause of heart disease and heart deaths among poor rural populations in Latin America (Rassi et al., 2010). An estimated 300,000 infected persons live in the United States, mostly immigrants from Latin America. A comparable number are living with this parasite in Europe (Gascon et al., 2010; Leslie, 2011).
T. cruzi infects a wide range of mammalian hosts where it establishes a chronic infection. During the initial acute phase, the parasite rapidly cycles between a replicative intracellular stage (amastigotes) and a non-replicative bloodstream stage (trypomastigotes), successfully disseminating throughout the body. The protozoan is capable of infecting diverse host cell types where it replicates freely within the cytoplasm (as opposed to within a vacuolar organelle, which is the case for the related parasites of the Leishmania genus). In an immunocompetent host, the infection is controlled by a mixed immune response involving both humoral and cellular effector mechanisms but, in the large majority of cases, the parasite is not eradicated leading to life-long infections. During the acute stage, lasting just a few weeks, persons have flu-like symptoms, and are rarely diagnosed or treated; this stage has low (ca. 5%) mortality and leads to an initially asymptomatic (“indeterminate”) phase, which for 60–70% of infected persons lasts for the rest of their lives. In the remaining ∼30–40% of individuals, chronic Chagas disease manifests within 1–3 decades, primarily involving the heart, gastrointestinal tract, or nervous system. Chagasic cardiomyopathy, which results from a chronic inflammatory process triggered and sustained by the persistence of the parasite (Urbina, 2010; Marin-Neto et al., 2007; Rassi et al., 2010), is associated with malignant arrhythmias, embolic events, and/or rapidly progressive congestive heart failure and death. Since persons are often infected as children, the morbidity and mortality from Chagas disease typically strikes during the prime adult years in people’s lives.
Two clinical drugs exist for treating Chagas disease, both developed empirically over 40 years ago. These are nitroheterocyclic compounds, benznidazole and nifurtimox, that act by generating free radicals or reductive stress in T. cruzi cells. When used during acute infection, they cure up to 80% of infections, but the vast majority of patients are not diagnosed until they are in the chronic stage where, unfortunately, parasitological cure rates with the drugs, as assessed by conventional serology, are less than 20%. Many clinicians are reluctant to use these drugs because of the unfavorable risk-to-benefit profiles due to side effects such as allergic dermopathy, vomiting, psychosis, and neuropathy (Urbina and Docampo, 2003; Urbina, 2010). Clinical data and animal models indicate that parasitological cures or reduction of the parasite burden of the patients are associated with improved clinical outcomes (Viotti et al., 2006, 2011). As a consequence, new drugs with greater antiparasitic activity and improved safety profiles would potentially make it possible to treat the scores of individuals harboring both acute and chronic T. cruzi infections, to prevent or mitigate the manifestations of chronic Chagas disease.
2. Sterol biosynthesis and CYP51 of T. cruzi
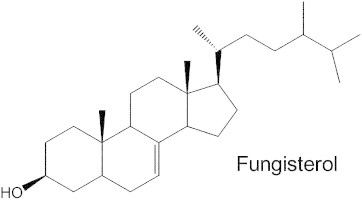
Sterols are essential lipid components of eukaryotic membranes. These molecules are important regulators of membrane physical properties, such as permeability and fluidity, and also have essential roles in aerobic metabolism, regulation of cell cycle, sterol uptake, and sterol transport (Daum et al., 1998). The initial steps in cholesterol biosynthesis also lead to the synthesis of other important molecules, including dolichol, ubiquinone, isopentyladenine, heme A, and prenylated proteins. The final products of sterol biosynthesis vary among eukaryotes with mammals producing cholesterol, while fungi plants and protozoa produce 24-alkyl sterols, with distinct modifications of both the steroid nucleus and the alkyl side chain for each phylogenetic group. The sterol biosynthesis pathway of T. cruzi epimastigotes is illustrated in Fig. 1. T. cruzi is similar to fungi in its sterol composition, with ergosterol (24-methyl-5,7,22-trien-3β-ol) and its 24-ethyl analog (24-ethyl-cholesta-5,7,22-trien-3β-ol) being the major mature sterols in the epimastigote stage (within the insect host) (Fig. 1) (Furlong, 1989; Korn et al., 1969; Urbina et al., 1998; Liendo et al., 1998). The major sterols produced by the amastigote stage (inside the mammalian host cells) are fungisterol (ergosta-7-en-3β-ol) and its 24-ethyl analog (24-ethyl-cholesta-7-en-3β-ol) (Liendo et al., 1999) (Fig. 2). Although T. cruzi incorporates its mammalian host sterols (primarily cholesterol) into its membranes, it has an essential requirement for de novo sterol synthesis for survival in all stages of its life cycle and is highly susceptible to sterol biosynthesis inhibitors (Liendo et al., 1998, 1999; Urbina et al., 1998).
Fig. 1.
