

Background: PINK1 null results in severe mitochondrial complex IV deficits.
Results: PINK1 regulates complex IV through interactions with Hsp60 upstream regulators, and ginsenoside Re can rescue complex IV deficits.
Conclusion: Complex IV deficiency was rescued by restoration of optimal NO signaling by ginsenoside Re.
Significance: NO-complex IV signaling provides potential for new therapeutics targeting mitochondrial dysfunction in PD.
Keywords: Mitochondria, Mitochondrial Diseases, Nitric Oxide, Parkinson's Disease, Pink1, Complex IV, Ginsenoside Re
Abstract
PINK1, linked to familial Parkinson's disease, is known to affect mitochondrial function. Here we identified a novel regulatory role of PINK1 in the maintenance of complex IV activity and characterized a novel mechanism by which NO signaling restored complex IV deficiency in PINK1 null dopaminergic neuronal cells. In PINK1 null cells, levels of specific chaperones, including Hsp60, leucine-rich pentatricopeptide repeat-containing (LRPPRC), and Hsp90, were severely decreased. LRPPRC and Hsp90 were found to act upstream of Hsp60 to regulate complex IV activity. Specifically, knockdown of Hsp60 resulted in a decrease in complex IV activity, whereas antagonistic inhibition of Hsp90 by 17-(allylamino) geldanamycin decreased both Hsp60 and complex IV activity. In contrast, overexpression of the PINK1-interacting factor LRPPRC augmented complex IV activity by up-regulating Hsp60. A similar recovery of complex IV activity was also induced by coexpression of Hsp90 and Hsp60. Drug screening identified ginsenoside Re as a compound capable of reversing the deficit in complex IV activity in PINK1 null cells through specific increases of LRPPRC, Hsp90, and Hsp60 levels. The pharmacological effects of ginsenoside Re could be reversed by treatment of the pan-NOS inhibitor l-NG-Nitroarginine Methyl Ester (l-NAME) and could also be reproduced by low-level NO treatment. These results suggest that PINK1 regulates complex IV activity via interactions with upstream regulators of Hsp60, such as LRPPRC and Hsp90. Furthermore, they demonstrate that treatment with ginsenoside Re enhances functioning of the defective PINK1-Hsp90/LRPPRC-Hsp60-complex IV signaling axis in PINK1 null neurons by restoring NO levels, providing potential for new therapeutics targeting mitochondrial dysfunction in Parkinson's disease.
Introduction
Mitochondria are tiny organelles inside eukaryotic cells, including neurons, that generate almost all energy in the form of ATP. Dysfunctional mitochondria have been proposed to play a major role in the pathogenesis of Parkinson disease (PD)3 (1). However, few effective therapeutic strategies are available to target mitochondrial dysfunction in PD, partly because of our lack of knowledge regarding the specific pathogenic mechanisms underlying mitochondrial dysfunction.
Human mutations in PINK1 (PARK6) have been linked to autosomal recessive forms of familial PD (2). PINK1 has serine/threonine kinase activity in mitochondria (2, 3). Functional analyses have revealed that PINK1 plays regulatory roles in various mitochondrial functions, including mitophagy and fusion-fission dynamics (4, 5). We previously reported a dramatic decrease in the activity of mitochondrial electron transport chain complex IV in PINK1 null dopaminergic neuronal cells in contrast to the normal levels of activity found in DJ-1 null dopaminergic neuronal cells (6). Other studies have also suggested that complex IV activity is affected by PINK1 gene knockdown or PINK1 null mutations (7, 8), suggesting an as-yet undefined role of PINK1 in the regulation of complex IV activity.
Complex IV (cytochrome c oxidase (COX), EC1.9.3.1) is the terminal enzyme of the mitochondrial respiratory chain. Complex IV is comprised of 13 subunits of dual genetic origin in mammals. The COX1, COX2, and COX3 subunits, encoded by the mitochondrial DNA, form the catalytic core of the enzyme, whereas the remaining 10 subunits are encoded by the nuclear DNA (9, 10). In yeast, complex IV biosynthesis requires the specific, sequential action of ∼20 nuclear-encoded assembly or accessory factors (11). More than half of these assembly factors are known to have human orthologs (9). Mutations in several of these factors are associated with autosomal recessive complex IV deficiency syndrome. For instance, complex IV-defective French-Canadian Leigh syndrome can be caused by mutations in the leucine-rich pentatricopeptide repeat-containing (LRPPRC), a complex IV assembly factor that regulates stabilization of the mitochondrial mRNAs MTCO1 and MTCO3 (12). Leigh syndrome with complex IV deficiency can result from more than 40 different mutations in SURF1, an assembly factor that regulates formation of the catalytic centers (13, 14). Mutations in other assembly factors cause fatal infantile hypertrophic cardiomyopathy with defective complex IV (15–17). However, little is known about regulation of the expression of these assembly factors and consequent effects on complex IV activity.
NO can mediate both physiological and pathological effects, depending on the physiological concentrations. Neuronal nitric oxide synthase (NOS) and endothelial NOS generate low amounts (0.2–2.0 nm) of NO persisting only for a few minutes, whereas inducible NOS produces high amounts (20–200 nm) of NO that can persist up to days (18–20). In neurodegenerative diseases, high concentrations of NO trigger apoptosis through the induction of O2− formation and subsequent generation of ONOO−, which inhibits and/or damages the mitochondrial ETC complexes, particularly complexes I and II, which possess iron-sulfur centers (21–23). Notably, NO reversibly inhibits complex IV activity by competing with O2 (24, 25). In contrast, long-term treatment of cell cultures with low concentrations of NO is known to induce mitochondrial biogenesis, which is mediated by a cGMP-dependent signaling pathway (26, 27). However, the pharmacological effects of low physiological concentrations of NO on dysfunctional mitochondria and the relevance of this signaling to dopaminergic neuronal survival remain to be elucidated in familial PD.
Development of disease-modifying drugs capable of restoring mitochondrial function remains a formidable challenge in PD and other mitochondria-associated diseases. Compounds with neuroprotective potential, including a few ordinary mitochondrial modulators such as coenzyme Q10 and creatine, are under investigation in clinical trials of PD to determine whether they can prevent continuous loss of dopaminergic neurons (28). Other compounds with potential for modulating mitochondria include natural products, such as the ginsenosides, that comprise the primary biologically active components of ginseng. These compounds have been shown to exert complex physiological functions, including preservation of mitochondrial integrity (29–32). However, the precise effects of ginsenosides on defective mitochondria under conditions of PD are not well understood.
This work is the first to identify the molecular basis underlying PINK1 null mutation-induced complex IV deficit. We found that significant loss of complex IV activity in PINK1 null dopaminergic neurons results from down-regulation of the Hsp60 and its upstream regulators, LRPPRC and Hsp90. PINK1 appears to regulate complex IV activity via specific interactions with LRPPRC and Hsp90. Importantly, complex IV deficiency in PINK1 null neuronal cells could be overcome by optimal treatment with ginsenoside Re. Treatment with ginsenoside Re specifically increased the expression levels of LRPPRC, Hsp90, and Hsp60 through activation of NO signaling in PINK1 null cells but not in the wild type cells.
