

Background: A computational model predicts the structure and gates of a nucleoside transporter.
Results: Residues in the predicted extracellular gate were substituted by cysteine pairs and cross-linked.
Conclusion: The predictions of the computational model for the extracellular gate are supported by experimental evidence.
Significance: Defining the gates of a transporter is of fundamental importance for understanding its structure and function.
Keywords: Computer Modeling, Membrane Transport, Mutagenesis Site-specific, Nucleoside Nucleotide Transport, Parasitology, Protein Cross-linking, Gating, Protein Structure and Function
Abstract
Equilibrative nucleoside transporters are a unique family of proteins that enable uptake of nucleosides/nucleobases into a wide range of eukaryotes and internalize a myriad of drugs used in the treatment of cancer, heart disease, AIDs, and parasitic infections. In previous work we generated a structural model for such a transporter, the LdNT1.1 nucleoside permease from the parasitic protozoan Leishmania donovani, using ab initio computation. The model suggested that aromatic residues present in transmembrane helices 1, 2, and 7 interact to form an extracellular gate that closes the permeation pathway in the inward-open conformation. Mutation of residues Phe-48TM1 and Trp-75TM2 abrogated transport activity, consistent with such prediction. In this study cysteine mutagenesis and oxidative cross-linking were combined to analyze proximity relationships of helices 1, 2, and 7 in LdNT1.1. Disulfide bond formation between introduced paired cysteines at the interface of such helices (A61CTM1/F74CTM2, A61CTM1/G350CTM7, and F74CTM2/G350CTM7) was analyzed by transport measurement and gel mobility shifts upon oxidation with Cu (II)-(1,10-phenanthroline)3. In all cases cross-linking inhibited transport. However, if LdNT1.1 ligands were included during cross-linking, inhibition of transport was reduced, suggesting that ligands moved the three gating helices apart. Moreover, all paired cysteine mutants exhibited a mobility shift upon oxidation, corroborating the formation of a disulfide bond. These data support the notion that helices 1, 2, and 7 constitute the extracellular gate of LdNT1.1, thus further validating the computational model and the previously demonstrated importance of F48TM1 and Trp-75TM2 in tethering together helices that are part of the gate.
Introduction
Naturally occurring nucleosides play central roles in cell metabolism as the precursors for a variety of biological molecules. In addition, nucleosides such as adenosine are ligands for various cell surface receptors and, hence, mediate multiple physiological processes such as vasodilation, neuromodulation, and intestinal motility (1). Moreover, many nucleoside analogs are currently used in anticancer, antiviral, and anti-protozoal therapies (2), and nucleoside transporters are also the target of drugs used in the treatment of cardiovascular ailments (3), thus highlighting the pharmacological role that nucleosides and nucleoside transporters play in disease.
De novo synthesis of purine and pyrimidine nucleosides is energetically costly; consequently, salvage and recycling of preformed nucleosides and nucleobases offer an alternative pathway for cells to accommodate their requirements for these metabolites. The Solute Carrier 29 (SLC29)2 family (2), referred to as “equilibrative nucleoside transporters” (ENTs), promotes the facilitated diffusion of nucleosides and nucleobases across biological membranes, although some protozoan members can couple substrate translocation to proton symport to mediate concentrative transport (4, 5). Purine nucleoside and nucleobase transporters are of particular importance in parasitic protozoa such as Leishmania, Trypanosoma, and Plasmodium, as none of these organisms is able to synthesize purines de novo and they are completely reliant upon uptake of preformed purines from their hosts via SLC29 permeases (6).
SLC29 family members from mammals and protozoa have been analyzed extensively by site-directed mutagenesis to define amino acids or structural components that are critical for transport function (7–23). However, there is no high resolution structure available for any SLC29 protein, and the absence of these permeases among archaea and bacteria precludes the use of these organisms for generation of purified protein for x-ray crystallography. In the absence of direct structural data, several groups have employed computational modeling to arrive at predicted structures for several SLC29 permeases from different parasitic protozoa (11, 24–26). Each of these models predicts that the relevant protein folds into a structure similar to that determined for bacterial major facilitator superfamily proteins such as the Escherichia coli lactose permease (LacY) (27) or glycerol phosphate transporter (GlpT) (28). Our recent ab initio computational studies (24) on the LdNT1.1 adenosine/pyrimidine nucleoside transporter from the parasitic protozoan Leishmania donovani captured this protein in a conformation that is “closed to the outside, open to the inside,” similar to the conformations observed in the crystal structures of LacY and glycerol phosphate transporter (GlpT). Analogous to these experimentally determined structures for bacterial permeases, the LdNT1.1 model predicted that transmembrane (TM) helices 1, 2, and 7 clustered together at the extracellular surface of the transporter to close off the permeation pathway and form an “extracellular gate.” The presence of such a gate is consistent with the commonly invoked “alternating access model” for membrane transport (29, 30) in which permeases alternate between one conformation in which an extracellular gate is closed and an intracellular gate is open (inward-open) and another conformation in which the extracellular gate is open and the intracellular gate is closed (outward-open). Thus, defining the gates of a permease is of fundamental importance for understanding its structure and function. For LdNT1.1, the model further identified that three aromatic residues located in TMs 1 (Phe-48), 2 (Trp-75), and 7 (Phe-346), might stack against each other to constitute a molecular clamp responsible for tethering these helices together when the extracellular gate is closed. Site-directed mutagenesis confirmed a critical role for Phe-48 and Trp-75 in transport (24), thus supporting the validity of this prediction.
Although computational models can provide substantial new insights into transporter structure and function, such as the proposed role for Phe-48 and Trp-75 in gating, they require stringent experimental validation to test their accuracy. To further test the validity of the LdNT1.1 ab initio model and in particular the postulated role of TMs 1, 2, and 7 in constituting the extracellular gate, we sought additional experimental evidence. Thus, in this study, we introduced cysteine pairs into TMs 1, 2, and 7 at locations near the extracellular surface of these helices that are predicted by the structural model to be in close proximity when the extracellular gate is closed. We subsequently employed disulfide cross-linking to determine whether the engineered cysteine residues come into close enough contact to be covalently linked to each other. This work accomplishes two objectives; 1) it experimentally tests the ab initio model and provides further evidence that the predicted topology of TM helices is essentially correct, and 2) it provides experimental evidence that TM helices 1, 2, and 7 constitute an extracellular gate that alternates between opened and closed conformations during the substrate transport cycle.
EXPERIMENTAL PROCEDURES
Chemicals, Materials, and Reagents
Restriction endonucleases and DNA-modifying enzymes were obtained from New England Biolabs, Inc., Roche Applied Science, or Invitrogen. Radiolabeled [2,8-3H]-adenosine (50 Ci/mmol) and [6,3-3H]glucose (25 Ci/mmol) were purchased from Moravek Biochemicals. Synthetic oligonucleotides were purchased from Invitrogen. Copper sulfate (CuSO4), 1,10-phenanthroline, N-ethylmaleimide, and Tris[2-carboxyethyl] phosphine hydrochloride (TCEP) were purchased from Sigma. 2-Sulfonatoethyl methanethiosulfonate sodium salt (MTSES) and 2-(trimethylammonium)ethylmethanethiosulfonate bromide (MTSET) were purchased from Biotium, Inc. (Hayward, CA). All other chemicals, materials, and reagents were of the highest grade commercially available.
Parasite Cell Cultures
L. donovani strains were propagated at 26 °C in RPMI medium (Invitrogen) containing 10% fetal bovine serum, 15 μg/ml hemin, and 100 μm xanthine. Null mutant Δldnt1/Δldnt2 (31) was supplemented with drugs against the integrated resistance markers (50 μg/ml hygromycin (Roche Applied Science) plus 50 μg/ml phleomycin (Research Products International)) as well as drugs that are cytotoxic to parasites expressing the wild type LdNT1.1 or LdNT2 transporters (1 μm tubercidin (Sigma) or 1 μm formycin B (Berry & Associates), respectively). Parasites transfected with pX63NeoRI constructs (described below) were selected and maintained in 100 μg/ml G418 (Invitrogen).