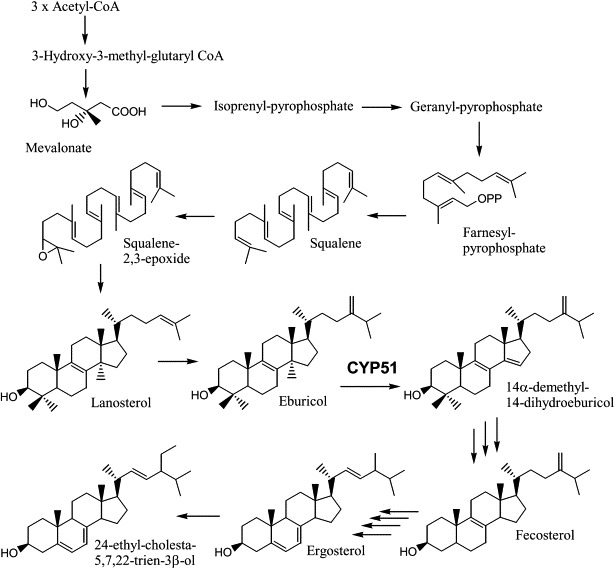
Sterol biosynthesis of T. cruzi epimastigotes (adapted from Lepesheva et al., 2011). The T. cruzi sterol-14-demethylase (labeled CYP51) prefers eburicol as substrate (Lepesheva et al., 2006).
Fig. 2.
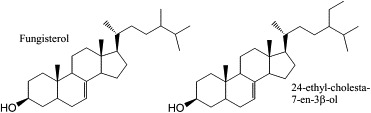
The main sterols of intracellular T. cruzi amastigotes: fungisterol and its 24-ethyl analog.
Synthesis of the major animal sterol (cholesterol) and the 24-alkyl-sterols in fungi, protozoa and plants, requires removal of the 14α-methyl groups from sterol precursors. The reaction is catalyzed by a microsomal cytochrome P450, the sterol C14-demethylase (CYP51). In mammals and yeast, where the substrate is lanosterol (Fig. 1), the enzyme is frequently called the lanosterol 14α-demethylase. In T. cruzi, new data suggests that the preferred substrate is not lanosterol, but rather eburicol (24-methylene-dihydrolanosterol) (Lepesheva et al., 2006) (Fig. 1). CYP51 specifically catalyzes the removal of the C14 methyl group from the sterol scaffold through three successive oxidations resulting in decarboxylation, releasing formic acid (Fischer et al., 1989). During catalysis, the active-site heme iron is reduced by a P450-reductase enzyme utilizing NADPH, from the resting ferric (Fe+++) state to active ferrous (Fe++) state; the resting state is regenerated in each cycle by the transfer of electrons to oxygen and the incorporation of this atom in the C14 substituent of the sterol substrate (Lepesheva et al., 2011). Inhibition of cytochrome P450 enzymes by azole drugs (discussed below) results from coordination of the azole nitrogen to the heme iron, with the lipophilic ligand attached to the azole occupying the binding site for the substrate. These inhibitors prevent both binding of the substrate and oxygen activation (Walker et al., 1993). Recent crystal structures of trypanosomal CYP51 bound to substrate and inhibitors have provided news insights into the details of the catalytic site of this enzyme (Chen et al., 2010; Lepesheva et al., 2010a,b; Hargrove et al., 2011).
3. History of azole drug testing on T. cruzi
Azole derivatives with selective activity against fungal CYP51 (devoid of significant activity against the human ortholog enzyme) have been used as first line antifungal drugs since the 1970s. The first description of activity of CYP51 inhibitors on T. cruzi was reported by Docampo and colleagues thirty years ago (Docampo, 1981). This report described growth inhibitory activity and ultrastructural alterations on T. cruzi cells produced by the topical antifungal azoles miconazole and econazole. Numerous studies followed over the years with the theme of testing antifungal CYP51 inhibitors against T. cruzi. The earlier generation imidazoles (e.g. ketoconazole, miconazole, clotrimazole) and triazoles (e.g. fluconazole and itraconazole) were found to have potent and selective in vitro activity, but were not curative in animal models of T. cruzi infection (Buckner, 2008). Referred to collectively as “azoles”, these drugs have profound effects on the sterol composition of cultured T. cruzi indicating that their mechanism of action is through disruption of sterol biosynthesis, presumably by binding to T. cruzi CYP51, since the block results in accumulation of 14-methylated sterols (Urbina et al., 1996, 1998; Liendo et al., 1999). Additional studies have shown direct binding of these compounds to purified recombinant T. cruzi CYP51 as well as inhibition of its enzymatic function in vitro (Buckner et al., 2003; Lepesheva et al., 2007).
