

Background: C-type lectin receptor signaling is involved in anti-fungal immunity. However, its signaling pathway is not fully characterized.
Results: We demonstrate that TRAF6 and TAK1 deficiency impair C. albicans induced NF-κB and MAPK activation and regulate inflammatory cytokine production.
Conclusion: TRAF6 and TAK1 are key components in the C-type lectin receptor signaling pathways and mediate immune responses against C. albicans.
Significance: This study identifies key mediators of the C-type lectin receptor signaling pathways.
Keywords: Candida albicans, Innate Immunity, Lectin, NF-kappa B (NF-KB), Pattern Recognition Receptor
Abstract
Tumor necrosis factor receptor-associated factor 6 (TRAF6) and TGFβ-activated kinase 1 (TAK1) are considered as key intermediates in Toll-like receptor (TLR) signaling. However, the role of TRAF6 and TAK1 in C-type lectin receptors (CLRs) in response to fungal infection has not been studied. In this study, we have utilized macrophages derived from TRAF6 knock-out mice and myeloid-specific TAK1-deficient mice and determined the role of TRAF6 and TAK1 in CLR-induced signal transduction events. We demonstrate that TRAF6 and TAK1 are required for NF-κB and JNK activation, and expression of proinflammatory cytokines in response to Candida albicans infection. Our results highlight TRAF6 and TAK1 as key components in the signaling cascade downstream of C-type lectin receptors and as critical mediators of the anti-fungal immune response. Therefore, our studies provide a mechanistic understanding of the host immune response to C. albicans, which has a significant impact for the development of anti-fungal therapeutics and in understanding risk-factors and determining susceptibility to C. albicans infection.
Introduction
Candida albicans is a dimorphic fungus that can transform from yeast to hyphal forms, causing lethal disseminated infections in immunocompromised patients. Host innate immune responses to fungi rely on pattern recognition receptors that recognize conserved groups of molecules called pathogen-associated molecular patterns, which are found in the fungal cell wall. Carbohydrates are major cell wall components in C. albicans. Therefore, host innate immune cells sense fungal pathogens mainly by engaging the C-type lectin receptors (CLRs).2 The CLR Dectin-1 can recognize carbohydrates such as β-glucan on the surface of the yeast form of C. albicans (1, 2), whereas Dectin-2 detects mannan moiety on the surface of C. albicans hyphae (3, 4). CLR signaling is initiated by the phosphorylation of immunoreceptor tyrosine-based activation motif (ITAM) like structures of themselves or on the adaptor proteins associated with CLRs, and the phosphorylation of immunoreceptor tyrosine-based activation motifs (ITAMs) leads to the activation of spleen tyrosine kinase (Syk) (5–7). Syk-coupled CLRs, including Dectin-1, Dectin-2, and Mincle, are involved in innate immune responses against fungal infection (3, 4, 8–10). The activation of Syk leads to the phosphorylation of phospholipase Cγ2 (11–13), which triggers the activation of downstream signaling pathways, including NF-κB and JNK.
Previous studies suggest that the adaptor protein CARD9 is an important signaling component downstream of Syk mediating NF-κB activation in the Dectin-1 and Dectin-2 signaling pathway (14–17). NF-κB activation is controlled by the IκB kinase (IKK) complex that phosphorylates IκBα and triggers the proteasome-mediated degradation of IκBα. The degradation of IκBα releases NF-κB from cytoplasmic compartment, and allows it to translocate into nucleus to regulate the expression of a variety of genes. Activation of IKK complex is dependent on both phosphorylation and ubiquitination of the IKK complex. Our previous studies indicate that CARD9 is not involved in regulating signal-induced phosphorylation of IKKα/β. Instead, it is selectively involved in ubiquitination of NEMO subunit in the IKK complex (16). However, how CARD9 is involved in the regulation of IKK ubiquitination is not fully determined.
TRAF6 is an E3 ubiquitin ligase that functions as a key regulator of multiple signaling pathways, such as MAPK, NF-κB, and PI3K/Akt, in response to microbial products and cytokines (19–24). However, it is not clear whether TRAF6 is involved in CLR-induced signaling in response to fungal stimulation. Earlier studies suggest that TRAF6 mediates CARMA1-Bcl10-MALT1 complex-induced NF-κB activation by regulating the ubiquitination of the IKK complex in lymphocytes (21, 25). Similar to CARMA1, CARD9 forms a complex with Bcl10-MALT1 in myeloid cells (26). Therefore, we hypothesized that TRAF6 might also be involved in C-type lectin receptor-induced NF-κB activation downstream of the CARD9-Bcl10-MALT1 complex.
It has been shown that TRAF6 interacts with TAK1 through the TAK1-associating protein TAB2 in response to various stimuli including TLR ligands, IL-1, and RANK ligand (27–29). TAK1 (transforming growth factor-β activated kinase-1) is a member of the mitogen-activated protein kinase kinase kinase family, which plays a pivotal role in adaptive and innate immune signaling (30, 31). TAK1 is activated by a diverse range of stimuli, such as ligands for Toll-like receptors (TLRs), tumor necrosis factor receptor (TNFR), and IL-1 receptor, which can lead to the activation of NF-κB and MAPK signaling pathways (30, 32, 33). Because TAK1 knock-out mice are embryonic lethal (33, 34), the function of TAK1 in myeloid cells has not been well characterized, and its role in the CLR signaling pathway has not been determined. Therefore, we addressed the functional importance of TAK1 and TRAF6 in NF-κB activation induced by the CLR signaling pathway in response to C. albicans, and we demonstrate that TRAF6 and TAK1 are required for CLR-induced NF-κB activation and play critical roles in anti-fungal innate immune responses.
