

Abstract
Aldo-keto reductase 1D1 (AKR1D1) deficiency, a rare but life-threatening form of bile acid deficiency, has not been previously described in China. Here, we describe the first two primary ∆4-3-oxosteroid 5β-reductase deficiency patients in Mainland China diagnosed by fast atom bombardment-mass spectroscopy of urinary bile acids and confirmed by genetic analysis. A high proportion of atypical 3-oxo-∆4-bile acids in the urine indicated a deficiency in ∆4-3-oxosteroid 5β-reductase. All of the coding exons and adjacent intronic sequence of the AKR1D1 gene were sequenced using peripheral lymphocyte genomic DNA of two patients and one of the patient’s parents. One patient exhibited compound heterozygous mutations: c.396C>A and c.722A>T, while the other was heterozygous for the mutation c.797G>A. Based on these mutations, a diagnosis of primary ∆4-3-oxosteroid 5β-reductase deficiency could be confirmed. With ursodeoxycholic acid treatment and fat-soluble vitamin supplements, liver function tests normalized rapidly, and the degree of hepatomegaly was markedly reduced in both patients.
Keywords: Primary ∆4-3-oxosteroid 5β-reductase gene, Cholestasis, Bile acid therapy, Aldo-keto reductase 1D1, Bile acid synthetic defects
INTRODUCTION
Primary ∆4-3-oxosteroid 5β-reductase deficiency (OMIM 235555, also called congenital bile acid synthesis defect type 2, CBAS 2) is a rare but potentially life-threatening form of bile acid deficiency that presents with neonatal progressive intrahepatic cholestasis and normal or slightly elevated γ-glutamyltransferase (GGT)[1]. In the patients described to date, it is noted that early diagnosis and primary bile acid treatment lead to progressive normalization of liver function and avoidance of liver transplantation[2].
The aldo-keto reductase 1D1 (AKR1D1) gene encodes the only known human ∆4-3-oxosteroid 5β-reductase, which catalyzes the reactions responsible for changes to the steroid nucleus of cholesterol in both major bile acid synthesis pathways[3]. Mutations in AKR1D1 lead to a deficiency of the bile acids that normally facilitate the detachment of γ-glutamyl-transpeptidase from the canalicular membrane. So, unlike other causes of cholestasis that present with markedly elevated GGT, CBAS 2 presents with normal or slightly elevated GGT[1].
To our knowledge, an accurate diagnosis of CBAS 2 requires AKR1D1 gene analysis, but only 8 reported cases of CBAS 2 worldwide have been diagnosed both biochemically and genetically[1,4]. To further the knowledge of the clinical features and therapeutic treatment of CBAS 2, here we describe two patients in Mainland China with progressive intrahepatic cholestasis in early infancy diagnosed by fast atom bombardment-mass spectroscopy (FAB-MS) analysis of urinary bile acids and genetic analysis.
CASE REPORT
Patients
A male patient (paitent 1) was delivered at term after an uneventful pregnancy by a caesarean delivery with a birth weight of 3100 g. The infant represented his mother’s third pregnancy; the first two pregnancies were terminated by abortion for social reasons. The patient’s parents were both healthy and non-consanguineous. He developed progressive jaundice from 10 d of life with dark urine. At the age of 4 mo, recurrent pruritus and white stool were noticed, and exploratory laparotomy was performed. Hepatic pathology showed marked giant transformation of hepatocytes, lobular disarray, hepatocellular bile stasis, mildly interlobular fibrosis, proliferation and inflammation. After treatment with traditional Chinese medicine for 2 mo, the pruritus was relieved, but the jaundice persisted. At the age of 11 mo, he was referred to the hospital. On physical examination, moderate jaundice was present. The abdomen was soft, and the liver was detected 2 cm below the xiphoid process and 4.5 cm below the right costal margin, while the spleen was detected 2 cm below the left costal margin. There were no dysmorphic features or cardiac murmur. Laboratory studies at 11 mo showed total bilirubin (TBil) 146 μmol/L (normal range: 5.1-17.1 μmol/L), direct bilirubin (DBil) 113 μmol/L (normal range: 0-6 μmol/L), alanine aminotransferase (ALT) 210 IU/L (normal range: 0-40 IU/L), aspartate aminotransferase (AST) 207 IU/L (normal range: 0-40 IU/L), alkaline phosphatase 162 IU/L (normal range: 42-383 IU/L), GGT 65 IU/L (normal range: 7-50 IU/L), total bile acids (TBA) 6.1 μmol/L (normal range: 0-10 μmol/L). Abdominal ultrasound showed hepatomegaly 4.5 cm below the right costal margin and splenomegaly 5 cm below the left costal margin.