Site-directed Mutagenesis and Plasmid Constructs
Mutagenesis was performed using the QuikChange® II XL site-directed mutagenesis kit, a polymerase chain reaction-based mutagenesis strategy (Stratagene). Single or paired double cysteines were introduced within either the wild type or the cysteine-less (cys-less) LdNT1.1 open reading frame templates that had been ligated into the EcoRI site of the episomal expression vector pX63NeoRI (12). The codons used to introduce the point mutations were A61C (GCC → TGC), F74C (TTC → TGC), and G350C (GGC → TGC). For each single and paired double cysteine mutant, two independent clones were isolated in parallel, and the presence of the mutations was verified by DNA sequencing at the Oregon Health and Science University Microbiology Research Core Facility using a model 377 Applied Biosystems automated fluorescence sequencer (PerkinElmer Life Sciences). Wild type, cys-less, and mutant LdNT1.1 pX63NeoRI constructs were transfected into transport-defective Δldnt1/Δldnt2 L. donovani promastigotes using standard electroporation conditions (32, 33). Transfectants were selected and expanded in liquid medium containing 100 μg/ml of neomycin analog G418 (Invitrogen).
Transport Assays
L. donovani promastigotes expressing the wild type and the cys-less LdNT1.1 transporters and the different single and paired double cysteine mutants were harvested in early-middle logarithmic phase (0.5–1.5 × 107 cells/ml), collected by centrifugation at 2000 rpm (835 × g) for 10 min at room temperature (∼25 °C), washed 2 times in phosphate-buffered saline (PBS: 138 mm NaCl, 8.1 mm Na2HPO4·7H20, 2.7 mm KCl, 1.5 mm KH2PO4 (pH 7.4)), and resuspended in PBS to a final density of 2–3 × 108 parasites/ml. The adenosine transport measurements (1 μm, 2.5 μCi/ml, 40s) depicted in Figs. 2–5, performed by the previously described oil-stop method (34), are the mean and S.D. of 3 independent experiments (n = 3), and values within a given experiment were the mean of triplicate measurements. For each mutant, two independent clones were isolated and tested in parallel to confirm each result (data not shown).
FIGURE 2.

A, B, and C, inhibition by CuPh of Ado influx by LdNT1.1 paired double cysteine mutants. Pairs of cysteines were introduced into the WT or cys-less LdNT1.1 background at the indicated positions. The mutant transporters were expressed in the L. donovani Δldnt1Δldnt2 null cell line, and the cells were assayed for uptake (40 s) of 1 μm [3H]Ado (A, C) and 10 μm [3H]Ado (B) at the room temperature (∼25 °C) after oxidation with the indicated concentrations of CuPh (10 min). A and B, whole cell uptake was measured in the absence (black bars) and in the presence (white bars) of 10 μm CuPh. Transport values are reported as percentage of the WT activity in the absence of CuPh (% of untreated WT). Asterisks indicate values that are significantly different (**, p < 0.05; ***, p < 0.01) from the non-treated samples (black bars) as determined by the two-tailed unpaired Student's t test. C, transport values at each CuPh concentration are reported as the percentage of activity in the absence of CuPh (% of untreated). D, immunoblot of membrane fractions (5 μg protein) probed with the anti-LdNT1.1 antibody (top) and the anti-α-tubulin antibody (bottom) as a loading control. The numbers at the left represent the position of the molecular weight markers, designated in kilodaltons.
FIGURE 3.
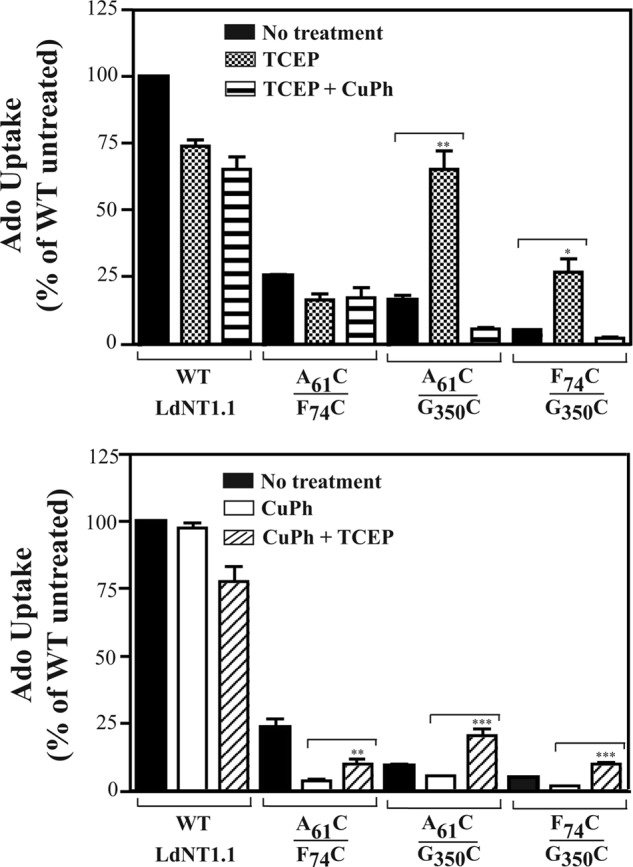
Effect of TCEP and CuPh on Ado influx by LdNT1.1 paired double cysteine mutants. Transport of [3H]Ado (1 μm, 40s) was measured at room temperature (∼25 °C) in Δldnt1Δldnt2 L. donovani parasites expressing the indicated transporter constructs after pretreatment for 30 min in the presence (crosshatched bars) and absence (black bars) of 5 mm TCEP followed by a 10-min incubation with 10 μm CuPh (horizontal striped bars, A) or after pretreatment for 10 min in the presence (white bars) and absence (black bars) of 10 μm CuPh followed by a 30-min incubation with 5 mm TCEP (diagonal striped bars; B). Asterisks indicate values that are significantly different (*, p < 0.05; **, p < 0.01; ***, p < 0.001) from the non-treated samples (black bars, A) or values that are significantly different (p < 0.01) from the samples treated with CuPh (white bars, B) as determined by the two-tailed unpaired Student's t test.
FIGURE 4.
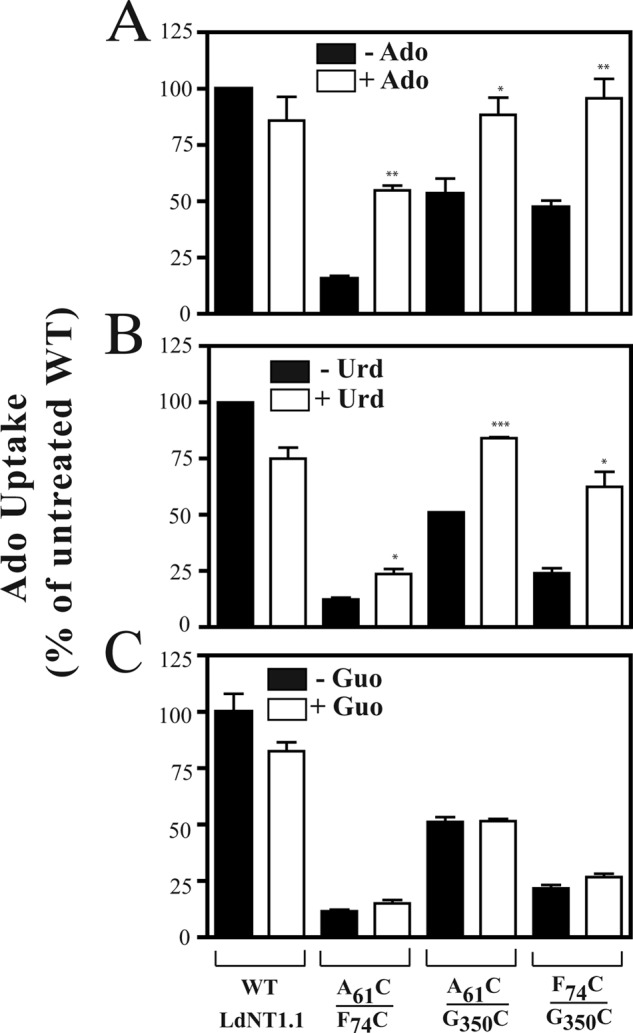
Effect of adding ligands during CuPh treatment on the transport activity of LdNT1.1 paired double cysteine mutants. L. donovani Δldnt1Δldnt2 parasites expressing the indicated transporter constructs were preincubated for 15 min in the presence (white bars) and absence (black bars) of 100 μm Ado (A), Urd (B), or Guo (C) followed by the addition of CuPh to a final concentration of 10 μm and incubation for another 10 min. Subsequently, transport of [3H]Ado (1 μm, 40s) was measured at room temperature (∼25 °C). Asterisks indicate values that are significantly different (*, p < 0.05; **, p < 0.01; ***, p < 0.001) from the samples with no ligand (black bars) as determined by the two-tailed unpaired Student's t test.
FIGURE 5.