As new azole drugs have been developed by pharmaceutical companies to combat fungal diseases, these in turn have been tested for activity against T. cruzi. The experimental azole drug, D0870 was the first to show cure of mice chronically infected with T. cruzi (Urbina et al., 1996). Unfortunately, D0870 was discontinued in clinical trials (of fungal infections) due to untoward side effects (Williams and Denning, 2001). The second-generation azole antifungal drug, voriconazole, has comparatively weak anti-T. cruzi activity (Buckner, 2008) and has not been pursued for clinical development. Posaconazole (Noxafil®, Merck; Fig. 3), on the other hand, is the most potent and efficacious azole drug against T. cruzi yet to be identified (Liendo et al., 1999; Urbina et al., 1998; Molina et al., 2000). Clinically approved in 2006 for the prophylaxis and treatment of invasive fungal infections due to its broad-spectrum antifungal activity and excellent safety profile, posaconazole could potentially be repurposed for use in Chagas disease, thereby avoiding substantial costs and risks of developing a new chemical entity. It is the only clinically approved azole drug that is curative in the chronic murine model of T. cruzi infection and has potent activity against benznidazole- and nifurtimox-resistant T. cruzi strains, even in immunocompromised animals (Molina et al., 2000). The in vitro potency of posaconazole (subnanomolar concentrations against intracellular amastigotes) is probably due to its very tight binding to T. cruzi CYP51, which involves direct interactions with 13 amino acid side chains in the active site and 12 in the hydrophobic substrate access channel and has, in fact, a stabilizing effect on the tertiary structure of the protein (Fig. 4) (Lepesheva et al., 2010a; Chen et al., 2010). These facts, combined with the favorable pharmacokinetic characteristics (including large volumes of distribution and long terminal half life) of the drug in humans and experimental animals account for the remarkable in vivo anti-T. cruzi activity when compared with other clinically used azoles. Furthermore, it has recently been demonstrated that posaconazole and other lipophilic drugs accumulate in the membranes of cells and, as a result, the effective local concentrations of these drugs that interact with their membrane-bound CYP51 targets are in fact much higher than their overall cell or tissue levels (Campoli et al., 2011). A recent case report from Spain described the successful use of posaconazole to treat a patient with chronic T. cruzi infection, compounded by immunosuppressive treatment required to control systemic lupus erythematosus, who had failed benznidazole chemotherapy (Pinazo et al., 2010). These data have led to two proof-of-concept phase II clinical studies with posaconazole for the specific treatment of Chagas disease: the first was launched in October 2010 at the Vall d’Hebron Hospital in Barcelona, Spain (http://www.clinicaltrials.gov/ct2/show/NCT01162967?term=posaconazole,+Chagas+disease&rank=1) and a second one, sponsored by Merck & Co (http://www.clinicaltrials.gov/ct2/show/NCT01377480?term=Chagas&rank=1) (Clayton, 2010), began recruiting patients in Argentina on the July 2011. Such studies will take us a big step closer to defining the role of this drug for treating Chagas disease. Unfortunately, posaconazole is an extremely expensive drug, so even if it is shown to be efficacious in humans, there is looming concern that it may be difficult to make it available to patients living in resource limited settings (Urbina, 2011).
Fig. 3.
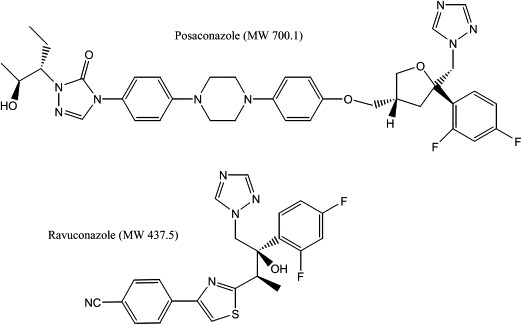
Structures of posaconazole and ravuconazole.
Fig. 4.
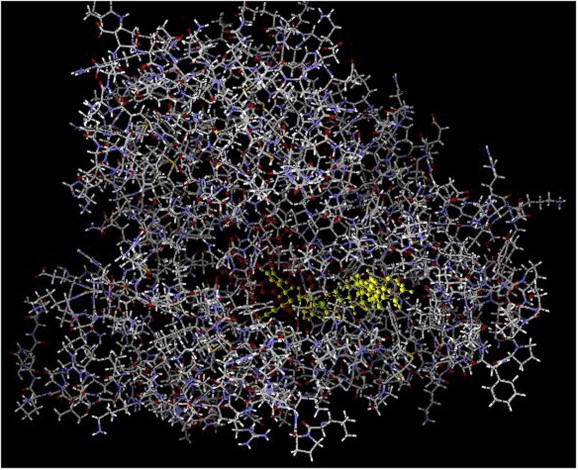
Crystal structure of T. cruzi CYP51 complexed with posaconazole, seen along the substrate access channel; protein in grey, posaconazole in bright green, heme in red. Figure prepared with Molegro Molecular Viewer using coordinates from Protein Data Bank code 3K1O (Lepesheva et al., 2010a). (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Other azole antifungal drugs that have been researched include albaconazole, TAK-187, and ravuconazole (Guedes et al., 2004; Urbina et al., 2003a,b; Corrales et al., 2005; Diniz et al., 2010). Ravuconazole (Eisai Co., Tokyo, Japan; Fig. 3) deserves special mention. It is highly active in vitro against T. cruzi (minimum inhibitory concentration of 1 nM against intracellular amastigotes), but has suppressive, rather than curative activity in murine (Urbina et al., 2003a) and canine (Diniz et al., 2010) models of Chagas disease, probably due to relatively short terminal plasma half-life of ravuconazole in mice (4.5 h) and dogs (8.8 h). However, the half-life is much longer in humans (4–8 days) that, combined with a large volume of distribution, make it possible that the tissue exposure levels would be high enough in humans to be curative. Based on these considerations, the Drugs for Neglected Diseases initiative (DNDi) and Eisai Co. are partnering in a phase II trial of a pro-drug of ravuconazole (E1224) (http://www.dndi.org/press-releases/532-eisai-and-dndi-enter-into-a-collaboration.html) (Clayton, 2010), which started in Bolivia in June 2011. Due to the simpler chemical structure, it is believed that cost of goods might be less of an issue for this candidate than for posaconazole.