EXPERIMENTAL PROCEDURES
Establishment and Culture of Dopaminergic Neuronal Cell Lines and Hsp60 KD Cell Lines
PINK1 null dopaminergic cell lines were established by breeding the previously described immortalizer transgenic mouse (33) with the PINK1 null mouse as reported previously (6). Briefly, at embryonic day 13.5, immortalized PINK1-null dopaminergic neuronal cells were established from the substantia nigra region of the double transgenic mouse embryos harboring both a TH9.0kb-SV40Tag-tsA58 transgene and the PINK1 null mutation. Dopaminergic neuronal cells were cultured in RF medium (DMEM supplemented with 10% fetal calf serum, 1% glucose, Penicillin-streptomycin and l-glutamine) at 33 °C with 5% CO2, as described previously (33). PINK1 null dopaminergic neuronal cells were further characterized at the permissive temperature (33 °C) for the expression of general neuronal markers TuJ1 and NeuN and a dopaminergic neuron-specific marker, tyrosine hydroxylase (TH), by immunoblot and RT-PCR analysis. To generate stable Hsp60 KD cells, wild-type SN4741 dopaminergic neuronal cells were transfected with a pSUPER.retro.puro vector (OligoEngine, Seattle, WA) containing Hsp60 shRNA (5′-CCG AAG ACG TTG ACG GAG A-3′) and pSUPER.retro.puro vector alone (for control cells). Stably transfected cell lines were selected by supplementing the culture medium with puromycin (1 μg/ml).
Plasmids, Gene Knockdown, and Transient Transfection Analysis
The pCMVTnT-PINK1-myc, pCDNA3-PINK1-FLAG, and pCDNA3.1-Hsp60-MycHis plasmids were provided by Dr. M. R. Cookson, Dr. Takahashi, and Dr. T. J. Corydon, respectively. Hsp90 and LRPPRC in pCDNA3 vector were purchased from Thermo Fisher Scientific Inc. To construct pFLAG-LRPPRC, the mouse LRPPRC cDNA was digested with EcoRI (5′) and KpnI (3′) and inserted into the pFLAG-CMV plasmid. For gene knockdown, the siRNA duplex was delivered using the TransIT-TKO transfection reagent (Mirus Bio Corp., Madison, WI), as described previously (6). siRNA oligonucleotides were made by Bioneer Corp. (Daejeon, Korea) and had the following sequences: siHsp60 sense, 5-r(CACUGUUCUGGCACGAUCU)d(TT)-3 and siHsp60 antisense, 5-r(AGAUCGUGCCAGAACAGUG)d(TT)-3. Transient transfection analyses were carried out as described previously (6).
Quantitative RT-PCR Analysis
Dopaminergic cells were used for total RNA isolation. cDNA was synthesized using 5 μg total RNA and RT-PCR kit (MP Biomedicals Inc.). Endogenous PINK1 mRNA levels were measured relative to β-actin by the standard curve method. Quantitative PCR was performed by AB7300 real-time PCR systems and DyNAmo HS SYBR Green quantitative PCR kit (Applied Biosystems and Thermo Fisher Scientific Inc.). The β-actin primer set (forward, 5′-TTC TTT GCA GCT CCT TCG TTG CCG-3′ and reverse, 5′-TGG ATG GCT ACG TAC ATG GCT GGG-3′) and the PINK1 primer set (forward, 5′-TGA TCT CAG ACT TTG GCT GCT-3′ and reverse, 5′-CCT TGG CCA TAG AAG GGA TT-3′) were used.
Chemicals
17-AAG was purchased from Selleck Chemicals LLC (Houston, TX). Puromycin was purchased from InvivoGen (San Diego, CA). Ginsenoside Re was purified as described below. All other chemicals were purchased from Sigma-Aldrich (St. Louis, MO).
Separation and Purification of Ginsenoside Re from Panax ginseng
Eight ginsenosides, including Re, were separated and purified from P. ginseng by high-speed counter-current chromatography coupled with an evaporative light-scattering detector (HSCCC-ELSD), as reported previously (34). Briefly, crude samples for HSCCC-ELSD separation were first purified from ginseng extract using a macroporous resin. Specifically, the extract was loaded onto a Diaion-HP20 column and fractionated using a methanol and water gradient elution. The ginsenosides-protopanaxadiol and protopanaxatriol fractions were subsequently eluted with 65 and 80% methanol and water gradient elution, respectively. These two fractions were then subjected to HSCCC-ELSD. The two-phase solvent system used for separation was composed of chloroform/methanol/water/isopropanol at a volume ratio of 4:3:2:1. Each fraction was collected and dried. Eight ginsenosides, including ginsenoside Re, were resolved, and the purity of each ginsenoside was greater than 98% as assessed by HPLC-ELSD.
Immunoprecipitation Assay
Transfected cells were incubated in lysis buffer (Thermo Fisher Scientific) containing protease inhibitors for 1 h on ice. Protein extracts were incubated with anti-FLAG M2-agarose (Sigma-Aldrich) or protein G plus agarose overnight at 4 °C. The precipitated beads were washed three times with PBS, and bound proteins were eluted from the beads by boiling in sample buffer (60 mm Tris-HCl (pH 6.8), 25% glycerol, 2% SDS, 14.4 mm β-mercaptoethanol, and 0.1% bromphenol blue). Eluted proteins were analyzed by 12% SDS-PAGE followed by LC/MS at the MS Core Facility (Ewha Womans University, Seoul, Korea) and immunoblot analysis.
Immunoblot Analysis
Cells were washed three times with PBS and incubated in cell lysis buffer (Cell Signaling Technology, Inc., Danvers, MA) containing 1 mm PMSF. Protein lysates were separated on 12% SDS-polyacrylamide gels and transferred to PVDF membranes. After blocking in 5% milk in TBS containing 0.1% Tween 20, the membranes were incubated overnight at 4 °C with primary antibodies. The following primary antibodies were used: anti-prohibitin, anti-GAPDH, anti-LRPPRC, anti-Hsp90, anti-Hsp60, anti-GRP78, and anti-actin (Santa Cruz Biotechnology, Inc., CA), anti-COX I (Abcam, Cambridge, UK), anti-COX IV and anti-GRP75 (Cell Signaling Technology, Inc.), anti-Myc (Clontech Laboratories, Inc., CA), anti-FLAG (Sigma-Aldrich), and anti-NDUFB6 (Invitrogen). After washing, the blots were probed with anti-goat, anti-mouse, or anti-rabbit IgG-HRP secondary antibodies and visualized by ECL (GE Healthcare), as described previously (6). Densitometric quantification of bands was performed using an image analyzer system and Multi Gauge version 2.3.
Immunocytochemistry
Cell cultures were prepared for immunocytochemistry by fixation with 4% paraformaldehyde in PBS for 15 min at room temperature, followed by permeabilization for 5 min with 0.1% Triton X-100 in PBS and three washes with PBS. Mitochondria were stained using MitoTracker (Invitrogen), and morphological changes were analyzed by confocal microscopy (Carl Zeiss, Germany).
Isolation of Mitochondria
Mitochondria were prepared as described previously (6). Briefly, cells were washed with ice-cold PBS and suspended in 10 mm ice-cold Tris buffer (pH 7.6) containing protease inhibitor mixture. Cells were mechanically disrupted using a 1-ml syringe, and, following the addition of ice-cold 1.5 m sucrose, centrifugation at 600 × g for 10 min at 2 °C. The supernatant was then centrifuged at 14,000 × g for 10 min at 2 °C, and the resulting pellet was washed and resuspended in 10 mm ice-cold Tris (pH 7.6) containing a protease inhibitor mixture. Some pellets were lysed for quantification, and the remainder was stored on ice until analysis.