EXPERIMENTAL PROCEDURES
Reagents and Antibodies
Antibodies against phospho-p38, p38, phospho-JNK, JNK, phospho-ERK, phospho-IKKα/β (Ser-176/180), phospho-Syk were purchased from Cell Signaling Technology; antibodies against ERK, IKKβ, ubiquitin, Bcl10, TAK1, and IκBα were from Santa Cruz Biotechnology. Antibodies against TRAF6 were purchased from Lifespan Biosciences for immunoprecipitation and Santa Cruz Biotechnology for Western blot. Zymosan was purchased from Sigma-Aldrich. N-Ethylmaleimide was purchased from Sigma-Aldrich. Fluorescence-conjugated monoclonal antibodies CD11b and F4/80 were purchased from BD Pharmingen or eBioscience. TNF-α, IL-10, IL-6, IL-1β, and IL-12p40 ELISA Ready-SET-GO kits were purchased from eBioscience.
Mice
The lysozyme M-Cre (LysM-Cre) knock-in mice (35) were purchased from The Jackson Laboratory (Bar Harbor, ME). Mice carrying the floxed allele of TAK1 (TAK1F/F) and the null allele of TAK1 (TAK1+/−) have been described previously (36) and were backcrossed to C57BL/6 at least six to nine generations at the time of the experiments. TAK1F/FLysM+/+ were crossed with TAK1+/+LysMcre/cre to generate offspring lacking TAK1 in the myeloid lineage (LysM-Cre- TAK1F/F) or heterozygote (TAK1F/+). LysM-Cre-TAK1F/F mice were also generated on a TNFR1-null background by mating with TNFR1-deficient mice obtained from The Jackson Laboratory. TRAF6 heterozygote mice (TRAF6+/−) were maintained on an ICR background, and TRAF6 heterozygote pairs were intercrossed to generate TRAF6-deficient mice (TRAF6−/−) that were sacrificed around day 12 for experiments. Mice were genotyped by ear clips and standard polymerase chain reaction. (Sequences are available upon request.) All mice were housed in a pathogen-free animal facility. All mouse experiments were approved by MD Anderson Cancer Center's institutional animal care and use committee. Mice were cared for in accordance with guidelines set forth by the American Association for Accreditation of Laboratory Animal Care and the U.S. Public Health Service Policy on Humane Care and Use of Laboratory Animals.
Bone Marrow-derived Macrophage (BMDM) Preparation
Primary cultures of BMDMs were prepared as described previously (13, 16). Briefly, bone marrow cells were harvested from the femurs and tibias of mice. Erythrocytes were removed from bone marrow samples by subjecting the samples to hypotonic solution. Cells were cultured for 7 days in DMEM containing 20% FBS, 55 μm β-mercaptoethanol, streptomycin (100 μg/ml), penicillin (100 units/ml), and 30% conditioned media from L929 cells expressing macrophage colony-stimulating factor. Non-adherent cells were removed and cells were passaged every 3 days. After 1 week of cultures, flow cytometry analysis indicated that the harvested cell population contained 86–95% CD11b+F4/80+cells as assessed.
Candida albicans Preparation
C. albicans (strain SC5314) was kindly provided by Dr. Michael C. Lorenz (Department of Microbiology and Molecular Genetics, University of Texas Medical School at Houston). A single colony of C. albicans was grown overnight at 30 °C in yeast peptone dextrose media. The cells were washed three times with PBS and then used as live yeast. To generate hyphae, the washed yeast cells were resuspended in RPMI with 10% FCS and grown at 37 °C for 3 h. The hyphae were then used for live stimulations. For heat-inactivated yeast, yeast cells were heated at 65 °C for 1 h.
Western Blot and Immunoprecipitation
Cells were lysed in a buffer containing 50 mm HEPES (pH 7.4), 250 mm NaCl, 1% Nonidet P-40, 1 mm EDTA, 1 mm Na3VO4, 1 mm NaF, 1 mm phenylmethylsulfonyl fluoride, 1 mm dithiothreitol, and a protease inhibitor mixture (Roche Diagnostics). For ubiquitination studies, 5 mm N-ethylmaleimide was added to the lysis buffer. The cell lysates were subjected to SDS-PAGE and Western blotted or immunoprecipitated. The immunoprecipitates were washed with lysis buffer. Where indicated, immunoprecipitates were washed with lysis buffer containing 1 m urea. Proteins were eluted with 2× SDS loading buffer. After boiling for 10 min, the samples were fractionated on 10% SDS-PAGE and transferred to nitrocellulose membranes. Immunoblots were incubated with specific primary antibodies followed by horseradish peroxidase-conjugated secondary antibodies and were developed by the enhanced chemiluminescence method according to the manufacturer's instructions (Pierce).
EMSA
Nuclear proteins were extracted from 7 × 106 cells as described previously (13). The resultant nuclear protein (5 μg) was incubated with 32P-labeled NF-κB or Oct-1 probe (Promega) for 15 min at room temperature and then subjected to PAGE and exposed to x-ray films.