4. Anti-T. cruzi CYP51 inhibitors under development in academia
Although repurposing an approved clinical drug (or a candidate in clinical development) has obvious practical advantages, it remains to be seen whether this approach will ultimately deliver a Chagas drug that is sufficiently effective, safe, and affordable for widespread use in populations with high prevalence of Chagas disease. As a consequence, we continue to see work by the Chagas research community exploring other CYP51 inhibitors for anti-T. cruzi activity. To this end, scientists at University of Washington have been developing a class of imidazole containing molecules related to the investigational anticancer drug, tipifarnib (Hucke et al., 2005; Kraus et al., 2009). Tipifarnib is an inhibitor of human protein farnesyl transferase (PFT) that was found to have a remarkable in vitro activity against T. cruzi. It was determined that the activity of the compound against T. cruzi cells was much greater than its inhibitory effects on the T. cruzi PFT enzyme, and subsequent experiments demonstrated that the primary mechanism of action was a blockade of sterol biosynthesis via inhibition of CYP51 (Hucke et al., 2005). Since the inhibitory activity on PFT was unnecessary for its T. cruzi activity, analogs of tipifarnib were designed to minimize this activity (which could lead to deleterious side effects) while improving CYP51 inhibitory activity. Compounds were synthesized that demonstrated sub-nanomolar inhibitory activity on intracellular T. cruzi amastigotes and highly potent suppressive activity on T. cruzi infection in the mouse model (Kraus et al., 2009). The current lead compound (Fig. 5) combines the excellent pharmacokinetic properties of tipifarnib (i.e. oral bioavailability and good metabolic stability) with minimal PFT activity, and sub-nanomolar potency on T. cruzi (Kraus et al., 2010). Efficacy experiments in mice are in progress.
Fig. 5.
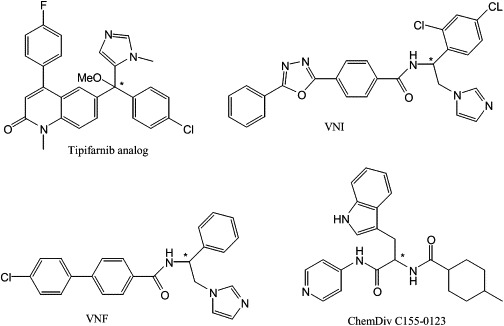
Structures of novel T. cruzi CYP51 inhibitors (see text). Asterix (*) indicates chiral center.
Working with the same strategic aim, the group led by Lepesheva and Waterman investigated a large variety of compounds as potential CYP51 inhibitors using an HTS spectroscopic assay that evaluates type II spectral responses to detect strong binders to the 6th coordination position of the heme iron (Lepesheva et al., 2007, 2008). However, it was found than in many cases the apparent binding parameters determined by spectral titration did not correlate with the inhibitory effects of the compounds on the reconstituted enzyme activity; thus, the spectral hits had to be confirmed using the biochemical assay (Lepesheva et al., 2007, 2008). The best results of this approach have been those resulting from a screen of a collection of azole derivatives of the Novartis Research Institute (Vienna, Austria), which led to the identification of two potent β-phenyl imidazoles, VNF (a.k.a SDZ285604) and VNI (Fig. 5), which produce a functionally irreversible inhibition of T. cruzi and T. brucei CYP51s at a 1:1 inhibitor ratio. Such compounds are remarkably selective for the Trypanosomatid enzymes (the human enzyme is only slightly affected by a 100-fold excess of the inhibitors) (Lepesheva et al., 2008) and VNF (Fig. 5) can eradicate T. cruzi ’s amastigote infection from cultured murine cardiomyocytes at 1 μM (Lepesheva et al., 2010b). Although such selective activity is promising, it remains to be seen whether VNF is as potent as the antifungal triazoles discussed above. Interestingly, it was also found that VNI binds to T. brucei CYP51 in a pose very similar to that of posaconazole bound to T. cruzi CYP51, although with less extensive interactions with the substrate access channel (Lepesheva et al., 2010b); these findings may provide insights on how to increase the potency of this class of compounds, while preserving their selectivity. No in vivo studies of VNF or VNI have been reported. Another approach by the same team led to the identification of a novel class of non-azole T. cruzi CYP51 inhibitors, based on the pyridine-indomethacin amide scaffold (Konkle et al., 2009; Lepesheva et al., 2008). Enzymatic studies showed that ChemDiv C155-0123 (Fig. 5) was a potent but reversible inhibitor of T. cruzi CYP51 (Lepesheva et al., 2008). However, the most potent compound of this series was only able to fully eradicate amastigotes from infected cardiomyocytes at 20 μM. Remarkably, ChemDiv C155-0123 was independently identified by the team of Podust and McKerrow, from a screen against Mycobacterium tuberculosis CYP51, as a potent inhibitor of T. cruzi CYP51 (Lepesheva et al., 2011; Chen et al., 2009). This compound was found to bind selectively to T. cruzi CYP51 and to eradicate T. cruzi amastigotes from infected mouse (J774) macrophages at 10 μM, with no toxicity for the host cells. In a subsequent study (Doyle et al., 2010), the same team reported that ChemDiv C155-0123 (referred to as LP10) inhibited the synthesis of endogenous sterol in T. cruzi epimastigotes and induced partial cures in a murine model of acute Chagas disease. If the ADME and toxicology profile of this compound or subsequent derivatives proves to be appropriate for human use it could become an alternative to the commercial antifungal azoles that now command the field.