Complex IV and Citrate Synthase Enzyme Assay
Complex IV and citrate synthase were analyzed as described previously (7). Briefly, complex IV activity was measured spectrophotometrically at 550 nm in a 240-μl reaction containing 30 mm potassium phosphate, 2.5 mm dodecylmaltoside, and 34 μm ferrocytochrome c (pH 7.4). Mitochondrial samples (1 μg/10 μl) were added to the buffer, and the absorbance was immediately measured at 30-s intervals for 5 min at 30 °C. Negative controls contained 1 mm potassium cyanide. Citrate synthase activity was measured spectrophotometrically at 412 nm in a 240-μl reaction containing 50 mm Tris-HCl (pH 7.5), 0.2 mm 5,5′-dithiobis-(2-nitrobenzoic acid), 0.1 mm acetyl-CoA, and 0.5 mm oxaloacetate. Mitochondrial samples (1 μg/10 μl) were preincubated in buffer without oxaloacetate for 5 min at 30 °C. Then, 2.5 μl of 50 mm oxaloacetate was added to start the reaction. The absorbance was measured at 30-s intervals for 5 min at 37 °C. For controls, an equivalent volume of water was added in place of oxaloacetate.
Statistics
Data are expressed as mean ± S.E. on the basis of data derived from three to eight independent experiments. Statistical analyses were performed using Sigmaplot, and significant differences were assessed using Student's t test. When necessary, multiple comparisons were made using one-way analysis of variance.
RESULTS
Loss of PINK1 Is Associated with Complex IV and Hsp60 Deficits
To investigate the molecular basis of loss of mitochondrial complex IV activity, we utilized wild-type SN4741 and PINK1 null dopaminergic neuronal cells that were generated in identical fashion, as established and characterized previously (6, 33). PINK1 null dopaminergic neuronal cells exhibit an accumulation of defective mitochondria (6) and a significant decrease in complex IV activity (Fig. 1A). The decrease in complex IV activity appears to be specifically associated with loss of PINK1, as a DJ-1 null mutation had no effect on complex IV activity (6). These results indicate that the wild-type and PINK1 null dopaminergic neuronal models provide an ideal system to investigate the mechanism underlying decrease in complex IV activity under PINK1 null condition.
FIGURE 1.

PINK1 null dopaminergic neuronal cells exhibit decreases in complex IV activity and levels of Hsp60, Hsp90, and LRPPRC. A, a significant reduction (∼60%) in mitochondrial complex IV activity was observed in PINK1 null dopaminergic neuronal cells compared with wild-type SN4741 dopaminergic neuronal cells. Citrate synthase activity was used to normalize ETC activity. B, immunoblot (IB) analysis revealed that levels of the complex IV assembly factor LRPPRC and the mitochondrial chaperone Hsp60 were reduced by 22 and 40%, respectively, in PINK1 null cells compared with wild-type cells. C, levels of the TOM complex-binding chaperone Hsp90 were significantly decreased by 80% in PINK1 null dopaminergic cells. All values represent the mean ± S.E. from three independent experiments. ***, p < 0.01. ADU, arbitrary defined unit.
To determine whether altered expression of mitochondria-related chaperones may underlie complex IV deficit in PINK1 null cells, expression levels of these proteins were compared between wild-type and PINK1 null dopaminergic neuronal cells. Results from this analysis revealed that levels of the mitochondrial chaperone Hsp60, the complex IV assembly factor LRPPRC (Fig. 1B), and the translocase of the outer membrane (TOM) complex binding chaperone Hsp90 (C) were reduced significantly in the PINK1 null dopaminergic neurons. Decreases in Hsp60, Hsp90, and LRPPRC did not occur in DJ-1 null dopaminergic neuronal cells (data not shown). Levels of other chaperones, such as Grp75 and Grp78, remained constant in all dopaminergic neuronal cells. On the basis of these observed decreases in chaperones and assembly factor levels, we next investigated whether Hsp60, LRPPRC, and Hsp90 are involved in the regulation of complex IV activity under normal and pathological conditions using wild-type SN4741 and PINK1 null dopaminergic neuronal cells, respectively.
Down-regulation of Hsp60 Results in Loss of Complex IV Activity
To assess the functional consequences of Hsp60 down-regulation in cells lacking PINK1, we generated SN4741 cells stably expressing Hsp60-targeted shRNA (Hsp60 KD cells) (Fig. 2A). We then analyzed the effect of loss of Hsp60 expression on complex IV activity. Results from this analysis demonstrated that Hsp60 KD cells (line #9) exhibited a more than 60% significant decrease in complex IV activity (Fig. 2B). In contrast, complex I and II activities were not affected by loss of Hsp60 (Fig. 2C). Thus, proper expression of Hsp60 is crucial to regulation of complex IV activity but not that of complexes I and II.
FIGURE 2.

Knockdown of Hsp60 results in a decrease in complex IV activity. A, Hsp60 KD cells were produced using an adenoviral shRNA system in wild-type SN4741 cells as described under “Experimental Procedures.” The largest decrease in Hsp60 expression (∼55%) of Hsp60 was observed in Hsp60 KD cell line #9. IB, immunoblot. B, analysis of the effects of Hsp60 expression level on complex IV activity revealed that complex IV activity was significantly decreased (more than 60%) in Hsp60 KD cells (line #9) but not complex I and II activity (C). These results suggest that Hsp60 expression is crucial to the maintenance of complex IV activity. All values represent the mean ± S.E. from a minimum of three independent experiments. ***, p < 0.01.
Up-regulation of LRPPRC Expression Increases Complex IV Activity in PINK1 Null Neuronal Cells
As expression of complex IV assembly factor LRPPRC was decreased by 30% in the PINK1 null condition (Fig. 1B), we investigated whether alteration in LRPPRC levels affected complex IV activity. First, LRPPRC was overexpressed in PINK1 null dopaminergic neuronal cells, and levels of complex IV activity and Hsp60 were analyzed. Increased LRPPRC expression (∼40%) resulted in a 25% increase in Hsp60 expression (Fig. 3A) and a 110% increase in complex IV activity (B). No changes were observed in the activity of complexes I and II (data not shown), suggesting that decreased LRPPRC levels specifically contributed to the complex IV deficit observed in PINK1 null cells. In contrast, KD of Hsp60 (line #9) did not affect the expression of LRPPRC (Fig. 3C), and the LRPPRC-induced effect was significantly reduced by Hsp60 KD (D), confirming LRPPRC as an upstream regulator of Hsp60 expression. Importantly, LRPPRC appears to interact with PINK1 (Fig. 3E) but not with Hsp60 (F), suggesting the potential regulatory role for PINK1 in LRPPRC function.
FIGURE 3.