RESULTS
TRAF6 Is Crucial for C. albicans Stimulation-induced NF-κB and JNK Activation
In TLR signaling, the adaptor molecule MyD88 activates TRAF6, which has E3 ubiquitin ligase activity. We were interested in determining whether TRAF6 is involved in CLR signaling in response to the clinically important pathogen C. albicans. To address this question, we first examined the stimulation-induced ubiquitination of TRAF6 following the stimulation of mouse bone marrow-derived macrophages with C. albicans hyphae, which are strong agonists for Dectin-2, a CLR that is coupled to the FcγR signaling complex (4). We found that immunoprecipitated TRAF6 was inducibly ubiquitinated following the stimulation (Fig. 1A) and formed a complex with Bcl10 and TAK1 (Fig. 1A). To determine whether the ubiquitination we observed is indeed TRAF6 ubiquitination or the ubiquitination of TRAF6-associated proteins, we used stringent washing conditions to disrupt any protein-protein interactions. We found that TRAF6-specific ubiquitination was observed upon C. albicans hyphae stimulation (Fig. 1B), suggesting that TRAF6 is functionally linked to Dectin-2 signaling.
FIGURE 1.

TRAF6 is ubiquitinated upon C. albicans stimulation and forms a complex with BCL10 and TAK1. WT BMDMs were stimulated with C. albicans hyphae (MOI of 1) for the indicated time points, and the cell lysates were immunoprecipitated with TRAF6 antibody-conjugated agarose. The immunoprecipitates were washed with lysis buffer (A) or lysis buffer containing 1 m urea (B). The immunoprecipitates and cell lysates were probed with the indicated antibodies. IP, immunoprecipitation.
We next evaluated NF-κB activation in TRAF6-deficient macrophages in response to this stimulation and found that the DNA-binding activity of NF-κB was significantly diminished in TRAF6-deficient macrophages (Fig. 2A), and the degradation of IκB was also defective in these cells (Fig. 2B). The regulation of the IKK complex requires the phosphorylation of IKKα/β subunits along with the K63-linked ubiquitination of IKKγ, leading to the full activation of the IKK complex (37, 38). Interestingly, we found that there was no detectable difference in the levels of IKKα/β phosphorylation between TRAF6-deficient and control mice (Fig. 2C), which suggests that TRAF6 regulates the ubiquitination but not the phosphorylation of IKK complex. This is consistent with the above observation that TRAF6 is associated with the Bcl10-containing complex (Fig. 1A) and that the Bcl10-CARD9 complex regulates IKK ubiquitination but not IKKα/β phosphorylation (16).
FIGURE 2.
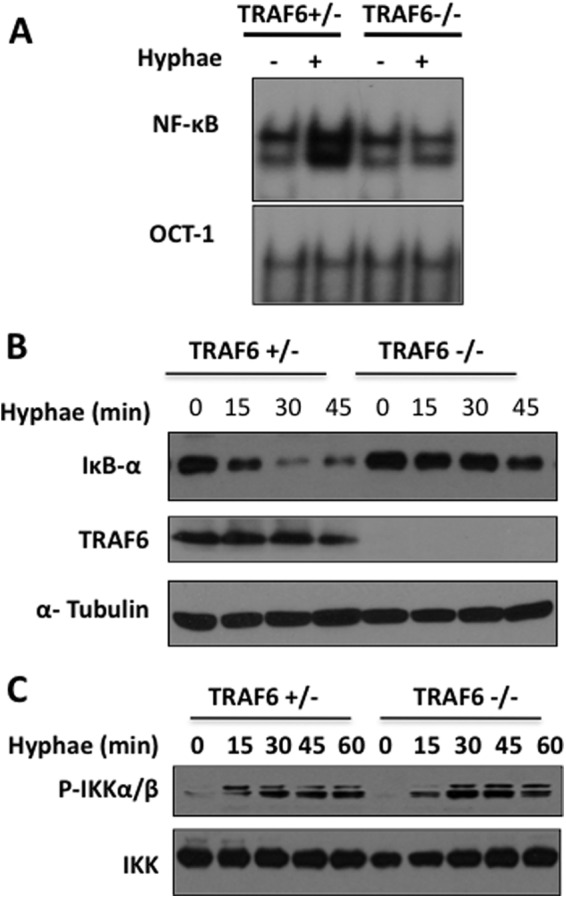
NF-κB activation induced by C. albicans hyphae is TRAF6-dependent. A, WT and TRAF6-deficient BMDMs were either untreated or treated with C. albicans hyphae (MOI of 1) for 90 min. The nuclear extracts were prepared from these cells and then subjected to the electrophoretic mobility shift assay using 32P-labeled NF-κB or OCT-1 probe. B, WT and TRAF6-deficient BMDMs were stimulated with C. albicans hyphae for the indicated time points, and IκB-α was examined using the indicated antibody. C, BMDMs were stimulated with C. albicans hyphae for the indicated time points. Total cell lysates were prepared from these cells and then subjected to Western blotting.
We next examined whether TRAF6-deficiency might affect MAPK activation. In response to hyphae, JNK but not ERK activation was partially impaired in TRAF6-deficient macrophages (Fig. 3A). Because NF-κB and JNK activation control the expression of various proinflammatory cytokines, we next examined the expression of IL-6 and IL-12p40 in wild-type and TRAF6-deficient BMDM following C. albicans stimulation. Although C. albicans stimulation induced robust production of proinflammatory cytokines IL-6 and IL-12 in wild-type macrophages, the production of these cytokines was significantly defective in the absence of TRAF6 (Fig. 3B).