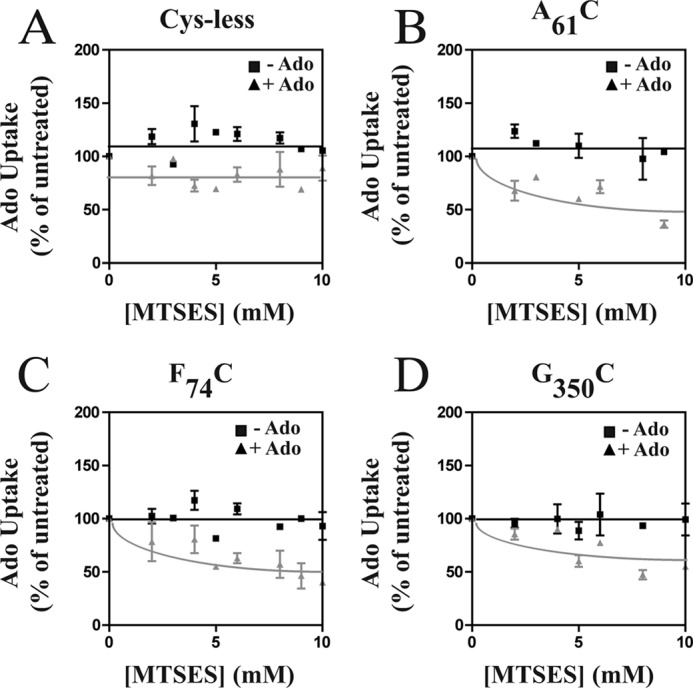
Sensitivity of A61CTM1, F74CTM2, and G350CTM7 cys-less LdNT1.1 mutant permeases to MTSES inactivation in the presence or absence of 1 mm Ado. L. donovani Δldnt1Δldnt2 parasites individually expressing A61CTM1, F74CTM2, and G350CTM7 in the cys-less LdNT1.1 background were treated for 10 min with the indicated concentrations of MTSES in the presence (triangles) or absence (squares) of 1 mm adenosine. [3H]Ado transport (1 μm, 40 s) was measured at room temperature (∼25 °C) after washing the cells free of MTSES and adenosine.
Preparation of Plasma Membranes
Transfected L. donovani parasites in early-middle logarithmic phase (0.5–1.5 × 107 cells/ml, 100 ml) were collected by centrifugation at 2000 rpm (835 × g) for 10 min at room temperature and washed 2 times with PBS. Plasma membranes were prepared as described previously (35), and the pellets were dissolved in 50–100 μl of resuspension buffer (20 mm Tris-HCl, 100 mm NaCl, 2 mm CaCl2, and 2% SDS (pH 7.4)) supplemented with protease inhibitor mixture (Complete, Hoffmann-La Roche) and were stored at −80 °C as aliquots for further use. Protein quantification was determined using the Bio-Rad DC Protein Assay kit (Bio-Rad). Membrane preparations used for cross-linking were diluted in the aforementioned buffer at a protein concentration of 1–2 mg/ml.
In Vivo and in Vitro Disulfide Cross-linking
Both in vivo (transfected parasites) and in vitro (plasma membranes) disulfide cross-linking reactions were performed at room temperature (∼25 °C). To induce the formation of disulfide bonds, transfected parasites collected by centrifugation and resuspended in PBS (500 μl, 2–3 × 108 cells/ml) and/or membrane preparations (5–20 μg of protein; 1–2 mg/ml) were incubated with the Cu(II)-(1,10 phenanthroline)3 oxidant catalyst (CuPh) before transport measurements or SDS-PAGE, respectively. The CuPh stock solution was freshly prepared for each experiment by mixing 4 μl of 1.25 m 1,10-phenanthroline in H2O:EtOH (1:1) with 6 μl of 250 mm CuSO4. CuPh working solutions were prepared then by diluting the stock solution with PBS to the desired concentration. CuPh cross-linking efficiency was found to be optimal at 10 μm/10 min for the experiments in vivo (Fig. 2C) and 20–50 μm/15 min for the oxidation in vitro.
For preparations of cells to be subsequently tested for transport function, disulfide formation was stopped by the addition of 10 mm EDTA, and parasites were washed 3 times with PBS and resuspended in 500 μl of PBS before measuring transport. For monitoring disulfide formation by immunoblotting, cross-linked membranes were diluted into 4× NuPAGE® LDS sample buffer (Invitrogen) supplemented with 10 mm EDTA and analyzed by SDS/PAGE as described below.
For studies of substrate-induced conformational changes, L. donovani parasites or cell membrane preparations were preincubated with the indicated concentration of ligand (100 μm adenosine, uridine, or guanosine) at room temperature (∼25 °C) for 15 min followed by oxidation with CuPh as described above.
In Vivo Reduction of Spontaneously Formed Disulfide Bonds with TCEP
Transfected L. donovani parasites, collected by centrifugation and resuspended in PBS (500 μl, 2–3 × 108 cells/ml), were incubated with the reducing agent TCEP at room temperature (∼25 °C) before transport measurements. TCEP efficiency (preserving cell viability) was found to be optimal at 5 mm/30 min. Parasites were then washed 3 times with PBS and resuspended in 500 μl of PBS before measuring transport.
In Vivo Methanethiosulfonate (MTS) Modification
Standard MTS treatments were performed for substituted single cysteine mutants within the cys-less LdNT1.1 template and the cys-less LdNT1.1 permease that served as negative control. L. donovani promastigotes were collected by centrifugation, washed 3 times with cold PBS, and resuspended in the same buffer (2–3 × 108 parasites/ml). Aliquots of 100 μl (per triplicate) were incubated for 10 min with the reported concentrations of MTSES (0–15 mm) and MTSET (0–1 mm) in PBS buffer at room temperature (∼25 °C). To assess the effect of LdNT1.1 substrates on MTS modification, aliquots of 100 μl (per triplicate) were incubated in parallel with adenosine (100 μm–1 mm final concentration) for 15 min before adding the MTS reagent. MTS reactions conducted in the presence or absence of ligand were terminated by diluting the parasites (10-fold) in ice-cold PBS. Subsequent washes and uptake assays were performed as described above. Control samples received vehicle (PBS) only. Additional controls were performed by incubating the samples with adenosine without subsequent exposure to the MTS derivatives. For each individual mutant and for the cys-less control, the extent of the MTS reaction was quantified by comparison of the uptake in MTS-treated and non-treated samples. These values were expressed as percentages of residual activity after MTS modification in the presence or absence of the ligand adenosine.
Immunoblot Analysis
To determine the mobility of cross-linked LdNT1.1 mutants, 5–20 μg of cell membranes were mixed with the corresponding volume of 4× NuPAGE®LDS sample buffer supplemented with 10 mm EDTA in the absence (non-reducing) or presence (reducing) of DTT or TCEP (5 mm) and heated at 70 °C for 10 min. Samples were resolved by electrophoresis on 10% gradient NuPAGE® Novex Bis-Tris gels (Invitrogen) without NuPAGE® reducing agent using the XCell SureLock™ Mini-Cell system (Invitrogen). Subsequently, proteins were electrotransferred under denaturing conditions onto nitrocellulose membranes (Protran, Whatman, GmbH, Germany) using the XCell II™ Blot Module (Invitrogen). Nitrocellulose membranes were then blocked with 5% fat-free milk in PBS (pH 7.4) containing 0.2% Tween 20 (PBS-T) (overnight at 4 °C). After a wash with PBS-T (5 min, room temperature), membranes were incubated with rabbit polyclonal anti Leishmania mexicana NT1-loop VII (Gln236-Lys331) antibody as previously detailed (36) (dilution 1:3,000 in PBS-T with 5% fat-free milk) for 1 h at room temperature. The blots were then washed 3 times with PBS-T (10 min/wash) and incubated for 1 h at room temperature with anti-rabbit IgG antibody conjugated to horseradish peroxidase (dilution 1:10,000 in PBS-T with 5% fat-free milk). After one 10-min wash with PBS-T and two 10-min washes with PBS, proteins were visualized by using ECL detection reagents (Thermo Scientific) and exposure to scientific imaging film (Kodak BioMax MR film). When indicated, membranes were also probed with a mouse antibody against α-tubulin (Oncogene monoclonal Ab-1) at a 1:500 dilution in PBS-T with 5% fat-free milk and then with goat anti-mouse IgG conjugated to horseradish peroxidase (dilution 1:10,000 in PBS-T with 5% fat-free milk) as a control to normalize for protein loading.