Overexpression of LRPPRC improves complex IV deficits in PINK1 null cells. As LRPPRC levels were significantly decreased in PINK1 null cells, the functional effect of LRPPRC overexpression on complex IV activity was assessed. A, overexpression of LRPPRC (∼40% increase) in PINK1 null dopaminergic neuronal cells resulted in a 25% increase in Hsp60 expression and specifically increased complex IV activity by 110% (B). IB, immunoblot. C, in contrast, expression of LRPPRC was not affected by down-regulation of Hsp60 in Hsp60 KD cells (line #9), and the LRPPRC-induced effect was significantly reduced by Hsp60 KD (D). E, when the interaction between LRPPRC and FLAG-tagged PINK1 was analyzed in COS cells by immunoprecipitation (IP), LRPPRC appeared to interact with PINK1. F, LRPPRC did not appear to have a direct interaction with myc-tagged Hsp60. All values represent the mean ± S.E. from a minimum of three independent experiments. **, ##, and $$, p < 0.05. ADU, arbitrary defined unit.
Coexpression of Hsp90 and Hsp60 Can Restore Complex IV Activity in PINK1 Null Neuronal Cells
As Hsp90 levels were reduced by 80% in PINK1 null dopaminergic neuronal cells (Fig. 1C), we next investigated whether specific inhibition of Hsp90 with 17-AAG has a similar effect on complex IV activity in wild-type neuronal cells. As expected, both complex IV activity and Hsp60 levels exhibited a dose-dependent decrease in response to treatment with 17-AAG (Fig. 4A). However, 17-AAG treatment did not alter levels of Hsp90 and LRPPRC levels (Fig. 4B), implicating Hsp90 as another upstream regulator of Hsp60 expression involved in regulation of complex IV activity.
FIGURE 4.

Treatment with an Hsp90 inhibitor results in a dose-dependent decrease in complex IV activity. A, because Hsp90 levels were reduced by 80% in PINK1 null dopaminergic neuronal cells, the effects of specific inhibition of Hsp90 using 17-AAG were analyzed in wild-type neuronal cells. Both complex IV activity and Hsp60 levels were decreased significantly in 17-AAG-treated cells in a dose-dependent manner. IB, immunoblot. B, in contrast, 17-AAG treatment did not affect expression of Hsp90 and LRPPRC. C, we investigated whether overexpression of Hsp60 alone can overcome decreased expression of Hsp90 in PINK1 null cells. Hsp60 expression alone could not rescue decreases in complex IV activity. D, Hsp90 expression alone did not result in any notable recovery in complex IV activity. All values represent the mean ± S.E. from a minimum of three independent experiments. ***, p < 0.01. Mito, mitochondria; Cyto, cytosol.
Next, we tested whether overexpression of Hsp60 alone can overcome Hsp90 and/or LRPPRC deficiency in the PINK1 null condition. Either Hsp60 or Hsp90 expression alone did not result in any notable recovery in complex IV activity (Fig. 4, C and D), likely because of the decreased level of Hsp90 in PINK1 null dopaminergic neuronal cells. In contrast, coexpression of Hsp60 with Hsp90 was associated with a significant increase in complex IV activity (40%) in PINK1 null dopaminergic neuronal cells (Fig. 5A). Because the Hsp90 chaperone is known to assist in mitochondrial import of preproteins via the TOM70 receptor (35), we tested whether Hsp90 interacts with Hsp60 to elucidate a potential regulatory role for Hsp90. Indeed, Hsp90 was found to interact with Hsp60 (Fig. 5B). Furthermore, overexpression of Hsp90 was associated with increased mitochondrial accumulation of Hsp60 (Fig. 5C), confirming that both Hsp90 and Hsp60 are necessary for significant functional recovery from complex IV deficiency resulting from loss of PINK1.
FIGURE 5.

Coexpression of Hsp60 and Hsp90 rescues deficits in complex IV activity in PINK1 null neuronal cells. A, because Hsp60 overexpression alone could not restore complex IV activity, Hsp60 was coexpressed with Hsp90 in PINK1 null dopaminergic neuronal cells. Coexpression of Hsp60 and Hsp90 significantly restored levels of complex IV activity, resulting in a 40% increase in activity in the PINK1 null dopaminergic neuronal cells. IB, immunoblot. B, to elucidate a potential regulatory role for Hsp90, a potential interaction between Hsp90 and Hsp60-myc was assessed. Indeed, Hsp90 was found to interact with Hsp60. C, overexpression of untagged Hsp90 resulted in an increase in accumulation of Hsp60-myc in the mitochondria (Mito) of PINK1- null cells. Thus, sufficient expression of Hsp90 is required for recovery of complex IV deficits in PINK1 null cells. All values represent the mean ± S.E. from a minimum of three independent experiments. ***, p < 0.01. COS, designation of cell line.
Ginsenoside Re Rescues Complex IV Deficiency in PINK1 Null Neuronal Cells via NO Signaling
As no disease-modifying drug has been identified that is capable of restoring mitochondrial function in PD, we next investigated whether any natural compounds, including ginsenosides, could improve the complex IV deficit in PINK1 null dopaminergic neuronal cells. Importantly, treatment with ginsenoside Re at the optimal concentration (3 μm) exerted a significant functional recovery (∼72% increase) in complex IV activity in PINK1 null cells (Fig. 6, A and B). In contrast, at concentrations higher than 5 μm, ginsenoside Re instead decreased the residual complex IV activity. The functional recovery at the optimal concentration was accompanied by the enhanced expression of both Hsp90 and LRPPRC and, consequently, Hsp60 (Fig. 6, C and D). The effect of ginsenoside Re was significantly reduced by Hsp60 KD (Fig. 6E). In contrast to its effects in PINK1 null neurons, in wild-type dopaminergic neuronal cells, the same dose (3 μm) of ginsenoside Re resulted in a mild inhibition (∼13% decrease) in complex IV activity (Fig. 6F), accompanied by small decreases in LRPPRC and Hsp60 levels (17 and 10%, respectively) (G). Ginsenoside Re had no effect on the expression of PINK1 mRNA (Fig. 6H). These results suggest that ginsenoside Re-induced increases in complex IV activity are specific to both the PINK1 null pathogenic condition and the optimal concentration (3 μm).
FIGURE 6.

Ginsenoside Re rescues complex IV deficits in PINK1 null cells. In our drug screening for agents capable of restoring defective mitochondrial function in the PINK1 null dopaminergic neuronal cell model, we identified ginsenoside Re as an agent capable of rescuing decreased complex IV activity. A, treatment with 3 μm ginsenoside Re resulted in the greatest functional recovery (∼72% increase) of the complex IV activity in PINK1 null cells (B). At concentrations greater than 5 μm, ginsenoside Re decreased residual complex IV activity. C and D, ginsenoside Re-induced recovery of complex IV activity was accompanied by increased expression of Hsp90, LRPPRC, and Hsp60. IB, immunoblot. E, the effect of ginsenoside Re was significantly reduced by Hsp60 KD. F, in wild-type SN4741 cells, treatment with 3 μm ginsenoside Re resulted in mild inhibition (∼13%) of complex IV activity. G, decreases in complex IV activity in ginsenoside Re-treated wild-type cells were accompanied by small decreases in LRPPRC and Hsp60 levels (17 and 10%, respectively). H, ginsenoside Re had no effect on the expression of PINK1 mRNA. All values represent the mean ± S.E. from a minimum of three independent experiments. ***, p < 0.01; **, ##, and $$, p < 0.05. NT, no treatment; Mito, mitochondria.