5. Prospects for CYP51 inhibitors for Chagas disease
Future treatments for Chagas disease will need to address the glaring inadequacies of the current therapeutics, benznidazole and nifurtimox. First and foremost, these drugs have cure rates as low as 20% for patients with chronic Chagas disease, the stage when the vast majority of individuals are diagnosed. Second, the side effect profile of current drugs makes completing full courses of treatment difficult due to patient intolerance. Third, the effectiveness of these drugs in immune compromised Chagas patients (e.g. organ transplant recipients or AIDS patients) is less than ideal. The outcomes of the ongoing phase II clinical trials of posaconazole and E1224 for Chagas disease will begin to provide answers to whether CYP51 inhibitors will be an improvement upon the existing drugs. Based on clinical experience in patients with fungal infections, posaconazole is likely to be much better tolerated than the existing anti-Chagas drugs. Whether the anti-parasitic activity of posaconazole in humans will be equivalent or better than that observed in experimental animals remains the key unanswered question (although the case report of the patient in Spain with chronic Chagas disease compounded by immunosuppression cured with posaconazole certainly points in this direction) and will not likely be fully resolved with the current phase II studies. Nonetheless, such studies mark a belated but important step for advancing our knowledge of CYP51 inhibitors for human T. cruzi infection.
If posaconazole proves to be successful in these trials and follow up studies, it is likely to revolutionize the approach to treating Chagas disease. In particularly, huge number of patients with chronic asymptomatic infection, who are not currently receiving therapy because of widespread disdain for current drugs, will likely become candidates for anti-T. cruzi therapy to prevent the development of the serious pathological manifestations resulting from the persistence of the parasite in the infected organs. Then the issue of affordability and availability will become paramount. As indicated above the high cost of posaconazole may limit its use in resource-limited regions unless its distribution is subsidized, while for the case of ravuconazole (E1224) the price issue will probably be less important (http://www.dndi.org/press-releases/532-eisai-and-dndi-enter-into-a-collaboration.html). For this reason, alternative, less expensive CYP51 inhibitors, described above, may be necessary to address the needs of patients.
If posaconazole falls short in the clinical trials, will this be the end of CYP51 inhibitors for Chagas disease? It certainly will be a setback, but hopefully the results will guide future action to address the shortcomings. Obviously, issues such as optimal dosing and duration of treatment are likely to be sorted out in subsequent clinical investigations. If satisfactory outcomes are not observed, this could be because of sub-optimal pharmacokinetics, such as inadequate penetration into key tissues harboring parasites. These findings would suggest the need to identify alternative CYP51 inhibitors with better-optimized pharmacokinetic properties. On the other hand, if CYP51 inhibitors are inadequate as monotherapy, could they serve as a component in combination chemotherapy for Chagas disease? Unpublished data indicate that combinations of posaconazole and benznidazole have synergistic activity in animal models (Bahia et al., 2011). A combination strategy could potentially allow for higher cure rates than with either drug alone, and potentially with lower doses or shorter treatment courses for better tolerability and lower cost. Other investigational drugs (such as ergosterol biosynthesis inhibitors acting at a different step of the biosynthetic pathway, e.g. squalene synthase (Urbina et al., 2004), could also be partnered with posaconazole or another CYP51 inhibitor for potentially synergistic activity that could give it the boost it may need. As with many other infectious diseases (e.g., malaria, TB, HIV), combination chemotherapy is becoming standard of care to optimize effectiveness and limit the development of resistance.
In conclusion, CYP51 inhibitors have been a topic of interest for Chagas disease researchers for over 30 years. Small scale, poorly controlled clinical studies with earlier generation antifungal drugs (fluconazole, ketoconazole, and itraconazole) left more questions than answers (Apt et al., 1998; Urbina and Docampo, 2003). Fortunately, we are moving into a new era where highly potent and safe anti-T. cruzi CYP51 inhibitors (posaconazole and E1224) will be studied in well controlled trials in Chagas patients to provide data that will inform future development for this class of compounds. It is an exciting time for this field.
Acknowledgements
Funding to Buckner from NIH (grant AI070218) and to Urbina by the Venezuelan Institute of Scientific Research.
Contributor Information
Frederick S. Buckner, Email: fbuckner@u.washington.edu.
Julio A. Urbina, Email: jurbina@mac.com.
References
- Apt W., Aguilera X., Arribada A., Perez C., Miranda C., Sanchez G., Zulantay I., Cortes P., Rodriguez J., Juri D. Treatment of chronic Chagas’ disease with itraconazole and allopurinol. Am. J. Trop. Med. Hyg. 1998;59:133–138. doi: 10.4269/ajtmh.1998.59.133. [DOI] [PubMed] [Google Scholar]
- Bahia, M.T. 2011. Anti-T. cruzi activities of benznidazole and posaconazole, used alone or in combination, in a murine model of acute Chagas disease. 27th Meeting on Applied Research on Chagas Disease, Uberaba, MG, Brazil.