RESULTS
Construction and Functional Analysis of Paired Double Cysteine Mutants
In a previous study we arrived at a structural model for the L. donovani LdNT1.1 permease that predicted that TM helices 1, 2, and 7 cluster together at the extracellular face of the transporter and close off the permeation pathway (24). To further validate this model and to investigate dynamic interactions between the extracellular halves of these helices, paired double cysteine mutations (A61CTM1/F74CTM2, A61CTM1/G350CTM7, and F74CTM2/G350CTM7) at the interface between TMs 1, 2, and 7 (Fig. 1, left) were introduced into the wild type LdNT1.1 background, as described under “Experimental Procedures.” According to the model, the distance between the Cαs of Ala-61TM1/Phe-74TM2, Ala-61TM1/Gly-350TM7, and Phe-74TM2/Gly-350TM7 is predicted to be 12, 7, and 8Å, respectively.
FIGURE 1.

Ab initio computational model of LdNT1.1. Left, indicated are residues that were mutated to cysteines. Tan cylinders represent predicted TM helices and are numbered 1–11. Residues at the extracellular termini of helices 1, 2, and 7 that were mutated to cysteines are indicated by space filling models: pink is Ala-61TM1, green is Phe-74TM2, and red is Gly-350TM7. The view is from the extracellular surface toward the interior, indicating that the ab initio model predicted an inward-open conformation. The figure was generated using PyMol. A suggestive model for the outward-open conformation (right) is given by rotating the N-terminal domain (helices 1–6) and the C-terminal domain (helices 7–11) 38° around an axis (red line) parallel to the lipid bilayer, as explained in “Results.”
The mutant permeases were then expressed in the Δldnt1Δldnt2 L. donovani double null mutant that is genetically deficient in the LdNT1.1, LdNT1.2, and LdNT2 genes and consequently provides a null background for transport of nucleosides (31). The ability of each transgenic mutant permease to mediate ligand translocation was evaluated by uptake assays using 1 μm [3H]adenosine, a natural substrate of LdNT1.1. As shown in Figs. 2, A and B, and 3, A and B, black bars, all three paired double cysteine mutants exhibited low albeit measurable transport activity (∼2–25% of the adenosine accumulated by the wild type transporter, depending on the experiment and on the mutant), and therefore, the proximity relationships between the selected cysteine residues and by extension between the TMs to which they belong were further analyzed by adenosine uptake after treatment with the oxidative catalyst CuPh. The immunoblot in Fig. 2D demonstrates that a principal reason for reduced transport activity of each mutant is the decreased level of LdNT1.1 protein expressed compared with wild type permease, although innate differences in transport efficiency may also contribute to decreased transport activity.
Effect of in Vivo Disulfide Bond Formation on LdNT1.1 Activity
Characterization of disulfide formation in vivo has the advantage that the protein of interest can be probed in its native cellular environment under physiological conditions. CuPh is a weak oxidizing agent known to catalyze the oxidation of adjacent thiol groups. As shown in Fig. 2, A–C, preincubation with CuPh (10 μm, 10 min) had no effect on the wild type LdNT1.1 activity, but it strongly inhibited adenosine influx (∼10-fold) in Δldnt1Δldnt2 L. donovani parasites transfected with the A61CTM1/F74CTM2 double mutant construct and also resulted in the modest impairment of paired double replacements F74CTM2/G350CTM7 (∼4-fold) and A61CTM1/G350CTM7 (∼2-fold). Transport sensitivity to inactivation by CuPh may be interpreted as evidence for cross-linking the two sulfhydryl groups, presumably because the disulfide bond imposes restrictions on the protein conformational changes necessary for transport (37–39). This result implies that the relevant cysteines and their associated helices 1, 2, and 7 are indeed in close proximity, as predicted by the computational model (24). Glucose transport was not affected under the experimental conditions reported (data not shown), thus confirming that the observed impairment in adenosine uptake is specific to the mutant LdNT1.1 proteins and not due to some unrelated modification that the mutant cell lines may have undergone upon treatment with CuPh. Furthermore, uptake assays performed at 10 μm adenosine (Ado), ∼10 times the Km value (34), showed very similar results for CuPh-induced inhibition of transport (Fig. 2B). This result indicates that cysteine cross-linking reduces the Vmax of the permease and does not simply increase the Km for Ado.
Oxidation with CuPh also weakly inhibited single cysteine mutants F74CTM2 (∼1.5-fold) and G350CTM7 (∼3-fold) (data not shown), raising the possibility that a partial cross-linking of these cysteine residues to an endogenous cysteine (Cys-36TM1, Cys-143TM4, Cys-144TM4, Cys-260loopTM6–7, or Cys-377TM7) might have occurred. To prove that each one of the engineered paired cysteines in A61CTM1/F74CTM2, A61CTM1/G350CTM7, and F74CTM2/G350CTM7 cross-link to each other, identical cysteine replacements were re-introduced into the cys-less LdNT1.1 background (a fully functional LdNT1.1 mutant protein in which all five native cysteine residues were substituted with alanine) (12). Cys-less F74CTM2/G350CTM7 led to an inactive permease and, therefore, was excluded from the study. On the other hand, cys-less A61CTM1/F74CTM2 (∼20% of residual activity versus the cys-less LdNT1.1) and cys-less A61CTM1/G350CTM7 (∼5% of residual activity versus the cys-less LdNT1.1) resulted in substantial inhibition of transport activity by incubation with CuPh (∼6- and ∼2-fold, respectively; Fig. 2A, inset). The observed changes indicate that the cross-linking and CuPh sensitivity reported in Fig. 2, A and B, are likely specific for the introduced cysteines of these two mutants.
TCEP Reduction Experiments Further Support Close Approach of TM7 to TMs 1 and 2
The low and variable basal levels of catalyzed transport, particularly exhibited by paired double cysteine mutants A61CTM1/G350CTM7 and F74CTM2/G350CTM7 (Fig. 2, A and B, and Fig. 3, A and B, black bars) as well as their modest sensitivity to inhibition by CuPh (Fig. 2, A–C) suggested that these cysteine pairs might be cross-linked before application of the oxidizing agent. To further investigate this possibility, we attempted to reverse any spontaneous disulfide bridges by pretreatment with TCEP, a sulfhydryl reducing compound that is used as a substitute for dithiothreitol (DTT) to specifically reduce disulfide bonds of proteins and peptides over a wider range of pH values (pH 2–9) (40).
Preincubation with TCEP (5 mm, 30 min) stimulated A61CTM1/G350CTM7 and F74CTM2/G350CTM7 adenosine uptake by an average of ∼5-fold (4.8 + 1.06, n = 5) (Fig. 3A, crosshatched bars), indicating that reduction of the spontaneously formed disulfide bonds in these mutants releases the permease from an inhibited state. Moreover, upon prior activation by TCEP, the enhanced activity of the newly reduced transporters was fully decreased to basal levels by oxidation with CuPh (Fig. 3A, horizontal striped bars), supporting the notion that the relevant cysteine pairs were already partially cross-linked before treatment with the reducing agent. Notably, glucose uptake remained unaffected upon TCEP reduction (data not shown), and the effects of TCEP on adenosine transport were only observed for the specified paired double cysteine mutants and not for the equivalent single cysteine mutants (data not shown), indicating that the results reported are indeed specific and that G350CTM7 must lie in close proximity to A61CTM1 and F74CTM2 when the permease is in the outward-closed conformation.
In addition, oxidation of each double cysteine mutant with CuPh followed by reduction with TCEP (Fig. 3B, diagonal striped bars) 1) substantially restored uptake activity lost upon oxidation alone (A61CTM1/F74CTM2) or 2) increased Ado uptake above the level of untreated transporter (A61CTM1/G350CTM7 and F74CTM2/G350CTM7). These results indicate that loss of transport upon cysteine cross-linking is reversible upon reduction of cross-linked residues and further support the notion that the A61C/G350C and F74C/G350C cysteine pairs are partially cross-linked in the untreated state and thus exhibit enhanced uptake upon reduction with TCEP.
Cysteine Cross-linking Can Be Modulated by Substrates
The alternating access model (29, 30) and several experimental studies suggest that LacY and related transmembrane permeases exist in conformations that alternately expose the binding site either to the extracellular or the cytoplasmic side of the membrane (27, 41–43). Transporter ligands might, therefore, influence cross-linking by altering the conformation of the permease so that the outer gate, constituted by TM helices 1, 2, and 7 in LdNT1.1, is opened a greater fraction of time. To obtain a suggestive model for the outward-open conformation of LdNT1.1, we relied upon its predicted topological similarity (24) to LacY and related permeases such as the fucose transporter FucP. Comparisons of the crystal structures of FucP in the outward-open conformation (44) and LacY in the inward-open conformation (27) suggest that both transporters convert between these alternate conformations by a ∼38° rigid body rotation of their N- and C-terminal domains about an axis parallel to the membrane bilayer. Fig. 1 shows such a rotation applied to the LdNT1.1 inward-open computational model (left) to obtain an approximate model for the outward-open conformation (right). One important consequence of this suggested conformational change is that the three residues highlighted in Fig. 1 move much farther apart in the outward-open structure, predicting that cross-linking between the substituted cysteine pairs would be impaired by ligand-induced conformational alterations.