We next examined whether an NO signaling mechanism underlies ginsenoside Re-induced rescue of complex IV activity, as ginsenoside Re has been shown to enhance NO production in cultured cells (36, 37). To this end, we tested whether the presence of the pan-NOS inhibitor L-NAME blocked the ginsenoside Re-induced effect on complex IV activity. As we predicted, the pharmacological recovery of complex IV activity caused by ginsenoside Re was completely abolished by treatment with L-NAME (Fig. 7A) in addition to inhibiting up-regulation of LRPPRC and Hsp60 (B). Together, these results suggest that NO signaling mediates the pharmacological rescue of complex IV activity induced by treatment with ginsenoside Re under PINK1 null conditions.
FIGURE 7.

The pharmacological effects of ginsenoside Re are mediated by NO signaling. Because ginsenoside Re is known to enhance NO production, we tested whether treatment with the pan-NOS inhibitor L-NAME could interfere with ginsenoside Re-mediated rescue of complex IV activity (A). Indeed, the restoration of complex IV activity by ginsenoside Re in PINK1 null cells was completely abolished by treatment with L-NAME. B, L-NAME treatment inhibited ginsenoside Re-induced up-regulation of both LRPPRC and Hsp60. All values represent the mean ± S.E. from a minimum of three independent experiments. IB, immunoblot. ** and $$, p < 0.05. NT, no treatment; Mito, mitochondria.
Finally, we confirmed the role of NO signaling in the rescue of complex IV activity by testing whether treatment with the NO donor sodium nitroprusside (SNP) could reproduce the pharmacological effect of ginsenoside Re on complex IV activity. Indeed, an optimal dose of the NO donor (20 nm, Fig. 8A) reproduced the rescue effect of ginsenoside Re, enhancing complex IV activity and increasing expression of Hsp90, LRPPRC, and Hsp60 (B–D). In wild-type cells, consistent with results with ginsenoside Re, the same dose of SNP (20 nm) moderately inhibited complex IV activity by 26% and decreased LRPPRC and Hsp60 levels by 20 and 25%, respectively (Fig. 8, E and F). These results indicate that the rescue effect of an optimized ginsenoside Re dose (3 μm) could be reproduced only by low-level treatment with SNP (20 nm) in PINK1 null cells. Together, these data suggest that recovery of complex IV activity by both agents is a PINK1 null-specific phenomenon, likely occurring through restoration of dysfunctional NO signaling caused by loss of PINK1. In contrast, identical treatments result in perturbation of NO signaling in wild-type cells.
FIGURE 8.

Treatment with an NO donor can reproduce the effects of ginsenoside Re on complex IV deficits in PINK1 null cells. To further confirm the involvement of NO signaling in ginsenoside Re-mediated restoration of complex IV activity, the NO donor SNP was tested to determine whether it could reproduce the pharmacological effects of ginsenoside Re at a specific concentration. A, only low levels of NO (20 nm) reproduced the beneficial effects of ginsenoside Re, enhancing complex IV activity (B). Restoration of complex IV activity by SNP was accompanied by the increased expression of Hsp90 (C), LRPPRC, and Hsp60 (D). IB, immunoblot. However, in wild-type cells, as expected, treatment with 20 nm SNP moderately inhibited complex IV activity by 26% (E) and decreased LRPPRC and Hsp60 levels by 20 and 25%, respectively (F), similar to the effects of ginsenoside Re. All values represent the mean ± S.E. from a minimum of three independent experiments. **, p < 0.05. NT, no treatment; Mito, mitochondria.
DISCUSSION
In the mitochondria, PINK1 is known as a key regulator of mitophagy and fusion-fission dynamics. Additionally, we have identified a novel mitochondrial role for PINK1 in the regulation of complex IV activity. Loss of PINK1 resulted in a severe complex IV deficit caused by the significant down-regulation of a specific subset of mitochondria-associated chaperones, including Hsp60, Hsp90, and LRPPRC, and consequent defective assembly of complex IV subunits. The complex IV deficit could be restored substantially by treatment with an optimized dose of ginsenoside Re, which resulted in up-regulation of Hsp60, Hsp90, and LRPPRC. This pharmacological rescue was abolished by NOS inhibition and could be reproduced by activation of NO signaling in PINK1 null dopaminergic neurons. Together, these novel findings implicate PINK1 in regulation of complex IV activity through interactions with two Hsp60 upstream regulators: LRPPRC and Hsp90. Moreover, controlled levels of NO signaling activated by ginsenoside Re may rescue the defective PINK1-LRPPRC/Hsp90-Hsp60-complex IV pathway under PINK1 null conditions.
Although PINK1 localizes to the mitochondria, the mechanisms by which it affects mitochondrial function are largely unknown. Our previous report suggests that PINK1 may regulate mitochondrial complex IV activity (6). To investigate the direct regulatory role of PINK1 in complex IV activity, we first analyzed the expression of mitochondria-related chaperones and complex IV assembly factors such as Hsp60, Hsp90, Grp78, Grp75, LRPPRC, SURF1, and SCO1 in wild-type and PINK1 null dopaminergic neurons and assessed the interactions of these proteins with PINK1. The mitochondrial chaperone Hsp60, the complex IV assembly factor LRPPRC, and the TOM complex binding chaperone Hsp90 were most significantly down-regulated by loss of PINK1, as shown in Fig. 1, B and C. Furthermore, both LRPPRC and Hsp90 interacted with PINK1, consistent with previous results from proteomic analysis of PINK1-interacting proteins (38). Thus, PINK1 may regulate complex IV activity by direct modulation of LRPPRC and Hsp90 levels, although the precise mechanisms underlying this pathway require further investigation. We next investigated the pathological significance of LRPPRC, Hsp90, or Hsp60 down-regulation on complex IV activity. First, we demonstrated the specific role of Hsp60 in regulation of complex IV activity by Hsp60 gene KD. Results from this experiment demonstrate that maintenance of proper levels of Hsp60 is crucial to regulation of complex IV activity but not to that of complexes I and II, consistent with previous observations, including ours. For example, we demonstrated previously specific down-regulation of both complex IV activity and Hsp60 levels in PINK1 null cells (6). Moreover, in an Alzheimer's disease model, β-amyloid was found to selectively inhibit complex IV activity, which could be specifically restored by Hsp60 (39). Hsp60 also increased complex IV activity in a cardiac myocyte model of ischemia/reperfusion injury (40). The specific regulatory role of Hsp60 on complex IV activity was further confirmed by both up-regulation or down-regulation of Hsp60 via the manipulation of its upstream regulators LRPPRC and Hsp90. For example, overexpression of LRPPRC resulted in up-regulation of Hsp60 expression and complex IV activity, whereas the pharmacological inhibition of Hsp90 resulted in down-regulation of Hsp60 expression and complex IV activity. However, neither LRPPRC nor Hsp90 affects the expression of the other, suggesting independent roles in regulation of Hsp60 (Figs. 3A and 4B). Furthermore, each upstream regulator appears to increase the stability of Hsp60 in mitochondria. For instance, the Hsp90 chaperone, which has been shown to play a role in mitochondrial import via the TOM70 receptor (35), is also associated with an increase in mitochondrial accumulation of Hsp60 (Fig. 5C). In the case of LRPPRC, Hsp60 levels were also increased by its overexpression (Fig. 3A). LRPPRC was recently characterized as an essential regulator of mitochondrial mRNA stability that modulates ETC enzyme activity via the posttranscriptional regulation (41), suggesting a potential regulatory role for LRPPRC in Hsp60 mRNA stability. The most consistent effect of mitochondrial ETC enzymes in Alzheimer's disease is the deficiency of complex IV without complex I and II deficits (42). In contrast, the complex IV deficits in PINK1 null condition are accompanied by the simultaneous decreases in complex I and II activity. Thus, the PINK1 null mutation appears to cause the broader defects in mitochondria, whereas Aβ in Alzheimer's disease leads to a rather specific impairment in complex IV (43), probably via binding with heme-a in complex IV (44). However, it is of interest to see whether the same Hsp90/LRPPRC-Hsp60 signaling axis is another target of Aβ in Alzheimer's disease. In summary, we propose that reduced Hsp60 levels lead to decreased complex IV activity and that increased Hsp60 levels are necessary for a functional recovery from the complex IV deficit present in the PINK1 null condition. Thus, the optimal functional recovery of complex IV activity requires the simultaneous up-regulation of both Hsp60 and its upstream regulators, Hsp90 and/or LRPPRC, which may be important to maintain increased stability of Hsp60 in mitochondria.