Another male patient (paitent 2) born at term after an uneventful pregnancy by a caesarean delivery with a birth weight of 3950 g was the first child of a non-consanguineous couple. The mother had hepatitis B and the father was healthy. He developed mild jaundice from 3 d of life and did not receive any medicine. At the age of 11 wk, he was referred to our hospital. On physical examination, mild jaundice was present. The abdomen was soft, and the liver was 4 cm below the right costal margin and 3 cm below the xiphoid process. The spleen was not felt. There were no dysmorphic features or cardiac murmur. Laboratory studies showed TBil 96.1 μmol/L (normal range: 5.1-17.1 μmol/L), DBil 64.4 μmol/L (normal range: 0-6 μmol/L), ALT 176 IU/L (normal range: 0-40 IU/L), AST 183 IU/L (normal range: 0-40 IU/L), GGT 33 IU/L (normal range: 7-50 IU/L), TBA 3 μmol/L (normal range: 0-10 μmol/L). Abdominal ultrasound showed hepatomegaly 4.5 cm below the right costal margin and splenomegaly 2 cm below the left costal margin.
Urine bile acid analysis
For patient 1, the first urine sample was taken at 11 mo of age when ursodeoxycholic acid (UDCA) (150 mg/d) was discontinued for 7 d. The FAB-MS analysis showed that the largest bile acid peaks were consistent with the presence of residual amounts of ursodeoxycholic acid metabolite (m/z 528), and the atypical bile acids of dihydroxy-oxo-cholenoic acids present in the glycine conjugate (m/z 460) and taurine conjugate (m/z 510) forms and monohydroxy-oxo-cholenic acids presents in the glycine conjugate (m/z 444) and the taurine conjugate (m/z 494) forms. Peaks attributable to the glycine and taurine conjugates of chenodeoxycholic acid and cholic acid (m/z 448, 464, 498, and 514) were present in traces or undetectable above background[5]. A second urine sample was taken at 17 mo of age, when the patient was treated with UDCA (250 mg/d). Liver function test (LFT) was normal, with slightly elevated aminotransferase. The FAB-MS analysis showed that the largest bile acid peaks were consistent with ursodeoxycholic acid metabolite (m/z 528), a dihydroxy-oxo-cholenoic acid present as the glycine conjugate (m/z 460) and a monohydroxy-oxo-cholenic acid present as the glycine conjugate (m/z 444).
For the second patient, a urine sample was taken at 3 mo of age, when he was treated with UDCA (100 mg/d). LFT had improved but was not completely normal. The FAB-MS analysis was similar to that for patient 1.
Genetic analysis
The study protocol conforms to the ethical guidelines of the Declaration of Helsinki of 1975. With the approval by the Ethics Committee on human research of the Children’s Hospital of Fudan University and the informed consent of the parents, 1 mL of whole blood was drawn from the patient and his parents. To confirm the diagnosis and establish the molecular basis of the disorder, genomic DNA was isolated from the white blood cells. Then, we sequenced all of the coding exons and adjacent intronic sequence of the AKR1D1 gene. Patient 1 was found to be heterozygous for c.797G>A in exon 7, which leads to an amino acid substitution of arginine by glutamine at amino acid position 266 (p.R266Q) (Figure 1). Patient 2 was revealed to be compound heterozygous for c.396C>A (nonsense mutation) and c.722A>T (p.D241V), the former from his father and the latter from his mother (Figure 2).