To determine the effect of ligands for LdNT1.1 on cysteine cross-linking, we examined whether incubation of the paired double cysteine mutants A61CTM1/F74CTM2, A61CTM1/G350CTM7, and F74CTM2/G350CTM7 with adenosine and/or uridine, two of the ligands for LdNT1.1, resulted in an altered degree of disulfide cross-linking. Fig. 4, A and B, illustrate that when the medium was supplemented with 100 μm Ado or uridine (Urd), respectively, the inhibition by CuPh was significantly impaired. This result is consistent with a conformational change associated with transport that opens the outer gate formed by helices 1, 2, and 7 and moves the relevant cysteine residues apart during a portion of the transport cycle. The addition of 100 μm guanosine (Guo), a ligand for LdNT2 (45) but not for LdNT1.1, did not significantly change the extent of inhibition by CuPh (Fig. 4C), as expected if the observed reduction in cysteine cross-linking is due to conformational changes that occur as a result of alternating access for substrates during the transport cycle.
However, an alternate explanation for the ability of adenosine and uridine to inhibit cross-linking of cysteine pairs could be that these ligands simply reduce accessibility of these residues to CuPh either by directly blocking the cysteines or by inducing a conformational change that occludes the residues. To rule out this possibility, we first introduced the single cysteine mutants A61CTM1, F74CTM2, and G350CTM7 into the cys-less LdNT1.1 permease and then measured their reactivity with the sulfhydryl modifying reagents MTSES (negatively charged) and MTSET (positively charged) in the presence and absence of adenosine. For all three introduced cysteine residues, the addition of adenosine increased the reactivity of the sulfhydryl with MTSES (measured as a reduction in uptake of [3H]adenosine after reaction with MTSES; gray lines in Fig. 5, B, C, and D, compared with black lines). Analogous experiments with MTSET showed no effect of adenosine upon reactivity of F74CTM2 or G350CTM7 and increased reactivity of A61CTM1 (data not shown). Hence, rather than blocking accessibility of each cysteine residue with sulfhydryl-reactive reagents, adenosine either had no effect or it increased reactivity. These results rule out the possibility that Ado decreases cysteine cross-linking simply by reducing accessibility of cysteine residues to reactants. Indeed the increased reactivity of each substituted cysteine residue with MTSES supports the prediction that helices 1, 2, and 7 move apart upon binding of adenosine (Fig. 1) to make these residues more accessible to reactants. Hence, the substrates for LdNT1.1 do likely inhibit cross-linking of the introduced cysteine pairs by inducing a conformational change that moves the cysteine residues apart from one another. Similar cysteine reactivity experiments have been employed by others to confirm that substrates or substrate analogs decrease cross-linking of cysteine residues by inducing conformational separation between relevant helices in serotonin (46) and glutamate (47, 48) transporters.
Cross-linking and Mobility Shift of Mutant Transporters with Substituted Cysteine Pairs
Plasma membrane preparations from transfected Δldnt1Δldnt2 parasites expressing paired double cysteine mutant transporters were subjected to cross-linking, SDS-polyacrylamide gel electrophoresis (SDS-PAGE), and immunoblot analysis as described under “Experimental Procedures.” Disulfide bond formation was assessed as a change in mobility on SDS-PAGE under non-reducing conditions. This method of monitoring cross-linking has been employed for other transporters (49–51) based on the premise that an intramolecular disulfide bond will prevent complete unfolding of the protein under denaturing conditions, thus affecting protein mobility.
As anticipated, CuPh did not induce a shift in mobility on SDS-PAGE for the wild type LdNT1.1 transporter (∼45 kDa). However, significantly decreased mobility was observed for all three paired double cysteine mutants upon oxidation with CuPh (20 μm, 15 min) under non-reducing conditions (Fig. 6A, lane 2). CuPh treatment of A61CTM1/G350CTM7 and F74CTM2/G350CTM7 resulted in a single band of decreased mobility (∼50 kDa versus ∼45 kDa), whereas oxidation of A61CTM1/F74CTM2 consistently yielded several bands (∼45–55 kDa versus ∼45 kDa). We do not know the origin of the multiple shifted bands. A background band of ∼55 kDa is present before (lane 1) and in many cases after oxidation (lanes 2–4), but it does not appear to result from cross-linking as it is present with and without either TCEP or CuPh. Furthermore, the bands of decreased mobility cannot be explained by cross-linking of oligomeric LdNT1.1. Cross-linking of membrane fractions with 1% formaldehyde caused a shift of the LdNT1.1 band from ∼45 to ∼70 kDa (data not shown), suggesting that the permease probably exists as a dimer in the membrane, but this dimer migrates at a position considerably above that of the ∼50-kDa bands generated by CuPh cross-linking.
FIGURE 6.
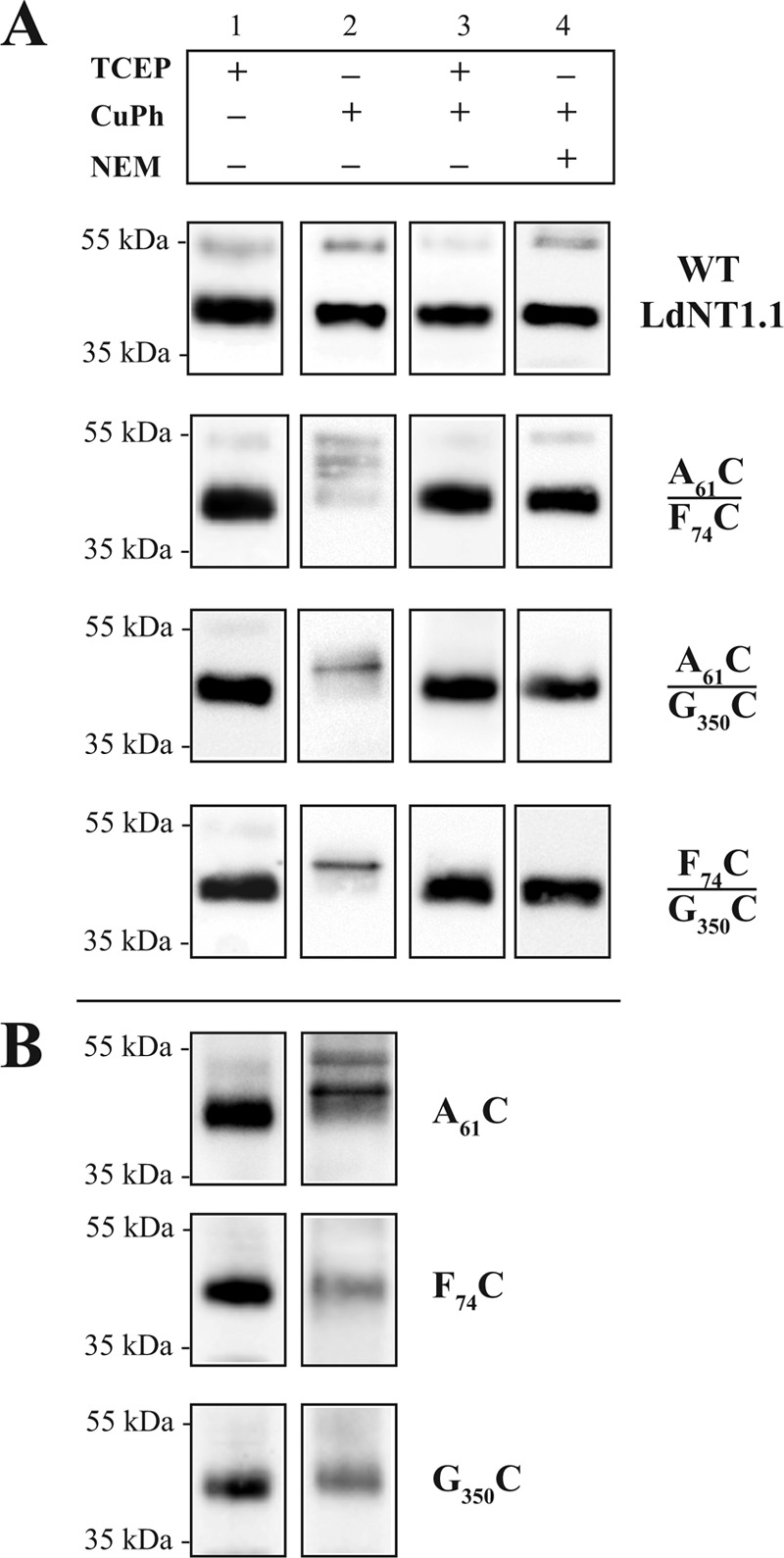
Cross-linking of LdNT1.1 double-paired cysteine mutants (A) and LdNT1.1 single cysteine mutants monitored by mobility shifts (B). Crude membrane preparations from transfected Δldnt1Δldnt2 L. donovani parasites were oxidized (lane 2) with 20 μm CuPh for 15 min at room temperature (∼25 °C) or reduced (lane 1) by the addition of 5 mm TCEP to the sample loading buffer. Subsequently, the membranes were subjected to SDS-PAGE as described under “Experimental Procedures.” Due to aggregation upon oxidation, samples in lane 2 contain three times the protein loaded onto the rest of the lanes. For comparison, different amounts of each mutant were loaded to match the level of expression equivalent to 5 μg of the WT permease. LdNT1.1 bands were detected by immunoblotting using an anti-LdNT1.1 antibody and chemiluminescence. A, double-paired cysteine mutant membranes in lane 2 show mobility shifts after cross-linking with CuPh that were reversed upon the addition of TCEP (lane 3). Lane 4 shows the effect of masking the SH groups by N-ethylmaleimide before oxidation with CuPh. B, effect of CuPh oxidation of single Cys mutants in an otherwise wild type transporter.