Although dysfunctional mitochondria having long been implicated in the pathogenesis of PD, few mitochondria-targeted therapies are available, largely because of the lack of appropriate phenotypic models and specific druggable targets for mitochondrial deficits (28). In this study, we have identified a natural product, ginsenoside Re, as an agent that can mediate recovery of complex IV activity in the PINK1 null dopaminergic neuronal cell model (6). Notably, treatment of ginsenoside Re at the optimum concentration (3 μm) resulted in a significant functional recovery of complex IV activity, which was associated with up-regulation of the Hsp60 upstream regulators Hsp90 and LRPPRC and, consequently, Hsp60 in PINK1 null cells (Fig. 6). The pharmacological effect of ginsenoside Re was a PINK1 null-specific phenomenon, as ginsenoside Re moderately inhibited complex IV activity and decreased LRPPRC and Hsp90 expression in wild-type dopaminergic neuronal cells. Importantly, we found that NO signaling was required for the pharmacological action of ginsenoside Re and that treatment with an NO donor (SNP) at the optimal concentration (20 nm), but not at high concentrations (50–100 nm), reproduced the same effects as ginsenoside Re, restoring complex IV activity and up-regulating Hsp90, LRPPRC, and Hsp60 in PINK1 null cells. However, in wild-type cells, the same dose of SNP (20 nm) moderately inhibited complex IV activity, which could be due to the toxic damaging effect and altered NO signaling by the presence of excessive NO. In fact, NO contains both antioxidant and prooxidant activities, which may affect mitochondrial function and neuronal survival. For example, NO can regulate the mitochondrial reactive oxygen species detoxification systems through PGC-1α for cell survival (45). Several NO donors also exert neuroprotection against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced dopaminergic toxicity by scavenging hydroxyl free radicals (46). In contrast, NO and its oxidation products, such as peroxinitrite, are known to contribute to mitochondrial damages and neurodegeneration in PD patients (47, 48). Thus, optimized ginsenoside Re and SNP treatments may restore NO signaling to appropriate levels to promote recovery from a complex IV deficit in PINK1 null cells, likely caused by dysregulation of NO signaling resulting from loss of PINK1.
In conclusion, for the first time, we have reported a novel regulatory role for PINK1 in the maintenance of mitochondrial complex IV activity, as depicted in a schematic diagram shown in Fig. 9. Two distinct Hsp60 upstream regulators, LRPPRC and Hsp90, that interact with PINK1 are significantly down-regulated in dopaminergic neuronal cells carrying a PINK1 null mutation, suggesting a novel role for PINK1 in regulation of LRPPRC and Hsp90 levels. Furthermore, the defective PINK1-LRPPRC/Hsp90-Hsp60-complex IV signaling axis in PINK1 null neuronal cells was rescued by restoration of optimal NO signaling by ginsenoside Re. This evidence supports the feasibility of using ginsenoside Re to promote the survival of ailing dopaminergic neurons in some types of familial PD. Further mechanistic studies underlying dysregulated NO signaling resulting from loss of PINK1 may help to develop disease-modifying therapies to specifically treat mitochondrial dysfunctions in PD.
FIGURE 9.
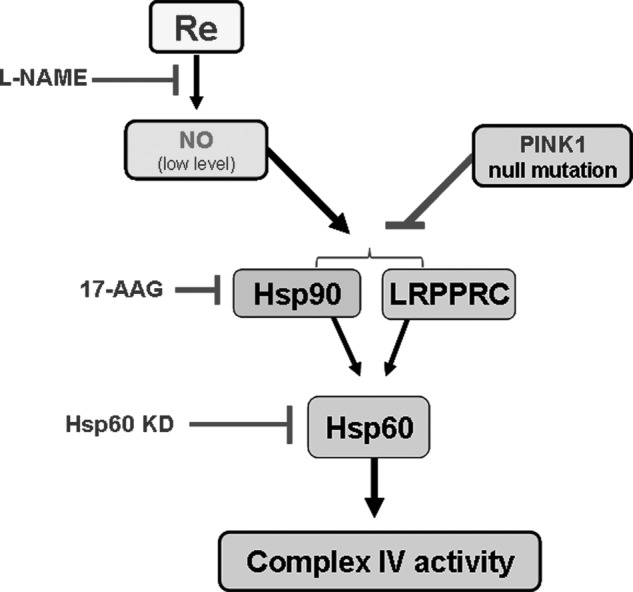
A schematic diagram describing a model for the defective PINK1-LRPPRC/Hsp90-Hsp60-complex IV signaling axis in PINK1 null cells. The PINK1 null mutation causes a severe deficit in complex IV activity as a result of down-regulation of the Hsp60 chaperone and its upstream regulators LRPPRC and Hsp90. Consistent with this model, Hsp60 KD and inhibition of Hsp90 by 17-AAG resulted in similar decreases in complex IV activity. In contrast, overexpression of the PINK1-interacting factor LRPPRC and coexpression of Hsp90 with Hsp60 increased complex IV activity. Decreases in complex IV activity could be rescued by treatment with the optimal concentration of ginsenoside Re, which was mediated through activation of NO signaling and, consequently, up-regulation of LRPPRC and Hsp90.
This work was supported in part by National Research Foundation Grant 2010-0009233 and National Core Research Center Program Grant 2012-0000952, Republic of Korea.
- PD
- Parkinson disease
- COX
- cytochrome c oxidase
- LRPPRC
- leucine-rich pentatricopeptide repeat-containing
- NO
- nitrous oxide
- KD
- knockdown
- HSCCC-ELSD
- high-speed counter-current chromatography coupled with an evaporative light-scattering detector
- 17-AAG
- 17-allylamino-17-demethoxygeldanamycin.