- Buckner F., Yokoyama K., Lockman J., Aikenhead K., Ohkanda J., Sadilek M., Sebti S., Van Voorhis W., Hamilton A., Gelb M.H. A class of sterol 14-demethylase inhibitors as anti-Trypanosoma cruzi agents. Proc. Natl. Acad. Sci. USA. 2003;100:15149–15153. doi: 10.1073/pnas.2535442100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckner F.S. Sterol 14-demethylase inhibitors for Trypanosoma cruzi infections. Adv. Exp. Med. Biol. 2008;625:61–80. doi: 10.1007/978-0-387-77570-8_6. [DOI] [PubMed] [Google Scholar]
- Campoli P., Al Abdallah Q., Robitaille R., Solis N.V., Fielhaber J.A., Kristof A.S., Laverdiere M., Filler S.G., Sheppard D.C. Concentration of antifungal agents within host cell membranes: a new paradigm governing the efficacy of prophylaxis. Antimicrob. Agents Chemother. 2011;55:5732–5739. doi: 10.1128/AAC.00637-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C.K., Doyle P.S., Yermalitskaya L.V., Mackey Z.B., Ang K.K., McKerrow J.H., Podust L.M. Trypanosoma cruzi CYP51 inhibitor derived from a Mycobacterium tuberculosis screen hit. PLoS Negl. Trop. Dis. 2009;3:e372. doi: 10.1371/journal.pntd.0000372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C.K., Leung S.S., Guilbert C., Jacobson M.P., McKerrow J.H., Podust L.M. Structural characterization of CYP51 from Trypanosoma cruzi and Trypanosoma brucei bound to the antifungal drugs posaconazole and fluconazole. PLoS Negl. Trop. Dis. 2010;4:e651. doi: 10.1371/journal.pntd.0000651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton J. Chagas Disease: Pushing Through the Pipeline. Nature. 2010;465:S12–S15. doi: 10.1038/nature09224. [DOI] [PubMed] [Google Scholar]
- Corrales M., Cardozo R., Segura M.A., Urbina J.A., Basombrio M.A. Comparative efficacies of TAK-187, a long-lasting ergosterol biosynthesis inhibitor, and benznidazole in preventing cardiac damage in a murine model of Chagas’ disease. Antimicrob. Agents Chemother. 2005;49:1556–1560. doi: 10.1128/AAC.49.4.1556-1560.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daum G., Lees N.D., Bard M., Dickson R. Biochemistry, cell biology and molecular biology of lipids of Saccharomyces cerevisiae. Yeast. 1998;14:1471–1510. doi: 10.1002/(SICI)1097-0061(199812)14:16<1471::AID-YEA353>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- Diniz L.F., Caldas I.S., Guedes P.M., Crepalde G., de Lana M., Carneiro C.M., Talvani A., Urbina J.A., Bahia M.T. Effects of ravuconazole treatment on parasite load and immune response in dogs experimentally infected with Trypanosoma cruzi. Antimicrob. Agents Chemother. 2010;54:2979–2986. doi: 10.1128/AAC.01742-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Docampo R. Biochemical and ultrastructural alterations produced by miconazole and econazole in Trypanosoma cruzi. Mol. Biochem. Parasitol. 1981;3:169–180. doi: 10.1016/0166-6851(81)90047-5. [DOI] [PubMed] [Google Scholar]
- Doyle P.S., Chen C.K., Johnston J.B., Hopkins S.D., Leung S.S., Jacobson M.P., Engel J.C., McKerrow J.H., Podust L.M. A nonazole CYP51 inhibitor cures Chagas’ disease in a mouse model of acute infection. Antimicrob. Agents Chemother. 2010;54:2480–2488. doi: 10.1128/AAC.00281-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer R.T., Stam S.H., Johnson P.R., Ko S.S., Magolda R.L., Gaylor J.L., Trzaskos J.M. Mechanistic studies of lanosterol 14 alpha-methyl demethylase: substrate requirements for the component reactions catalyzed by a single cytochrome P-450 isozyme. J. Lipid Res. 1989;30:1621–1632. [PubMed] [Google Scholar]
- Furlong S.T. Sterols of parasitic protozoa and helminths. Exp. Parasitol. 1989;68:482–485. doi: 10.1016/0014-4894(89)90134-3. [DOI] [PubMed] [Google Scholar]
- Gascon J., Bern C., Pinazo M.J. Chagas disease in Spain, the United States and other non-endemic countries. Acta Trop. 2010;115:22–27. doi: 10.1016/j.actatropica.2009.07.019. [DOI] [PubMed] [Google Scholar]
- Guedes P.M., Urbina J.A., de Lana M., Afonso L.C., Veloso V.M., Tafuri W.L., Machado-Coelho G.L., Chiari E., Bahia M.T. Activity of the new triazole derivative albaconazole against Trypanosoma (Schizotrypanum) cruzi in dog hosts. Antimicrob. Agents Chemother. 2004;48:4286–4292. doi: 10.1128/AAC.48.11.4286-4292.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hargrove, T.Y., Wawrzak, Z., Liu, J., Waterman, M.R., Nes, W.D., Lepesheva, G.I., 2011. Structural complex of sterol 14alpha-demethylase (CYP51) with 14alpha-methylenecyclopropyl-Δ7-24, 25-dihydrolanosterol. J. Lipid Res. (Epub ahead of print). [DOI] [PMC free article] [PubMed]