Figure 1.
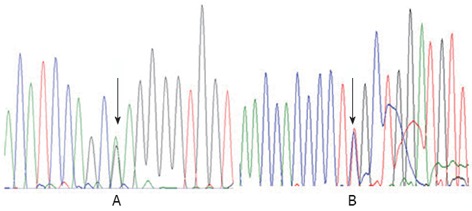
Genomic DNA sequences in exon 7 of the aldo-keto reductase 1D1 gene in patient 1. A: The arrow identified a heterozygote mutation for c.797G>A (p.R266Q) in this patient; B: The reverse strand sequence shows the same result.
Figure 2.
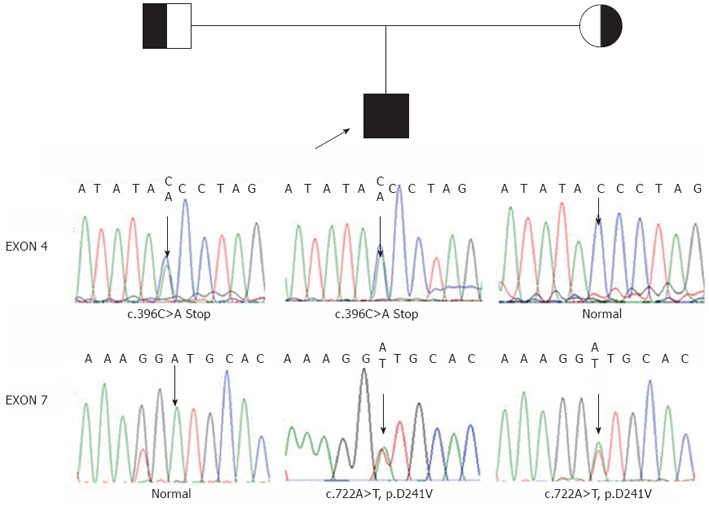
Pedigree for patient 2 shown with genomic DNA sequences in exons 4 and 7 of the aldo-keto reductase 1D1 gene in patient 2 and his parents. The arrow in exon 4 identified C/A in patient 2s and his father, but C in his mother. The arrow in exon 7 identified A/T in patient 2 and his mother, but T in his father. These represent compound heterozygote with c.396C>A (nonsene mutation) from his father and c.722A>T (p.D241V) from his mother.
Management and treatment
Patient 1 was treated with UDCA (40 mg/kg per day) for 5 mo. Liver transaminase concentration was normalized (Figure 3A), and the degree of hepatomegaly was markedly reduced. Jaundice has improved and disappeared. Recently, liver function tests showed TBil 6.7 μmol/L, DBil 2.9 μmol/L, ALT 8 IU/L, AST 16 IU/L, and GGT 10 IU/L.
Figure 3.
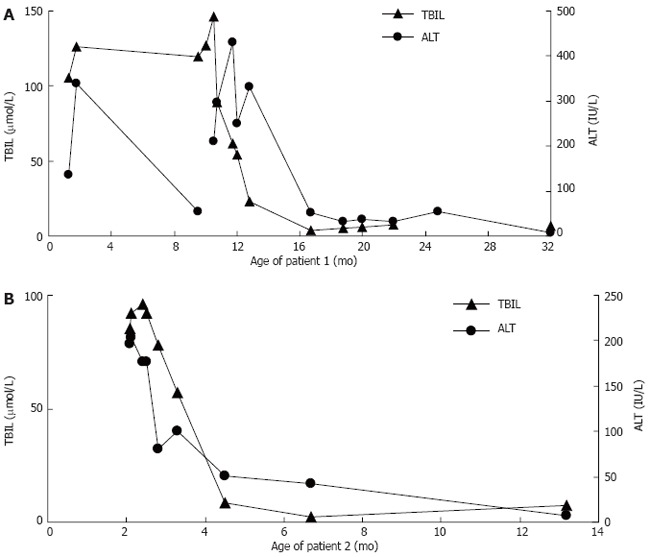
The responds of total bilirubin and alanine aminotransferase to treatment with ursodeoxycholic acid and/or chenodeoxycholic acid. A: Patient 1; B: Patient 2. TBIL: Total bilirubin; ALT: Alanine aminotransaminase.