REFERENCES
- 1. Schapira A. H. (2012) Targeting mitochondria for neuroprotection in Parkinson's disease. Antioxid. Redox Signal. 16, 965–973 [DOI] [PubMed] [Google Scholar]
- 2. Valente E. M., Abou-Sleiman P. M., Caputo V., Muqit M. M., Harvey K., Gispert S., Ali Z., Del Turco D., Bentivoglio A. R., Healy D. G., Albanese A., Nussbaum R., González-Maldonado R., Deller T., Salvi S., Cortelli P., Gilks W. P., Latchman D. S., Harvey R. J., Dallapiccola B., Auburger G., Wood N. W. (2004) Hereditary early-onset Parkinson's disease caused by mutations in PINK1. Science 304, 1158–1160 [DOI] [PubMed] [Google Scholar]
- 3. Sim C. H., Lio D. S., Mok S. S., Masters C. L., Hill A. F., Culvenor J. G., Cheng H. C. (2006) C-terminal truncation and Parkinson's disease-associated mutations down-regulate the protein serine/threonine kinase activity of PTEN-induced kinase-1. Hum. Mol. Genet. 15, 3251–3262 [DOI] [PubMed] [Google Scholar]
- 4. Narendra D., Tanaka A., Suen D. F., Youle R. J. (2008) Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 183, 795–803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yang Y., Ouyang Y., Yang L., Beal M. F., McQuibban A., Vogel H., Lu B. (2008) Pink1 regulates mitochondrial dynamics through interaction with the fission/fusion machinery. Proc. Natl. Acad. Sci. U.S.A. 105, 7070–7075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Shim J. H., Yoon S. H., Kim K. H., Han J. Y., Ha J. Y., Hyun D. H., Paek S. H., Kang U. J., Zhuang X., Son J. H. (2011) The antioxidant Trolox helps recovery from the familial Parkinson's disease-specific mitochondrial deficits caused by PINK1- and DJ-1-deficiency in dopaminergic neuronal cells. Mitochondrion 11, 707–715 [DOI] [PubMed] [Google Scholar]
- 7. Gegg M. E., Cooper J. M., Schapira A. H., Taanman J. W. (2009) Silencing of PINK1 expression affects mitochondrial DNA and oxidative phosphorylation in dopaminergic cells. PloS ONE 4, e4756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Liu W., Acín-Peréz R., Geghman K. D., Manfredi G., Lu B., Li C. (2011) Pink1 regulates the oxidative phosphorylation machinery via mitochondrial fission. Proc. Natl. Acad. Sci. U.S.A. 108, 12920–12924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Barrientos A., Gouget K., Horn D., Soto I. C., Fontanesi F. (2009) Suppression mechanisms of COX assembly defects in yeast and human. Insights into the COX assembly process. Biochim. Biophys. Acta 1793, 97–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Diaz F. (2010) Cytochrome c oxidase deficiency. Patients and animal models. Biochim. Biophys. Acta 1802, 100–110 [DOI] [PubMed] [Google Scholar]
- 11. Stiburek L., Zeman J. (2010) Assembly factors and ATP-dependent proteases in cytochrome c oxidase biogenesis. Biochim. Biophys. Acta 1797, 1149–1158 [DOI] [PubMed] [Google Scholar]
- 12. Mootha V. K., Lepage P., Miller K., Bunkenborg J., Reich M., Hjerrild M., Delmonte T., Villeneuve A., Sladek R., Xu F., Mitchell G. A., Morin C., Mann M., Hudson T. J., Robinson B., Rioux J. D., Lander E. S. (2003) Identification of a gene causing human cytochrome c oxidase deficiency by integrative genomics. Proc. Natl. Acad. Sci. U.S.A. 100, 605–610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Péquignot M. O., Dey R., Zeviani M., Tiranti V., Godinot C., Poyau A., Sue C., Di Mauro S., Abitbol M., Marsac C. (2001) Mutations in the SURF1 gene associated with Leigh syndrome and cytochrome c oxidase deficiency. Hum. Mutat. 17, 374–381 [DOI] [PubMed] [Google Scholar]
- 14. Zhu Z., Yao J., Johns T., Fu K., De Bie I., Macmillan C., Cuthbert A. P., Newbold R. F., Wang J., Chevrette M., Brown G. K., Brown R. M., Shoubridge E. A. (1998) SURF1, encoding a factor involved in the biogenesis of cytochrome c oxidase, is mutated in Leigh syndrome. Nat. Genet. 20, 337–343 [DOI] [PubMed] [Google Scholar]
- 15. Coenen M. J., van den Heuvel L. P., Ugalde C., Ten Brinke M., Nijtmans L. G., Trijbels F. J., Beblo S., Maier E. M., Muntau A. C., Smeitink J. A. (2004) Cytochrome c oxidase biogenesis in a patient with a mutation in COX10 gene. Ann. Neurol. 56, 560–564 [DOI] [PubMed] [Google Scholar]
- 16. Papadopoulou L. C., Sue C. M., Davidson M. M., Tanji K., Nishino I., Sadlock J. E., Krishna S., Walker W., Selby J., Glerum D. M., Coster R. V., Lyon G., Scalais E., Lebel R., Kaplan P., Shanske S., De Vivo D. C., Bonilla E., Hirano M., DiMauro S., Schon E. A. (1999) Fatal infantile cardioencephalomyopathy with COX deficiency and mutations in SCO2, a COX assembly gene. Nat. Genet. 23, 333–337 [DOI] [PubMed] [Google Scholar]
- 17. Stiburek L., Vesela K., Hansikova H., Hulkova H., Zeman J. (2009) Loss of function of Sco1 and its interaction with cytochrome c oxidase. Am. J. Physiol. Cell Physiol. 296, C1218–1226 [DOI] [PubMed] [Google Scholar]
- 18. Brown G. C. (2010) Nitric oxide and neuronal death. Nitric Oxide 23, 153–165 [DOI] [PubMed] [Google Scholar]
- 19. Hall C. N., Garthwaite J. (2009) What is the real physiological NO concentration in vivo? Nitric Oxide 21, 92–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lundberg J. O., Farkas-Szallasi T., Weitzberg E., Rinder J., Lidholm J., Anggåard A., Hökfelt T., Lundberg J. M., Alving K. (1995) High nitric oxide production in human paranasal sinuses. Nat. Med. 1, 370–373 [DOI] [PubMed] [Google Scholar]
- 21. Drapier J. C., Hibbs J. B., Jr. (1988) Differentiation of murine macrophages to express nonspecific cytotoxicity for tumor cells results in L-arginine-dependent inhibition of mitochondrial iron-sulfur enzymes in the macrophage effector cells. J. Immunol. 140, 2829–2838 [PubMed] [Google Scholar]
- 22. Guix F. X., Uribesalgo I., Coma M., Muñoz F. J. (2005) The physiology and pathophysiology of nitric oxide in the brain. Prog. Neurobiol. 76, 126–152 [DOI] [PubMed] [Google Scholar]
- 23. Lancaster J. R., Jr., Hibbs J. B., Jr. (1990) EPR demonstration of iron-nitrosyl complex formation by cytotoxic activated macrophages. Proc. Natl. Acad. Sci. U.S.A. 87, 1223–1227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Brown G. C., Bolaños J. P., Heales S. J., Clark J. B. (1995) Nitric oxide produced by activated astrocytes rapidly and reversibly inhibits cellular respiration. Neurosci. Lett. 193, 201–204 [DOI] [PubMed] [Google Scholar]