- Hucke O., Gelb M.H., Verlinde C.L., Buckner F.S. The protein farnesyltransferase inhibitor Tipifarnib as a new lead for the development of drugs against Chagas disease. J. Med. Chem. 2005;48:5415–5418. doi: 10.1021/jm050441z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konkle M.E., Hargrove T.Y., Kleshchenko Y.Y., von Kries J.P., Ridenour W., Uddin M.J., Caprioli R.M., Marnett L.J., Nes W.D., Villalta F., Waterman M.R., Lepesheva G.I. Indomethacin amides as a novel molecular scaffold for targeting Trypanosoma cruzi sterol 14alpha-demethylase. J. Med. Chem. 2009;52:2846–2853. doi: 10.1021/jm801643b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korn E.D., Von Brand T., Tobie E.J. The sterols of Trypanosoma cruzi and Crithidia fasciculata. Comp. Biochem. Physiol. 1969;30:601–610. doi: 10.1016/0010-406x(69)92137-9. [DOI] [PubMed] [Google Scholar]
- Kraus J.M., Verlinde C.L., Karimi M., Lepesheva G.I., Gelb M.H., Buckner F.S. Rational modification of a candidate cancer drug for use against Chagas disease. J. Med. Chem. 2009;52:1639–1647. doi: 10.1021/jm801313t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraus J.M., Tatipaka H.B., McGuffin S.A., Chennamaneni N.K., Karimi M., Arif J., Verlinde C.L., Buckner F.S., Gelb M.H. Second generation analogues of the cancer drug clinical candidate Tipifarnib for anti-chagas disease drug discovery. J. Med. Chem. 2010;53:3887–3898. doi: 10.1021/jm9013136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepesheva G.I., Zaitseva N.G., Nes W.D., Zhou W., Arase M., Liu J., Hill G.C., Waterman M.R. CYP51 from Trypanosoma cruzi: a phyla-specific residue in the B’ helix defines substrate preferences of sterol 14alpha-demethylase. J. Biol. Chem. 2006;281:3577–3585. doi: 10.1074/jbc.M510317200. [DOI] [PubMed] [Google Scholar]
- Lepesheva G.I., Ott R.D., Hargrove T.Y., Kleshchenko Y.Y., Schuster I., Nes W.D., Hill G.C., Villalta F., Waterman M.R. Sterol 14alpha-demethylase as a potential target for antitrypanosomal therapy: enzyme inhibition and parasite cell growth. Chem. Biol. 2007;14:1283–1293. doi: 10.1016/j.chembiol.2007.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepesheva G.I., Hargrove T.Y., Anderson S., Kleshchenko Y., Furtak V., Wawrzak Z., Villalta F., Waterman M.R. Structural insights into inhibition of sterol 14alpha-demethylase in the human pathogen Trypanosoma cruzi. J. Biol. Chem. 2010;285:25582–25590. doi: 10.1074/jbc.M110.133215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepesheva G.I., Hargrove T.Y., Kleshchenko Y., Nes W.D., Villalta F., Waterman M.R. CYP51: a major drug target in the cytochrome P450 superfamily. Lipids. 2008;43:1117–1125. doi: 10.1007/s11745-008-3225-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepesheva G.I., Park H.W., Hargrove T.Y., Vanhollebeke B., Wawrzak Z., Harp J.M., Sundaramoorthy M., Nes W.D., Pays E., Chaudhuri M., Villalta F., Waterman M.R. Crystal structures of Trypanosoma brucei sterol 14alpha-demethylase and implications for selective treatment of human infections. J. Biol. Chem. 2010;285:1773–1780. doi: 10.1074/jbc.M109.067470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepesheva G.I., Villalta F., Waterman M.R. Targeting Trypanosoma cruzi sterol 14alpha-demethylase (CYP51) Adv. Parasitol. 2011;75:65–87. doi: 10.1016/B978-0-12-385863-4.00004-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leslie M. Infectious diseases. A tropical disease hits the road. Science. 2011;333:934. doi: 10.1126/science.333.6045.934. [DOI] [PubMed] [Google Scholar]
- Liendo A., Lazardi K., Urbina J.A. In-vitro antiproliferative effects and mechanism of action of the bis-triazole D0870 and its S(−) enantiomer against Trypanosoma cruzi. J. Antimicrob. Chemother. 1998;41:197–205. doi: 10.1093/jac/41.2.197. [DOI] [PubMed] [Google Scholar]
- Liendo A., Visbal G., Piras M.M., Piras R., Urbina J.A. Sterol composition and biosynthesis in Trypanosoma cruzi amastigotes. Mol. Biochem. Parasitol. 1999;104:81–91. doi: 10.1016/s0166-6851(99)00129-2. [DOI] [PubMed] [Google Scholar]
- Marin-Neto J.A., Cunha-Neto E., Maciel B.C., Simoes M.V. Pathogenesis of chronic Chagas heart disease. Circulation. 2007;115:1109–1123. doi: 10.1161/CIRCULATIONAHA.106.624296. [DOI] [PubMed] [Google Scholar]