When CuPh-oxidized membranes of paired double cysteine mutants were incubated with either 5 mm DTT or TCEP before SDS-PAGE, all mutants exhibited the single ∼45-kDa band observed for the wild type permease in untreated or in CuPh-treated samples, indicating that the disulfide bonds could be readily reduced (Fig. 6A, lane 3). Furthermore, when the sulfhydryl groups were masked by preincubation with N-ethylmaleimide (Fig. 6A, lane 4), no intramolecular cross-linking was observed. We, therefore, conclude that the ∼50–55-kDa bands observed under oxidizing conditions are due to intramolecular cross-links, indicating that positions Ala-61TM1, Phe-74TM2, and Gly-350TM7 are spatially close to each other in the inward-open conformation.
To rule out the possibility that cross-links between the introduced Cys residues and endogenous Cys residues in LdNT1.1 could explain the observed loss of activity upon cross-linking (Fig. 2), we also examined each single Cys mutant before and after cross-linking (Fig. 6B). F74C and G350C did not exhibit any upward shift in mobility, suggesting that neither of these introduced Cys residues cross-links to an endogenous Cys. In contrast, the A61C mutant did reveal upward shifts that could be due to such cross-linking. Nonetheless, the activity measurements depicted in the inset to Fig. 2A, which were done with the A61C/F74C and A61C/G530C mutants in the cys-less background, demonstrate that the loss of activity upon CuPh-induced cross-linking cannot be due to such endogenous cross-links, which cannot occur in the cys-less background. For the F74C/G350C mutant, which was not stable in the cys-less background, the absence of cross-links for the two individually introduced Cys residues also argues against the loss of activity being due to cross-linking with endogenous cysteines.
We also tested whether the presence or absence of adenosine and/or uridine during cross-linking reduced the level the slower migrating bands that result from cross-linking. Although substrate-induced differences were evident when analyzing uptake (Fig. 4, +/− Ado or Urd), no clear decreases in cross-linking were observed by immunoblot analysis (data not shown). This result could reflect the greater quantitative sensitivity of uptake measurements compared with quantification of band intensities on immunoblots. Studies on other transporters (46) have also noted that ligand effects on cross-linking can be more accurately monitored by measuring inhibition of transport rather than cysteine cross-linking per se. In addition, examination of Cys cross-linking in transporters by Western blotting of isolated membranes may induce reversal of a substantial proportion of the cross-links originally introduced into intact cells (52). This discrepancy between the two distinct experimental formats, whole cells and isolated membranes, can thus result in quantitatively different observations when band shifts on Western blots are compared with uptake measurements employing intact cells.
DISCUSSION
Structure determination of eukaryotic integral membrane proteins by high resolution methods such as x-ray crystallography (53) and nuclear magnetic resonance (NMR) (54) is a challenging process. To date, none of the ENTs, a unique family of membrane proteins (the SLC29 family) that promote the uptake of nucleosides and/or nucleobases into a wide range of eukaryotes (2, 55, 56), has been crystallized. In previous work we proposed a structural model for the LdNT1.1 nucleoside permease from the parasitic protozoan L. donovani, a member of the SLC29 family, using ab initio computation (24). To test the hypothesis that helices 1, 2, and 7 of LdNT1.1 are firmly packed in the outward-closed conformation, three sets of cysteine pairs were placed on the predicted extracellular interfaces of these helices (A61CTM1/F74CTM2, A61CTM1/G350CTM7, and F74CTM2/G350CTM7) (Fig. 1), and each cysteine pair was subjected to oxidative cross-linking. Site-directed oxidative cross-linking is a powerful approach for estimating helix-packing and probing ligand-induced conformational changes, as cross-linking reactions can be performed before and after interaction with substrate.
As reported in the Results section, all three sets of paired cysteines showed impaired adenosine transport function upon exposure to the oxidation catalyst CuPh (Fig. 2, A and B). Two of the cysteine pairs, A61CTM1/G350CTM7 and F74CTM2/G350CTM7, appear to cross-link spontaneously, thus reducing their innate transport activity. The A61CTM1/F74CTM2 cysteine pair was not spontaneously cross-linked, but it could be covalently linked by treatment with CuPh (Fig. 2, A and B). The average separation between the Cαs of disulfide-bonded cysteine residues in proteins is 5.6 Å (57, 58), and they cannot be farther apart than 7 Å (59). The computational model predicts Cα distances between 7 and 12 Å for each of the residues that were converted to cysteine pairs, and the largest distance is predicted for the cysteine pair, A61CTM1/F74CTM2, that does not cross-link spontaneously. Given the potential flexibility of the protein and the fact that the model represents an approximation and not a high resolution structure, the proximity predictions of the model are in good accord with the experimentally observed ability to cross-link the three cysteine pairs. These results confirm that TM helices 1, 2, and 7 come into close proximity and thus provide significant experimental evidence that the computational model of LdNT1.1 does predict the correct distance constraints and disposition of helices in the membrane.
If helices 1, 2, and 7 constitute a gate that opens and closes during the transport cycle, one would predict that this cluster of helices would open and close in the presence of extracellular substrate. This substrate-induced conformational change would be expected to decrease the extent of cross-linking between the helices, as it would likely move the cysteine pairs too far apart to be cross-linked when the permease is in the outward-open confirmation. Supporting this prediction, when the three mutant permeases were incubated with CuPh in the presence of the ligands adenosine or uridine, a reduced level of inhibition of transport was observed in subsequent adenosine uptake assays (Fig. 4, A and B). These results are consistent with previous studies on the model permease Lac Y, where it has been shown by multiple independent biochemical and biophysical methods (42) that the opening probability of the periplasmic cavity depends on the presence of substrate. Thus, in the absence of a galactopyranoside, the transporter is primarily in the inward-open conformation, with a large cavity open to the cytoplasm and a tightly sealed periplasmic side. However, binding of substrate causes the closing of the inward-open cavity with opening of a complementary outward-open cavity (41). The effects of substrates on helix cross-linking in LdNT1.1 further support the notion that TM helices 1, 2, and 7 constitute the extracellular gate and, by extension, that the previously demonstrated importance of Phe-48TM1 and Trp-75TM2 in LdNT1.1 activity is due to their role in tethering together helices that are part of the gate.
Given the similarities in sequence and predicted folding between various members of the SLC29 family, the experimental confirmation offered here for the predicted structure and gating of LdNT1.1 is likely to be relevant for orthologs of this permease throughout the eukaryotic kingdom. Thus we predict that interactions between helices 1, 2, and 7 are likely to gate mammalian SLC29 permeases such as the human ENTs. Furthermore, in future investigations similar mutagenesis and cross-linking approaches may allow a definition of the structural components that constitute the intracellular gate of LdNT1.1, identifying at the molecular level two central structural components of the permease that mediate transport function.
The absence of a crystal structure for any SLC29 permease has represented an obstacle for understanding how these physiologically and pharmacologically relevant transporters function. This work illustrates that the computational model for LdNT1.1 can help to fill this void by providing experimentally testable structural predictions. These predictions, such as the identification of the extracellular gate, illuminate the function of this nucleoside transporter and by extension of the whole SCL29 family of permeases.