FIGURE 3.
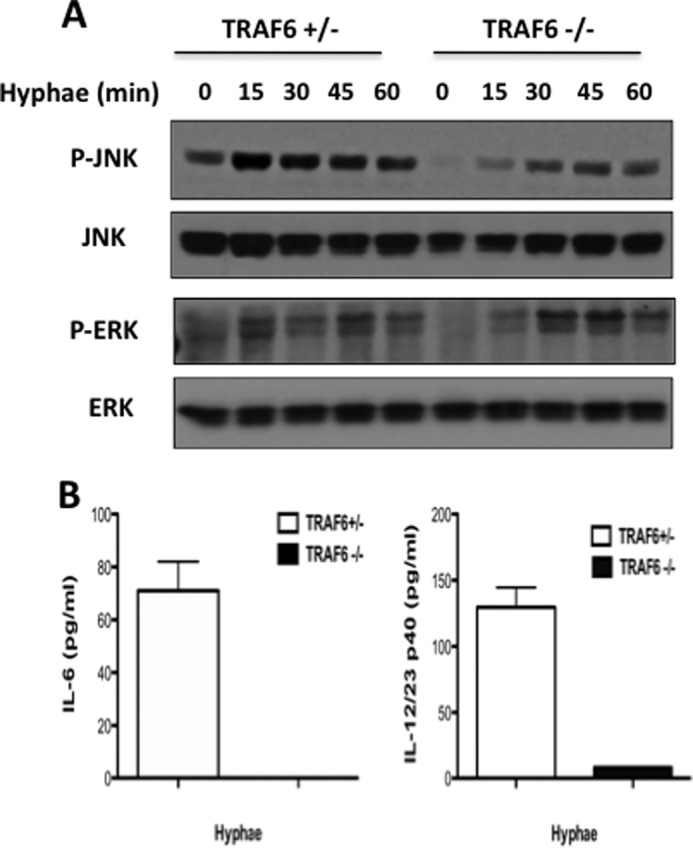
TRAF6 contributes to JNK but not IKKα/β phosphorylation. WT and TRAF6-deficient BMDMs were either untreated or treated with C. albicans hyphae (MOI of 1) for the indicated time points, and phosphorylation was examined using the indicated antibodies. B, BMDMs from heterozygous (white bars) and TRAF6-deficient (black bars) mice were stimulated overnight with C. albicans hyphae (MOI of 1). ELISA was used to measure the level of cytokines in these cultured media. The data are the means ± S.D. of triplicate wells and are representative of three independent experiments.
TAK1 Is Essential for NF-κB, JNK, and p38 Activation in Response to C. albicans
Because we found that TRAF6 is associated with TAK1 (Fig. 1A), we next determined the role of TAK1 in CLR signaling-induced NF-κB activation following fungal infection. To address this question, we obtained myeloid-specific TAK1-deficient mice by crossing TAK1-conditional knock-out mice (36) with LysM-Cre transgenic mice. However, we found that bone marrow cells from the myeloid-specific TAK1-deficient mice could not develop into macrophages (data not shown). To obtain TAK1-deficient macrophages, we further crossed myeloid-specific (LysM-Cre) TAK1-deficient mice with TNFR1-deficient mice. We were able to generate macrophages from bone marrow cells harvested from the resultant TAK1/TNFR1 double knock-out mice (Fig. 4A). Therefore, these cells provided us a genetic tool for determining the function of TAK1 in macrophages.
FIGURE 4.

NF-κB and MAPK activation induced by C. albicans hyphae and yeast are TAK1-dependent. A, flow cytometric analysis of BMDMs cultured with macrophage colony-stimulating factor (M-CSF) containing medium from LysMCre/+, TAK1+/+LysMcre/+TNFR1−/−, and TAK1F/−LysMcre/+TNFR1−/− mice with anti-F4/80, and anti-CD11b. B, LysMCre/+ (WT), TAK1+/+LysMcre/+TNFR1−/− (TNFR1-KO), TAK1F/−LysMcre/+TNFR1−/− (TNFR1-KO/TAK1-KO) BMDMs were either untreated or treated with C. albicans hyphae (MOI: 1) or heat-inactivated yeast (MOI: 5) for 90 min. The nuclear extracts were prepared from these cells and then subjected to the electrophoretic mobility shift assay using 32P-labeled NF-κB or OCT-1 probe. C, LysMCre/+ (WT), TAK1+/+LysMcre/+TNFR1−/− (TNFR1-KO), TAK1F/−LysMcre/+TNFR1−/− (TNFR1-KO/TAK1-KO) BMDMs were stimulated with C. albicans hyphae for the indicated time points. Total cell lysates were prepared from these cells and then subjected to Western blotting. Unstim., unstimulated.