Patient 2 was treated with UDCA (40 mg/kg per day) for 4 mo. The liver transaminase concentration was normalized, and the degree of hepatomegaly was markedly reduced. Jaundice has improved and disappeared. After FAB-MS analysis of urine bile acid, the patient was given chenodeoxycholic acid (CDCA, 25 mg/kg per day) instead of UDCA for 4 mo. After the CDCA was discontinued, the patient’s total bilirubin and alanine aminotransferase was normal (Figure 3). However, the patient was not normally responsive to both visual stimuli and auditory stimuli. Magnetic resonance imaging at the age of 13 mo showed cerebral dysplasia with abnormally low levels of white matter.
DISCUSSION
Primary ∆4-3-oxosteroid 5β-reductase deficiency, first reported by Setchell et al[6], is an extremely rare condition. We diagnosed two Chinese infants, both of whom presented with neonatal progressive intrahepatic cholestasis with markedly elevated serum conjugated bilirubin and aminotransferase but normal serum TBA and normal or slightly elevated GGT. Diagnosis of CBAS 2 was established by FAB-MS analysis of urinary bile acids and genetic analysis. According to clinical features and biochemical findings, bile acid synthesis deficiency was highly suspected[1,7,8]. However, analyses of the bile acid profile and AKR1D1 gene sequence were not available in our center at that time. Our patients were given a large dose of UDCA combined with a fat-soluble vitamin supplement, which was inadvertently found to produce a good response in our patients with CBAS 1.
The first urine sample of patient 1 was collected when UDCA (150 mg/d) was stopped 7 d, although there are several ions that may represent bile acid metabolites that are usually observed with UDCA therapy for metabolism of UDCA may be present, the profile was conspicuous by lack of normal primary bile acid conjugates that are characteristic of a bile acid synthetic defect involving a deficiency in the activity of the ∆4-3-oxosteroid 5β-reductase deficiency. The second urine sample of patient 1 and the urine sample of patient 2 were collected when UDCA tharapies were administered. The profiles were complicated by several ions that represent bile acid metabolites usually observed with UDCA therapy. Both were highly probable that these were primary defects in bile acid synthesis[6]. To definitively confirm a primary defect[4], we sequenced all the coding exons and adjacent intronic sequence of the AKR1D1 gene.
Three new mutations in AKR1D1 exons were detected in our patients, all of which altered the sequence and function of protein and were predicted to be disease causing by Mutation Taster (http://www.mutationtaster.org/). All of the altered amino acid loci are highly conserved among orthologous proteins in different species. There were two patients reported who were found with just one heterozygote mutation[4]. We sequenced all the coding exons and adjacent intronic sequence of the AKR1D1 gene, another heterozygous mutation might be outside the coding and splicing sequence or there might be a large DNA genomic aberration which couldn’t be found in Sanger sequencing[4,8]. Based on biochemical changes in urine bile acids and mutations in AKR1D1 gene, the diagnosis of CBAS 2 in both patients was confirmed.
UDCA is not recommended to be used in treatment of patients with the AKR1D1 deficiency, because it has no ability to downregulate endogenous bile acid synthesis and prevent continued synthesis of atypical and potentially hepatotoxic bile acid intermediates and their metabolities[9]. However, an analysis of the urinary bile acids of patient 1 showed that, even after liver function had normalized, large amounts of abnormal bile acids and UDCA remained. In addition, patient 2, who was treated by UDCA followed by CDCA, showed cerebral dysplasia with low levels of white matter. However, it would be difficult to determine whether this injury was due to the deficiency of delta 4-3-oxosteroid 5β-reductase and accumulation of abnormal bile acids in lipids in the cerebrum or intracranial space or to other metabolic complications.