- 25. Shen W., Hintze T. H., Wolin M. S. (1995) Nitric oxide. An important signaling mechanism between vascular endothelium and parenchymal cells in the regulation of oxygen consumption. Circulation 92, 3505–3512 [DOI] [PubMed] [Google Scholar]
- 26. Nisoli E., Clementi E., Paolucci C., Cozzi V., Tonello C., Sciorati C., Bracale R., Valerio A., Francolini M., Moncada S., Carruba M. O. (2003) Mitochondrial biogenesis in mammals. The role of endogenous nitric oxide. Science 299, 896–899 [DOI] [PubMed] [Google Scholar]
- 27. Nisoli E., Falcone S., Tonello C., Cozzi V., Palomba L., Fiorani M., Pisconti A., Brunelli S., Cardile A., Francolini M., Cantoni O., Carruba M. O., Moncada S., Clementi E. (2004) Mitochondrial biogenesis by NO yields functionally active mitochondria in mammals. Proc. Natl. Acad. Sci. U.S.A. 101, 16507–16512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Meissner W. G., Frasier M., Gasser T., Goetz C. G., Lozano A., Piccini P., Obeso J. A., Rascol O., Schapira A., Voon V., Weiner D. M., Tison F., Bezard E. (2011) Priorities in Parkinson's disease research. Nat. Rev. Drug Discov. 10, 377–393 [DOI] [PubMed] [Google Scholar]
- 29. Leung K. W., Yung K. K., Mak N. K., Chan Y. S., Fan T. P., Wong R. N. (2007) Neuroprotective effects of ginsenoside-Rg1 in primary nigral neurons against rotenone toxicity. Neuropharmacology 52, 827–835 [DOI] [PubMed] [Google Scholar]
- 30. Nah S. Y., Kim D. H., Rhim H. (2007) Ginsenosides. Are any of them candidates for drugs acting on the central nervous system? CNS Drug Rev. 13, 381–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tian J., Zhang S., Li G., Liu Z., Xu B. (2009) 20(S)-ginsenoside Rg3, a neuroprotective agent, inhibits mitochondrial permeability transition pores in rat brain. Phytother. Res. 23, 486–491 [DOI] [PubMed] [Google Scholar]
- 32. Ye R., Zhang X., Kong X., Han J., Yang Q., Zhang Y., Chen Y., Li P., Liu J., Shi M., Xiong L., Zhao G. (2011) Ginsenoside Rd attenuates mitochondrial dysfunction and sequential apoptosis after transient focal ischemia. Neuroscience 178, 169–180 [DOI] [PubMed] [Google Scholar]
- 33. Son J. H., Chun H. S., Joh T. H., Cho S., Conti B., Lee J. W. (1999) Neuroprotection and neuronal differentiation studies using substantia nigra dopaminergic cells derived from transgenic mouse embryos. J. Neurosci. 19, 10–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Shehzad O., Ha I. J., Park Y., Ha Y. W., Kim Y. S. (2011) Development of a rapid and convenient method to separate eight ginsenosides from Panax ginseng by high-speed counter-current chromatography coupled with evaporative light scattering detection. J. Sep. Sci. 34, 1116–1122 [DOI] [PubMed] [Google Scholar]
- 35. Young J. C., Hoogenraad N. J., Hartl F. U. (2003) Molecular chaperones Hsp90 and Hsp70 deliver preproteins to the mitochondrial import receptor Tom70. Cell 112, 41–50 [DOI] [PubMed] [Google Scholar]
- 36. Chen X., Salwinski S., Lee T. J. (1997) Extracts of Ginkgo biloba and ginsenosides exert cerebral vasorelaxation via a nitric oxide pathway. Clin. Exp. Pharmacol. Physiol. 24, 958–959 [DOI] [PubMed] [Google Scholar]
- 37. Zhang H., Zhou Q., Li X., Zhao W., Wang Y., Liu H., Li N. (2007) Ginsenoside Re promotes human sperm capacitation through nitric oxide-dependent pathway. Mol. Reprod. Dev. 74, 497–501 [DOI] [PubMed] [Google Scholar]
- 38. Rakovic A., Grunewald A., Voges L., Hofmann S., Orolicki S., Lohmann K., Klein C. (2011) PINK1-interacting proteins. Proteomic analysis of overexpressed PINK1. Parkinsons Dis., 153979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Veereshwarayya V., Kumar P., Rosen K. M., Mestril R., Querfurth H. W. (2006) Differential effects of mitochondrial heat shock protein 60 and related molecular chaperones to prevent intracellular β-amyloid-induced inhibition of complex IV and limit apoptosis. J. Biol. Chem. 281, 29468–29478 [DOI] [PubMed] [Google Scholar]
- 40. Lin K. M., Lin B., Lian I. Y., Mestril R., Scheffler I. E., Dillmann W. H. (2001) Combined and individual mitochondrial HSP60 and HSP10 expression in cardiac myocytes protects mitochondrial function and prevents apoptotic cell deaths induced by simulated ischemia-reoxygenation. Circulation 103, 1787–1792 [DOI] [PubMed] [Google Scholar]
- 41. Ruzzenente B., Metodiev M. D., Wredenberg A., Bratic A., Park C. B., Cámara Y., Milenkovic D., Zickermann V., Wibom R., Hultenby K., Erdjument-Bromage H., Tempst P., Brandt U., Stewart J. B., Gustafsson C. M., Larsson N. G. (2012) LRPPRC is necessary for polyadenylation and coordination of translation of mitochondrial mRNAs. EMBO J. 31, 443–456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kish S. J., Bergeron C., Rajput A., Dozic S., Mastrogiacomo F., Chang L. J., Wilson J. M., DiStefano L. M., Nobrega J. N. (1992) Brain cytochrome oxidase in Alzheimer's disease. J. Neurochem. 59, 776–779 [DOI] [PubMed] [Google Scholar]
- 43. Canevari L., Clark J. B., Bates T. E. (1999) β-Amyloid fragment 25–35 selectively decreases complex IV activity in isolated mitochondria. FEBS Lett. 457, 131–134 [DOI] [PubMed] [Google Scholar]
- 44. Atamna H., Boyle K. (2006) Amyloid-beta peptide binds with heme to form a peroxidase. Relationship to the cytopathologies of Alzheimer's disease. Proc. Natl. Acad. Sci. U.S.A. 103, 3381–3386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Borniquel S., Valle I., Cadenas S., Lamas S., Monsalve M. (2006) Nitric oxide regulates mitochondrial oxidative stress protection via the transcriptional coactivator PGC-1α. FASEB J. 20, 1889–1891 [DOI] [PubMed] [Google Scholar]
- 46. Mohanakumar K. P., Thomas B., Sharma S. M., Muralikrishnan D., Chowdhury R., Chiueh C. C. (2002) Nitric oxide. An antioxidant and neuroprotector. Ann. N.Y. Acad. Sci. 962, 389–401 [DOI] [PubMed] [Google Scholar]
- 47. Hunot S., Boissière F., Faucheux B., Brugg B., Mouatt-Prigent A., Agid Y., Hirsch E. C. (1996) Nitric oxide synthase and neuronal vulnerability in Parkinson's disease. Neuroscience 72, 355–363 [DOI] [PubMed] [Google Scholar]
- 48. Radi R., Cassina A., Hodara R. (2002) Nitric oxide and peroxynitrite interactions with mitochondria. Biol. Chem. 383, 401–409 [DOI] [PubMed] [Google Scholar]