To determine whether TAK1 is involved in CLR-induced signaling, we stimulated the TAK1-deficient macrophages generated above with both the yeast and hyphal forms of C. albicans and then examined the DNA-binding activity of NF-κB from these cells (Fig. 4B). We found that in response to the stimulation by both the yeast and hyphal forms of C. albicans, NF-κB activation was defective in macrophages from TAK1-deficient mice compared with those from wild-type TAK1 mice (Fig. 4B). This defect of NF-κB activation in TAK1-deficient macrophages is due to the defective phosphorylation of IKKα/β subunits of the IKK complex (Fig. 4C). This defect is different from TRAF6 deficiency (Fig. 2C), indicating that TAK1 and TRAF6 regulate IKK activation through different mechanisms.
Because TAK1 is suggested to regulate the MAPKs, we determined the role of TAK1 in mediating the activation of JNK, ERK, and p38. Although Syk could be effectively activated by the stimulation of C. albicans hyphae in TAK1/TNFR1-double knock-out macrophages, JNK, p38, and ERK activation were all defective in these cells (Fig. 4C), indicating that TAK1 is a master regulator for the activation of multiple kinases downstream of Syk in CLR signaling.
TAK1 Regulates Cytokines Necessary for T Helper Cell Differentiation in Response to C. albicans
To determine whether TAK1 deficiency can regulate the expression of cytokines important in inducing an adaptive immune response to fungal infections, we evaluated the levels of IL-6, IL-12, and TNF-α in TAK1/TNFR1-deficient macrophages compared with TNFR1−/− or wild-type macrophages, and found that TAK1 deficiency caused a decrease in proinflammatory cytokine production compared with TNFR1−/− and wild-type macrophages (Fig. 5, A–D). The cytokine IL-1β plays an important role in the differentiation of Th17 cells. Therefore, we examined the production of IL-1β in TAK1-deficient cells. Because IL-1β production in macrophages is a two-step process, stimulation of macrophages with hyphae alone cannot induce detectable levels of IL-1β (data not shown). Priming cells with zymosan prior to stimulation with hyphae was a necessary step to induce IL-1β production (Fig. 5, E and F). We measured the levels of IL-1β production in TAK1/TNFR1-deficient cells and found that TNFR1-deficient macrophages showed lower levels of IL-1β production (Fig. 5E), which may be due to the secondary effect of TNFα for IL-1β expression. Nevertheless, TAK1/TNFR1-deficient macrophages had much lower levels of IL-1β production compared with TNFR1-deficient macrophages (Fig. 5, E and F). Together, these results demonstrate that TAK1 plays an essential role in CLR-induced innate immune response.
FIGURE 5.

TAK1 contributes to cytokine induction by C. albicans stimulation. A–D, BMDMs from LysMCre/+ (WT, black bars), TAK1+/+LysMcre/+TNFR1−/− (TNFR1−/−, lined bars) and TAK1F/−LysMcre/+TNFR1−/− (TNFR1−/−TAK1−/−, white bars) mice were stimulated overnight with C. albicans hyphae (MOI of 1). ELISA was used to measure the level of TNFα (A), IL-6 (B), IL-12/23 p40 (C), and IL-10 (D) in these cultured media. E and F, above cells were primed with zymosan or left untreated then were stimulated overnight with C. albicans hyphae (MOI: 1). The data are the means ± S.D. of triplicate wells and are representative of three independent experiments. Unstim., unstimulated.
DISCUSSION
Stimulation of C-type lectin receptors on innate immune cells triggers the activation of signaling cascades that activates multiple transcriptional factors, including NF-κB, and leads to expression of various cytokines, which play essential roles for immune responses. In this study, we sought to understand the role of TAK1 and TRAF6 in mediating the innate immune response against the infection of opportunistic fungus C. albicans. We provide compelling genetic evidence that TRAF6 and TAK1 are key components in the CLR signaling pathway and mediate NF-κB activation in macrophages, in response to C. albicans stimulation. Our data also indicate that TRAF6 and TAK1 deficiency causes a decrease in JNK activation.
Previous studies showed a critical role for TRAF6 in TLR and CD40 receptor signaling (20). In TLR signaling, TRAF6 is linked to upstream signaling by forming a complex with Interleukin-1 receptor-associated kinase 1 (IRAK1) (39–41) and Interleukin-1 receptor-associated kinase 2 (IRAK2) (42, 43). In T cell receptor signaling, TRAF6 associates with its upstream CARMA1-BCL10-MALT1 complex to regulate IKK activation (21, 25, 44). In this study, we provide the genetic evidence that TRAF6 is also required for C-type lectin receptor-induced NF-κB activation. Our data suggest that similar to the TCR signaling pathway, TRAF6 is linked to its downstream signaling cascade through its association with a Bcl10-containing complex. Consistent with the observations found in CARD9 and Bcl10-deficient cells (16), TRAF6 deficiency does not affect the phosphorylation of IKKα/β, suggesting that TRAF6 is only involved in the regulation of IKK ubiquitination. In this study, we found that TRAF6 itself also undergoes a signal-induced ubiquitination in response to fungal stimulation. However, how TRAF6 is inducibly ubiquitinated remains to be determined. One possibility is its auto-ubiquitination (23).
Our study also provides genetic evidence that TAK1 is required for CLR-induced NF-κB activation in response to fungal infection. Previous studies suggest that TAK1-binding proteins (TAB), TAB2, and TAB3 are required for TAK1 activation. In the IL-1, RANKL, and TNF signaling pathways, the functional interaction between TAK1 and TAB2/TAB3 is crucial for mediating IKK and MAPK signaling (45). Further studies will be needed to investigate whether TAB2 and TAB3 mediate C-type lectin receptor signaling and whether or not TAB2 and TAB3 play redundant functions in this signaling.