Acknowledgments
We thank our colleague Marco Sanchez for advice and extensive discussions concerning the study presented herein. We also thank Johannes Elferich for generating the rotational model shown in Fig. 1.
This work was supported, in whole or in part, by National Institutes of Health Grant AI44138 (to S. M. L.). This work was also supported by National Science Foundation Grant 0746589 (to U. S.).
- SLC
- solute carrier family
- ENT
- equilibrative nucleoside transporter
- LdNT
- L. donovani nucleoside transporter
- TM
- transmembrane
- LacY
- lactose permease
- TCEP
- Tris[2-carboxyethyl] phosphine hydrochloride
- CuPh
- Cu(II)-(1,10 phenanthroline)3
- MTS
- methanethiosulfonate
- MTSES
- 2-sulfonatoethyl-methanethiosulfonate sodium salt
- MTSET
- 2-(trimethylammonium) methanethiosulfonate bromide
- cys-less
- cysteine-less
- Bis-Tris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol.
REFERENCES
- 1. Rose J. B., Coe I. R. (2008) Physiology of nucleoside transporters. Back to the future…. Physiology 23, 41–48 [DOI] [PubMed] [Google Scholar]
- 2. Kong W., Engel K., Wang J. (2004) Mammalian nucleoside transporters. Curr. Drug Metab. 5, 63–84 [DOI] [PubMed] [Google Scholar]
- 3. King A. E., Ackley M. A., Cass C. E., Young J. D., Baldwin S. A. (2006) Nucleoside transporters. From scavengers to novel therapeutic targets. Trends Pharmacol. Sci. 27, 416–425 [DOI] [PubMed] [Google Scholar]
- 4. Stein A., Vaseduvan G., Carter N. S., Ullman B., Landfear S. M., Kavanaugh M. P. (2003) Equilibrative nucleoside transporter family members from Leishmania donovani are electrogenic proton symporters. J. Biol. Chem. 278, 35127–35134 [DOI] [PubMed] [Google Scholar]
- 5. Ortiz D., Sanchez M. A., Koch H. P., Larsson H. P., Landfear S. M. (2009) An acid-activated nucleobase transporter from Leishmania major. J. Biol. Chem. 284, 16164–16169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hammond D. J., Gutteridge W. E. (1984) Purine and pyrimidine metabolism in the Trypanosomatidae. Mol. Biochem. Parasitol. 13, 243–261 [DOI] [PubMed] [Google Scholar]
- 7. Visser F., Sun L., Damaraju V., Tackaberry T., Peng Y., Robins M. J., Baldwin S. A., Young J. D., Cass C. E. (2007) Residues 334 and 338 in transmembrane segment 8 of human equilibrative nucleoside transporter 1 are important determinants of inhibitor sensitivity, protein folding, and catalytic turnover. J. Biol. Chem. 282, 14148–14157 [DOI] [PubMed] [Google Scholar]
- 8. Valdés R., Liu W., Ullman B., Landfear S. M. (2006) Comprehensive examination of charged intramembrane residues in a nucleoside transporter. J. Biol. Chem. 281, 22647–22655 [DOI] [PubMed] [Google Scholar]
- 9. Visser F., Zhang J., Raborn R. T., Baldwin S. A., Young J. D., Cass C. E. (2005) Residue 33 of human equilibrative nucleoside transporter 2 is a functionally important component of both the dipyridamole and nucleoside binding sites. Mol. Pharmacol. 67, 1291–1298 [DOI] [PubMed] [Google Scholar]
- 10. Visser F., Baldwin S. A., Isaac R. E., Young J. D., Cass C. E. (2005) Identification and mutational analysis of amino acid residues involved in dipyridamole interactions with human and Caenorhabditis elegans equilibrative nucleoside transporters. J. Biol. Chem. 280, 11025–11034 [DOI] [PubMed] [Google Scholar]
- 11. Arastu-Kapur S., Arendt C. S., Purnat T., Carter N. S., Ullman B. (2005) Second-site suppression of a nonfunctional mutation within the Leishmania donovani inosine-guanosine transporter. J. Biol. Chem. 280, 2213–2219 [DOI] [PubMed] [Google Scholar]
- 12. Valdés R., Vasudevan G., Conklin D., Landfear S. M. (2004) Transmembrane domain 5 of the LdNT1.1 nucleoside transporter is an amphipathic helix that forms part of the nucleoside translocation pathway. Biochemistry 43, 6793–6802 [DOI] [PubMed] [Google Scholar]
- 13. SenGupta D. J., Unadkat J. D. (2004) Glycine 154 of the equilibrative nucleoside transporter, hENT1, is important for nucleoside transport and for conferring sensitivity to the inhibitors nitrobenzylthioinosine, dipyridamole, and dilazep. Biochem. Pharmacol. 67, 453–458 [DOI] [PubMed] [Google Scholar]
- 14. Arastu-Kapur S., Ford E., Ullman B., Carter N. S. (2003) Functional analysis of an inosine-guanosine transporter from Leishmania donovani. The role of conserved residues, aspartate 389 and arginine 393. J. Biol. Chem. 278, 33327–33333 [DOI] [PubMed] [Google Scholar]
- 15. SenGupta D. J., Lum P. Y., Lai Y., Shubochkina E., Bakken A. H., Schneider G., Unadkat J. D. (2002) A single glycine mutation in the equilibrative nucleoside transporter gene, hENT1, alters nucleoside transport activity and sensitivity to nitrobenzylthioinosine. Biochemistry 41, 1512–1519 [DOI] [PubMed] [Google Scholar]
- 16. Visser F., Vickers M. F., Ng A. M., Baldwin S. A., Young J. D., Cass C. E. (2002) Mutation of residue 33 of human equilibrative nucleoside transporters 1 and 2 alters sensitivity to inhibition of transport by dilazep and dipyridamole. J. Biol. Chem. 277, 395–401 [DOI] [PubMed] [Google Scholar]
- 17. Paproski R. J., Visser F., Zhang J., Tackaberry T., Damaraju V., Baldwin S. A., Young J. D., Cass C. E. (2008) Mutation of Trp29 of human equilibrative nucleoside transporter 1 alters affinity for coronary vasodilator drugs and nucleoside selectivity. Biochem. J. 414, 291–300 [DOI] [PubMed] [Google Scholar]
- 18. Yao S. Y., Ng A. M., Vickers M. F., Sundaram M., Cass C. E., Baldwin S. A., Young J. D. (2002) Functional and molecular characterization of nucleobase transport by recombinant human and rat equilibrative nucleoside transporters 1 and 2. Chimeric constructs reveal a role for the ENT2 helix 5–6 region in nucleobase translocation. J. Biol. Chem. 277, 24938–24948 [DOI] [PubMed] [Google Scholar]
- 19. Sundaram M., Yao S. Y., Ng A. M., Cass C. E., Baldwin S. A., Young J. D. (2001) Equilibrative nucleoside transporters. Mapping regions of interaction for the substrate analogue nitrobenzylthioinosine (NBMPR) using rat chimeric proteins. Biochemistry 40, 8146–8151 [DOI] [PubMed] [Google Scholar]
- 20. Yao S. Y., Sundaram M., Chomey E. G., Cass C. E., Baldwin S. A., Young J. D. (2001) Identification of Cys-140 in helix 4 as an exofacial cysteine residue within the substrate-translocation channel of rat equilibrative nitrobenzylthioinosine (NBMPR)-insensitive nucleoside transporter rENT2. Biochem. J. 353, 387–393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Vasudevan G., Ullman B., Landfear S. M. (2001) Point mutations in a nucleoside transporter gene from Leishmania donovani confer drug resistance and alter substrate selectivity. Proc. Natl. Acad. Sci. U.S.A. 98, 6092–6097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hyde R. J., Cass C. E., Young J. D., Baldwin S. A. (2001) The ENT family of eukaryote nucleoside and nucleobase transporters. Recent advances in the investigation of structure/function relationships and the identification of novel isoforms. Mol. Membr. Biol. 18, 53–63 [PubMed] [Google Scholar]
- 23. Sundaram M., Yao S. Y., Ng A. M., Griffiths M., Cass C. E., Baldwin S. A., Young J. D. (1998) Chimeric constructs between human and rat equilibrative nucleoside transporters (hENT1 and rENT1) reveal hENT1 structural domains interacting with coronary vasoactive drugs. J. Biol. Chem. 273, 21519–21525 [DOI] [PubMed] [Google Scholar]
- 24. Valdés R., Arastu-Kapur S., Landfear S. M., Shinde U. (2009) An ab initio structural model of a nucleoside permease predicts functionally important residues. J. Biol. Chem. 284, 19067–19076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Papageorgiou I., De Koning H. P., Soteriadou K., Diallinas G. (2008) Kinetic and mutational analysis of the Trypanosoma brucei NBT1 nucleobase transporter expressed in Saccharomyces cerevisiae reveals structural similarities between ENT and MFS transporters. Int. J. Parasitol. 38, 641–653 [DOI] [PubMed] [Google Scholar]