- Molina J., Martins-Filho O., Brener Z., Romanha A.J., Loebenberg D., Urbina J.A. Activities of the triazole derivative SCH 56592 (posaconazole) against drug-resistant strains of the protozoan parasite Trypanosoma (Schizotrypanum) cruzi in immunocompetent and immunosuppressed murine hosts. Antimicrob. Agents Chemother. 2000;44:150–155. doi: 10.1128/aac.44.1.150-155.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinazo M.J., Espinosa G., Gallego M., Lopez-Chejade P.L., Urbina J.A., Gascon J. Successful treatment with posaconazole of a patient with chronic Chagas disease and systemic lupus erythematosus. Am. J. Trop. Med. Hyg. 2010;82:583–587. doi: 10.4269/ajtmh.2010.09-0620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rassi A., Jr., Rassi A., Marin-Neto J.A. Chagas disease. Lancet. 2010;375:1388–1402. doi: 10.1016/S0140-6736(10)60061-X. [DOI] [PubMed] [Google Scholar]
- Urbina J. New insights in Chagas’s disease treatment. Drug Future. 2011;35:409–419. [Google Scholar]
- Urbina J.A. Specific chemotherapy of Chagas disease: relevance, current limitations and new approaches. Acta Trop. 2010;115:55–68. doi: 10.1016/j.actatropica.2009.10.023. [DOI] [PubMed] [Google Scholar]
- Urbina J.A., Docampo R. Specific chemotherapy of Chagas disease: controversies and advances. Trends Parasitol. 2003;19:495–501. doi: 10.1016/j.pt.2003.09.001. [DOI] [PubMed] [Google Scholar]
- Urbina J.A., Payares B., Molina J., Sanoja C., Liendo A., Lazardi K., Piras M.M., Piras R., Wincker P., Ryley J.F. Cure of short- and long-term experimental Chagas’ disease using D0870. Science. 1996;273:969–971. doi: 10.1126/science.273.5277.969. [DOI] [PubMed] [Google Scholar]
- Urbina J.A., Payares G., Contreras L.M., Liendo A., Sanoja C., Molina J., Piras M., Piras R., Perez N., Wincker P., Loebenberg D. Antiproliferative effects and mechanism of action of SCH 56592 against Trypanosoma (Schizotrypanum) cruzi: in vitro and in vivo studies. Antimicrob. Agents Chemother. 1998;42:1771–1777. doi: 10.1128/aac.42.7.1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbina J.A., Payares G., Sanoja C., Lira R., Romanha A.J. In vitro and in vivo activities of ravuconazole on Trypanosoma cruzi, the causative agent of Chagas disease. Int. J. Antimicrob. Agents. 2003;21:27–38. doi: 10.1016/s0924-8579(02)00273-x. [DOI] [PubMed] [Google Scholar]
- Urbina J.A., Payares G., Sanoja C., Molina J., Lira R., Brener Z., Romanha A.J. Parasitological cure of acute and chronic experimental Chagas disease using the long-acting experimental triazole TAK-187. Activity against drug-resistant Trypanosoma cruzi strains. Int. J. Antimicrob. Agents. 2003;21:39–48. doi: 10.1016/s0924-8579(02)00274-1. [DOI] [PubMed] [Google Scholar]
- Urbina J.A., Concepcion J.L., Caldera A., Payares G., Sanoja C., Otomo T., Hiyoshi H. In vitro and in vivo activities of E5700 and ER-119884, two novel orally active squalene synthase inhibitors, against Trypanosoma cruzi. Antimicrob. Agents Chemother. 2004;48:2379–2387. doi: 10.1128/AAC.48.7.2379-2387.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viotti R., Vigliano C., Lococo B., Bertocchi G., Petti M., Alvarez M.G., Postan M., Armenti A. Long-term cardiac outcomes of treating chronic Chagas disease with benznidazole versus no treatment: a nonrandomized trial. Ann. Intern. Med. 2006;144:724–734. doi: 10.7326/0003-4819-144-10-200605160-00006. [DOI] [PubMed] [Google Scholar]
- Viotti R., Vigliano C., Alvarez M.G., Lococo B., Petti M., Bertocchi G., Armenti A., De Rissio A.M., Cooley G., Tarleton R., Laucella S. Impact of aetiological treatment on conventional and multiplex serology in chronic Chagas disease. PLoS Negl. Trop. Dis. 2011;5:e1314. doi: 10.1371/journal.pntd.0001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker K.A., Kertesz D.J., Rotstein D.M., Swinney D.C., Berry P.W., So O.Y., Webb A.S., Watson D.M., Mak A.Y., Burton P.M. Selective inhibition of mammalian lanosterol 14 alpha-demethylase: a possible strategy for cholesterol lowering. J. Med. Chem. 1993;36:2235–2237. doi: 10.1021/jm00067a022. [DOI] [PubMed] [Google Scholar]
- Williams K.J., Denning D.W. Termination of development of D0870. J. Antimicrob. Chemother. 2001;47:720–721. doi: 10.1093/oxfordjournals.jac.a002691. [DOI] [PubMed] [Google Scholar]