Although TAK1 is associated with TRAF6, these two proteins appear to regulate different modifications of the IKK complex. TAK1 deficiency impairs CLR-induced IKK phosphorylation, whereas TRAF6 deficiency does not affect this phosphorylation. Although it is not clear how TAK1 is activated in the CLR signaling pathway, a recent study suggests that TAK1 may be regulated by PKCδ-dependent phosphorylation in response to fungal infection (18). However, it remains to be determined whether PKCδ directly phosphorylates TAK1 and whether this phosphorylation is required for TAK1 activation. On the basis of recent reports and our own results, we propose that signals from C-type lectin receptors branch into two signaling cascades at PKCδ level, leading to IKK complex activation (Fig. 6). In the first cascade, activation of the phospholipase Cγ2-PKCδ-TAK1 axis leads to the phosphorylation of IKKα/β, while in a parallel cascade, CARD9-Bcl10-TRAF6 recruits the IKK complex leading to ubiquitination of IKKγ/NEMO. The combination of phosphorylation and ubiquitination confers full activity to the IKK complex.
FIGURE 6.

Proposed model for the signaling pathway mediating NF-κB activation induced by C-type lectin receptors. In macrophages, recognition of C. albicans by CLRs leads to the phosphorlation of Syk and subsequently activates phospholipase C (PLC) γ2. Activated PKCδ can phosphorylate CARD9, which, may lead to the recruitment of BCL10-TRAF6-TAK1. TAK1 induces the phosphorylation of IKKα/β, whereas TRAF6 leads to the ubiquitination of Nemo. The combination of Nemo ubiquitination and IKKα/β phosphorylation can lead to the degradation of IκBα and subsequent NF-κB activation. Ub, ubiquitin.
This work was supported in part by National Institutes of Health Grants RO1AI050848 and RO1GM065899 (to X. L.).
- CLR
- C-type lectin receptor
- Syk
- spleen tyrosine kinase
- IKK
- IκB kinase
- TLR
- Toll-like receptor
- TNFR
- tumor necrosis factor receptor
- BMDM
- bone marrow-derived macrophage
- TRAF6
- tumor necrosis factor receptor-associated factor 6
- TAK1
- TGFβ-activated kinase 1
- MOI
- multiplicity of infection.
REFERENCES
- 1. Brown G. D., Taylor P. R., Reid D. M., Willment J. A., Williams D. L., Martinez-Pomares L., Wong S. Y., Gordon S. (2002) Dectin-1 is a major β-glucan receptor on macrophages. J. Exp. Med. 196, 407–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Brown G. D., Herre J., Williams D. L., Willment J. A., Marshall A. S., Gordon S. (2003) Dectin-1 mediates the biological effects of β-glucans. J. Exp. Med. 197, 1119–1124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sato K., Yang X. L., Yudate T., Chung J. S., Wu J., Luby-Phelps K., Kimberly R. P., Underhill D., Cruz P. D., Jr., Ariizumi K. (2006) Dectin-2 is a pattern recognition receptor for fungi that couples with the Fc receptor γ chain to induce innate immune responses. J. Biol. Chem. 281, 38854–38866 [DOI] [PubMed] [Google Scholar]
- 4. Saijo S., Ikeda S., Yamabe K., Kakuta S., Ishigame H., Akitsu A., Fujikado N., Kusaka T., Kubo S., Chung S. H., Komatsu R., Miura N., Adachi Y., Ohno N., Shibuya K., Yamamoto N., Kawakami K., Yamasaki S., Saito T., Akira S., Iwakura Y. (2010) Dectin-2 recognition of α-mannans and induction of Th17 cell differentiation is essential for host defense against Candida albicans. Immunity 32, 681–691 [DOI] [PubMed] [Google Scholar]
- 5. Mócsai A., Ruland J., Tybulewicz V. L. (2010) The SYK tyrosine kinase: a crucial player in diverse biological functions. Nat. Rev. Immunol. 10, 387–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kerrigan A. M., Brown G. D. (2011) Syk-coupled C-type lectins in immunity. Trends Immunol. 32, 151–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kingeter L. M., Lin X. (2012) C-type lectin receptor-induced NF-κB activation in innate immune and inflammatory responses. Cell Mol. Immunol. 9, 105–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Taylor P. R., Tsoni S. V., Willment J. A., Dennehy K. M., Rosas M., Findon H., Haynes K., Steele C., Botto M., Gordon S., Brown G. D. (2007) Dectin-1 is required for β-glucan recognition and control of fungal infection. Nat. Immunol. 8, 31–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Robinson M. J., Osorio F., Rosas M., Freitas R. P., Schweighoffer E., Gross O., Verbeek J. S., Ruland J., Tybulewicz V., Brown G. D., Moita L. F., Taylor P. R., Reis e Sousa C. (2009) Dectin-2 is a Syk-coupled pattern recognition receptor crucial for Th17 responses to fungal infection. J. Exp. Med. 206, 2037–2051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wells C. A., Salvage-Jones J. A., Li X., Hitchens K., Butcher S., Murray R. Z., Beckhouse A. G., Lo Y. L., Manzanero S., Cobbold C., Schroder K., Ma B., Orr S., Stewart L., Lebus D., Sobieszczuk P., Hume D. A., Stow J., Blanchard H., Ashman R. B. (2008) The macrophage-inducible C-type lectin, mincle, is an essential component of the innate immune response to Candida albicans. J. Immunol. 180, 7404–7413 [DOI] [PubMed] [Google Scholar]