- 26. Baldwin S. A., McConkey G. A., Cass C. E., Young J. D. (2007) Nucleoside transport as a potential target for chemotherapy in malaria. Curr. Pharm. Des. 13, 569–580 [DOI] [PubMed] [Google Scholar]
- 27. Abramson J., Smirnova I., Kasho V., Verner G., Kaback H. R., Iwata S. (2003) Structure and mechanism of the lactose permease of Escherichia coli. Science 301, 610–615 [DOI] [PubMed] [Google Scholar]
- 28. Huang Y., Lemieux M. J., Song J., Auer M., Wang D. N. (2003) Structure and mechanism of the glycerol-3-phosphate transporter from Escherichia coli. Science 301, 616–620 [DOI] [PubMed] [Google Scholar]
- 29. Jardetzky O. (1966) Simple allosteric model for membrane pumps. Nature 211, 969–970 [DOI] [PubMed] [Google Scholar]
- 30. Kavanaugh M. P. (1998) Neurotransmitter transport. Models in flux. Proc. Natl. Acad. Sci. U.S.A. 95, 12737–12738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Liu W., Boitz J. M., Galazka J., Arendt C. S., Carter N. S., Ullman B. (2006) Functional characterization of nucleoside transporter gene replacements in Leishmania donovani. Mol. Biochem. Parasitol. 150, 300–307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. LeBowitz J. H., Coburn C. M., McMahon-Pratt D., Beverley S. M. (1990) Development of a stable Leishmania expression vector and application to the study of parasite surface antigen genes. Proc. Natl. Acad. Sci. U.S.A. 87, 9736–9740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. LeBowitz J. H. (1994) Transfection experiments with Leishmania. Methods Cell. Biol. 45, 65–78 [DOI] [PubMed] [Google Scholar]
- 34. Vasudevan G., Carter N. S., Drew M. E., Beverley S. M., Sanchez M. A., Seyfang A., Ullman B., Landfear S. M. (1998) Cloning of Leishmania nucleoside transporter genes by rescue of a transport-deficient mutant. Proc. Natl. Acad. Sci. U.S.A. 95, 9873–9878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ortiz D., Valdés R., Sanchez M. A., Hayenga J., Elya C., Detke S., Landfear S. M. (2010) Purine restriction induces pronounced translational up-regulation of the NT1 adenosine/pyrimidine nucleoside transporter in Leishmania major. Mol. Microbiol. 78, 108–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Detke S. (2007) TOR-induced resistance to toxic adenosine analogs in Leishmania brought about by the internalization and degradation of the adenosine permease. Exp. Cell. Res. 313, 1963–1978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Leighton B. H., Seal R. P., Watts S. D., Skyba M. O., Amara S. G. (2006) Structural rearrangements at the translocation pore of the human glutamate transporter, EAAT1. J. Biol. Chem. 281, 29788–29796 [DOI] [PubMed] [Google Scholar]
- 38. Zomot E., Zhou Y., Kanner B. I. (2005) Proximity of transmembrane domains 1 and 3 of the γ-aminobutyric acid transporter GAT-1 inferred from paired cysteine mutagenesis. J. Biol. Chem. 280, 25512–25516 [DOI] [PubMed] [Google Scholar]
- 39. Brocke L., Bendahan A., Grunewald M., Kanner B. I. (2002) Proximity of two oppositely oriented reentrant loops in the glutamate transporter GLT-1 identified by paired cysteine mutagenesis. J. Biol. Chem. 277, 3985–3992 [DOI] [PubMed] [Google Scholar]
- 40. Gray W. R. (1993) Disulfide structures of highly bridged peptides. A new strategy for analysis. Protein Sci. 2, 1732–1748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Smirnova I., Kasho V., Kaback H. R. (2011) Lactose permease and the alternating access mechanism. Biochemistry 50, 9684–9693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kaback H. R., Smirnova I., Kasho V., Nie Y., Zhou Y. (2011) The alternating access transport mechanism in LacY. J. Membr. Biol. 239, 85–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Stroud R. M. (2007) Transmembrane transporters. An open and closed case. Proc. Natl. Acad. Sci. U.S.A. 104, 1445–1446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Dang S., Sun L., Huang Y., Lu F., Liu Y., Gong H., Wang J., Yan N. (2010) Structure of a fucose transporter in an outward-open conformation. Nature 467, 734–738 [DOI] [PubMed] [Google Scholar]
- 45. Carter N. S., Drew M. E., Sanchez M., Vasudevan G., Landfear S. M., Ullman B. (2000) Cloning of a novel inosine-guanosine transporter gene from Leishmania donovani by functional rescue of a transport-deficient mutant. J. Biol. Chem. 275, 20935–209341 [DOI] [PubMed] [Google Scholar]
- 46. Tao Z., Zhang Y. W., Agyiri A., Rudnick G. (2009) Ligand effects on cross-linking support a conformational mechanism for serotonin transport. J. Biol. Chem. 284, 33807–33814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Crisman T. J., Qu S., Kanner B. I., Forrest L. R. (2009) Inward-facing conformation of glutamate transporters as revealed by their inverted-topology structural repeats. Proc. Natl. Acad. Sci. U.S.A. 106, 20752–20757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zhang X., Qu S. (2011) Proximity of transmembrane segments 5 and 8 of the glutamate transporter GLT-1 inferred from paired cysteine mutagenesis. PLOS One 6, e21288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Qiu Z., Nicoll D. A., Philipson K. D. (2001) Helix packing of functionally important regions of the cardiac Na+-Ca2+ exchanger. J. Biol. Chem. 276, 194–199 [DOI] [PubMed] [Google Scholar]
- 50. Kubo Y., Konishi S., Kawabe T., Nada S., Yamaguchi A. (2000) Proximity of periplasmic loops in the metal-Tetracycline/H+ antiporter of Escherichia coli observed on site-directed chemical cross-linking. J. Biol. Chem. 275, 5270–5274 [DOI] [PubMed] [Google Scholar]
- 51. Loo T. W., Clarke D. M. (2000) The packing of the transmembrane segments of human multidrug resistance P-glycoprotein is revealed by disulfide cross-linking analysis. J. Biol. Chem. 275, 5253–5256 [DOI] [PubMed] [Google Scholar]
- 52. Jiang J., Shrivastava I. H., Watts S. D., Bahar I., Amara S. G. (2011) Large collective motions regulate the functional properties of glutamate transporter trimers. Proc. Natl. Acad. Sci. U.S.A. 108, 15141–15146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. White S. H. (2004) The progress of membrane protein structure determination. Protein Sci. 13, 1948–1949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Opella S. J., Marassi F. M. (2004) Structure determination of membrane proteins by NMR spectroscopy. Chem. Rev. 104, 3587–3606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Young J. D., Yao S. Y., Sun L., Cass C. E., Baldwin S. A. (2008) Human equilibrative nucleoside transporter (ENT) family of nucleoside and nucleobase transporter proteins. Xenobiotica 38, 995–1021 [DOI] [PubMed] [Google Scholar]
- 56. Baldwin S. A., Beal P. R., Yao S. Y., King A. E., Cass C. E., Young J. D. (2004) The equilibrative nucleoside transporter family, SLC29. Pflugers Arch. 447, 735–743 [DOI] [PubMed] [Google Scholar]
- 57. Schmidt B., Ho L., Hogg P. J. (2006) Allosteric disulfide bonds. Biochemistry 45, 7429–7433 [DOI] [PubMed] [Google Scholar]
- 58. Krovetz H. S., VanDongen H. M., VanDongen A. M. (1997) Atomic distance estimates from disulfides and high-affinity metal-binding sites in a K+ channel pore. Biophys. J. 72, 117–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Loo T. W., Bartlett M. C., Clarke D. M. (2004) Disulfide cross-linking analysis shows that transmembrane segments 5 and 8 of human P-glycoprotein are close together on the cytoplasmic side of the membrane. J. Biol. Chem. 279, 7692–7697 [DOI] [PubMed] [Google Scholar]