In conclusion, we report the first two documented cases of CBAS 2 in Mainland China. Although rare, primary 3-oxo-delta 4-steroid 5β-reductase deficiency should be considered in patients with neonatal cholestatic jaundice with normal TBA and normal or slightly elevated GGT.
Footnotes
Supported by National Natural Science Foundation of China, No. 81070281
Peer reviewer: Silvana Zanlungo, Professor, Department of Gastroenterology, Pontificia Universidad Católica de Chile, Marcoleta 367, Casilla 114-D, Santiago, Chile
S- Editor Gou SX L- Editor A E- Editor Zhang DN
References
- 1.Clayton PT. Disorders of bile acid synthesis. J Inherit Metab Dis. 2011;34:593–604. doi: 10.1007/s10545-010-9259-3. [DOI] [PubMed] [Google Scholar]
- 2.Gonzales E, Gerhardt MF, Fabre M, Setchell KD, Davit-Spraul A, Vincent I, Heubi JE, Bernard O, Jacquemin E. Oral cholic acid for hereditary defects of primary bile acid synthesis: a safe and effective long-term therapy. Gastroenterology. 2009;137:1310–1320.e1-3. doi: 10.1053/j.gastro.2009.07.043. [DOI] [PubMed] [Google Scholar]
- 3.Drury JE, Mindnich R, Penning TM. Characterization of disease-related 5beta-reductase (AKR1D1) mutations reveals their potential to cause bile acid deficiency. J Biol Chem. 2010;285:24529–24537. doi: 10.1074/jbc.M110.127779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ueki I, Kimura A, Chen HL, Yorifuji T, Mori J, Itoh S, Maruyama K, Ishige T, Takei H, Nittono H, et al. SRD5B1 gene analysis needed for the accurate diagnosis of primary 3-oxo-Delta4-steroid 5beta-reductase deficiency. J Gastroenterol Hepatol. 2009;24:776–785. doi: 10.1111/j.1440-1746.2008.05669.x. [DOI] [PubMed] [Google Scholar]
- 5.Lemonde HA, Custard EJ, Bouquet J, Duran M, Overmars H, Scambler PJ, Clayton PT. Mutations in SRD5B1 (AKR1D1), the gene encoding delta(4)-3-oxosteroid 5beta-reductase, in hepatitis and liver failure in infancy. Gut. 2003;52:1494–1499. doi: 10.1136/gut.52.10.1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Setchell KD, Suchy FJ, Welsh MB, Zimmer-Nechemias L, Heubi J, Balistreri WF. Delta 4-3-oxosteroid 5 beta-reductase deficiency described in identical twins with neonatal hepatitis. A new inborn error in bile acid synthesis. J Clin Invest. 1988;82:2148–2157. doi: 10.1172/JCI113837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sundaram SS, Bove KE, Lovell MA, Sokol RJ. Mechanisms of disease: Inborn errors of bile acid synthesis. Nat Clin Pract Gastroenterol Hepatol. 2008;5:456–468. doi: 10.1038/ncpgasthep1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sumazaki R, Nakamura N, Shoda J, Kurosawa T, Tohma M. Gene analysis in delta 4-3-oxosteroid 5 beta-reductase deficiency. Lancet. 1997;349:329. doi: 10.1016/s0140-6736(05)62828-0. [DOI] [PubMed] [Google Scholar]
- 9.Setchell KD, Heubi JE. Defects in bile acid biosynthesis--diagnosis and treatment. J Pediatr Gastroenterol Nutr. 2006;43 Suppl 1:S17–S22. doi: 10.1097/01.mpg.0000226386.79483.7b. [DOI] [PubMed] [Google Scholar]