- 11. Xu S., Huo J., Lee K. G., Kurosaki T., Lam K. P. (2009) Phospholipase Cγ2 is critical for Dectin-1-mediated Ca2+ flux and cytokine production in dendritic cells. J. Biol. Chem. 284, 7038–7046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tassi I., Cella M., Castro I., Gilfillan S., Khan W. N., Colonna M. (2009) Requirement of phospholipase C-γ2 (PLCγ2) for Dectin-1-induced antigen presentation and induction of TH1/TH17 polarization. Eur. J. Immunol. 39, 1369–1378 [DOI] [PubMed] [Google Scholar]
- 13. Gorjestani S., Yu M., Tang B., Zhang D., Wang D., Lin X. (2011) Phospholipase Cγ2 (PLCγ2) is key component in Dectin-2 signaling pathway, mediating anti-fungal innate immune responses. J. Biol. Chem. 286, 43651–43659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gross O., Gewies A., Finger K., Schäfer M., Sparwasser T., Peschel C., Förster I., Ruland J. (2006) Card9 controls a non-TLR signalling pathway for innate anti-fungal immunity. Nature 442, 651–656 [DOI] [PubMed] [Google Scholar]
- 15. Hsu Y. M., Zhang Y., You Y., Wang D., Li H., Duramad O., Qin X. F., Dong C., Lin X. (2007) The adaptor protein CARD9 is required for innate immune responses to intracellular pathogens. Nat. Immunol. 8, 198–205 [DOI] [PubMed] [Google Scholar]
- 16. Bi L., Gojestani S., Wu W., Hsu Y. M., Zhu J., Ariizumi K., Lin X. (2010) CARD9 mediates dectin-2-induced IκBα kinase ubiquitination leading to activation of NF-κB in response to stimulation by the hyphal form of Candida albicans. J. Biol. Chem. 285, 25969–25977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yamasaki S., Ishikawa E., Sakuma M., Hara H., Ogata K., Saito T. (2008) Mincle is an ITAM-coupled activating receptor that senses damaged cells. Nat. Immunol. 9, 1179–1188 [DOI] [PubMed] [Google Scholar]
- 18. Strasser D., Neumann K., Bergmann H., Marakalala M. J., Guler R., Rojowska A., Hopfner K. P., Brombacher F., Urlaub H., Baier G., Brown G. D., Leitges M., Ruland J. (2012) Syk kinase-coupled C-type lectin receptors engage protein kinase C-σ to elicit Card9 adaptor-mediated innate immunity. Immunity 36, 32–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cao Z., Xiong J., Takeuchi M., Kurama T., Goeddel D. V. (1996) TRAF6 is a signal transducer for interleukin-1. Nature 383, 443–446 [DOI] [PubMed] [Google Scholar]
- 20. Kobayashi T., Walsh P. T., Walsh M. C., Speirs K. M., Chiffoleau E., King C. G., Hancock W. W., Caamano J. H., Hunter C. A., Scott P., Turka L. A., Choi Y. (2003) TRAF6 is a critical factor for dendritic cell maturation and development. Immunity 19, 353–363 [DOI] [PubMed] [Google Scholar]
- 21. Sun L., Deng L., Ea C. K., Xia Z. P., Chen Z. J. (2004) The TRAF6 ubiquitin ligase and TAK1 kinase mediate IKK activation by BCL10 and MALT1 in T lymphocytes. Mol. Cell 14, 289–301 [DOI] [PubMed] [Google Scholar]
- 22. Yang W. L., Wang J., Chan C. H., Lee S. W., Campos A. D., Lamothe B., Hur L., Grabiner B. C., Lin X., Darnay B. G., Lin H. K. (2009) The E3 ligase TRAF6 regulates Akt ubiquitination and activation. Science 325, 1134–1138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lamothe B., Besse A., Campos A. D., Webster W. K., Wu H., Darnay B. G. (2007) Site-specific Lys-63-linked tumor necrosis factor receptor-associated factor 6 auto-ubiquitination is a critical determinant of I kappa B kinase activation. J. Biol. Chem. 282, 4102–4112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lamothe B., Campos A. D., Webster W. K., Gopinathan A., Hur L., Darnay B. G. (2008) The RING domain and first zinc finger of TRAF6 coordinate signaling by interleukin-1, lipopolysaccharide, and RANKL. J. Biol. Chem. 283, 24871–24880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. McCully R. R., Pomerantz J. L. (2008) The protein kinase C-responsive inhibitory domain of CARD11 functions in NF-κB activation to regulate the association of multiple signaling cofactors that differentially depend on Bcl10 and MALT1 for association. Mol. Cell Biol. 28, 5668–5686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hara H., Saito T. (2009) CARD9 versus CARMA1 in innate and adaptive immunity. Trends Immunol. 30, 234–242 [DOI] [PubMed] [Google Scholar]
- 27. Mizukami J., Takaesu G., Akatsuka H., Sakurai H., Ninomiya-Tsuji J., Matsumoto K., Sakurai N. (2002) Receptor activator of NF-κB ligand (RANKL) activates TAK1 mitogen-activated protein kinase kinase kinase through a signaling complex containing RANK, TAB2, and TRAF6. Mol. Cell Biol. 22, 992–1000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Takaesu G., Kishida S., Hiyama A., Yamaguchi K., Shibuya H., Irie K., Ninomiya-Tsuji J., Matsumoto K. (2000) TAB2, a novel adaptor protein, mediates activation of TAK1 MAPKKK by linking TAK1 to TRAF6 in the IL-1 signal transduction pathway. Mol. Cell 5, 649–658 [DOI] [PubMed] [Google Scholar]
- 29. Skaug B., Jiang X., Chen Z. J. (2009) The role of ubiquitin in NF-κB regulatory pathways. Annu. Rev. Biochem. 78, 769–796 [DOI] [PubMed] [Google Scholar]
- 30. Adhikari A., Xu M., Chen Z. J. (2007) Ubiquitin-mediated activation of TAK1 and IKK. Oncogene 26, 3214–3226 [DOI] [PubMed] [Google Scholar]
- 31. Eftychi C., Karagianni N., Alexiou M., Apostolaki M., Kollias G. (2012) Myeloid TAKI [corrected] acts as a negative regulator of the LPS response and mediates resistance to endotoxemia. PLoS One 7, e31550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kishimoto K., Matsumoto K., Ninomiya-Tsuji J. (2000) TAK1 mitogen-activated protein kinase kinase kinase is activated by autophosphorylation within its activation loop. J. Biol. Chem. 275, 7359–7364 [DOI] [PubMed] [Google Scholar]
- 33. Shim J. H., Xiao C., Paschal A. E., Bailey S. T., Rao P., Hayden M. S., Lee K. Y., Bussey C., Steckel M., Tanaka N., Yamada G., Akira S., Matsumoto K., Ghosh S. (2005) TAK1, but not TAB1 or TAB2, plays an essential role in multiple signaling pathways in vivo. Genes Dev. 19, 2668–2681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jadrich J. L., O'Connor M. B., Coucouvanis E. (2006) The TGF β activated kinase TAK1 regulates vascular development in vivo. Development 133, 1529–1541 [DOI] [PubMed] [Google Scholar]
- 35. Clausen B. E., Burkhardt C., Reith W., Renkawitz R., Förster I. (1999) Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res. 8, 265–277 [DOI] [PubMed] [Google Scholar]
- 36. Xie M., Zhang D., Dyck J. R., Li Y., Zhang H., Morishima M., Mann D. L., Taffet G. E., Baldini A., Khoury D. S., Schneider M. D. (2006) A pivotal role for endogenous TGF-β-activated kinase-1 in the LKB1/AMP-activated protein kinase energy-sensor pathway. Proc. Natl. Acad. Sci. U.S.A. 103, 17378–17383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ninomiya-Tsuji J., Kishimoto K., Hiyama A., Inoue J., Cao Z., Matsumoto K. (1999) The kinase TAK1 can activate the NIK-I kappaB as well as the MAP kinase cascade in the IL-1 signalling pathway. Nature 398, 252–256 [DOI] [PubMed] [Google Scholar]
- 38. Wang C., Deng L., Hong M., Akkaraju G. R., Inoue J., Chen Z. J. (2001) TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature 412, 346–351 [DOI] [PubMed] [Google Scholar]
- 39. Qian Y., Commane M., Ninomiya-Tsuji J., Matsumoto K., Li X. (2001) IRAK-mediated translocation of TRAF6 and TAB2 in the interleukin-1-induced activation of NFκB. J. Biol. Chem. 276, 41661–41667 [DOI] [PubMed] [Google Scholar]
- 40. Takaesu G., Ninomiya-Tsuji J., Kishida S., Li X., Stark G. R., Matsumoto K. (2001) Interleukin-1 (IL-1) receptor-associated kinase leads to activation of TAK1 by inducing TAB2 translocation in the IL-1 signaling pathway. Mol. Cell Biol. 21, 2475–2484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jiang Z., Ninomiya-Tsuji J., Qian Y., Matsumoto K., Li X. (2002) Interleukin-1 (IL-1) receptor-associated kinase-dependent IL-1-induced signaling complexes phosphorylate TAK1 and TAB2 at the plasma membrane and activate TAK1 in the cytosol. Mol. Cell Biol. 22, 7158–7167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Keating S. E., Maloney G. M., Moran E. M., Bowie A. G. (2007) IRAK-2 participates in multiple toll-like receptor signaling pathways to NFkappaB via activation of TRAF6 ubiquitination. J. Biol. Chem. 282, 33435–33443 [DOI] [PubMed] [Google Scholar]
- 43. Lin S. C., Lo Y. C., Wu H. Nature 465, 885–890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Shambharkar P. B., Blonska M., Pappu B. P., Li H., You Y., Sakurai H., Darnay B. G., Hara H., Penninger J., Lin X. (2007) Phosphorylation and ubiquitination of the IκB kinase complex by two distinct signaling pathways. EMBO J. 26, 1794–1805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Besse A., Lamothe B., Campos A. D., Webster W. K., Maddineni U., Lin S. C., Wu H., Darnay B. G. (2007) TAK1-dependent signaling requires functional interaction with TAB2/TAB3. J. Biol. Chem. 282, 3918–3928 [DOI] [PMC free article] [PubMed] [Google Scholar]