

Background: PAX2, a transcription factor, plays a role in embryogenesis.
Results: Knockdown of PAX2 inhibits colon cancer cells proliferation and xenograft growth. PAX2 activates AP-1, leading to increased expression of cyclin D1.
Conclusion: PAX2 promotes proliferation of colon cancer cells via AP-1.
Significance: PAX2 exhibits oncogenic properties in colon cancer cells and may serve as a target for cancer therapy.
Keywords: AP1 Transcription Factor, Colorectal Cancer, Proliferation, Signal Transduction, Tumor, PAX2
Abstract
Paired box (PAX) 2, a transcription factor, plays a critical role in embryogenesis. When aberrantly expressed in adult tissues, it generally exhibits oncogenic properties. However, the underlying mechanisms remain unclear. We reported previously that the expression of PAX2 was up-regulated in human colon cancers. However, the role of PAX2 in colon cancer cells has yet to be determined. The aim of this study is to determine the function of PAX2 in colon cancer cells and to investigate the possible mechanisms underlain. We find that knockdown of PAX2 inhibits proliferation and xenograft growth of colon cancer cells. Inhibition of PAX2 results in a decreased expression of cyclin D1. Expression of cyclin D1 is found increased in human primary colon malignant tumors, and its expression is associated with that of PAX2. These data indicate that PAX2 is a positive regulator of expression of cyclin D1. We find that knockdown of PAX2 inhibits the activity of AP-1, a transcription factor that induces cyclin D1 expression, implying that PAX2 induces cyclin D1 through AP-1. PAX2 has little effect on expression of AP-1 members including c-Jun, c-Fos, and JunB. Our data show that PAX2 prevents JunB from binding c-Jun and enhances phosphorylation of c-Jun, which may elevate the activity of AP-1. Taken together, these results suggest that PAX2 promotes proliferation of colon cancer cells through AP-1.
Introduction
Paired box (PAX) genes encode a set of transcription factors, and nine members of the family have been identified in mammals (1). They are regulators of tissue development and cellular differentiation in embryos, acting to promote cell proliferation, migration, and survival (1, 2). Generally, expression of PAX genes attenuates when development is complete. PAX genes are named for the paired box DNA binding domain that is common to all the family members. The family is classified into subgroups I–IV. The genes that comprise subgroups II (PAX2, PAX5, and PAX8) and III (PAX3 and PAX7) are expressed in cancers and are therefore useful as tumor markers (2). Knockdown of their expression leads to tumor cell death, suggesting their potential involvement in tumor development.
PAX2 is a critical regulator of embryogenesis. It may exhibit oncogenic properties if aberrantly expressed in adult tissues. The abnormal re-expression of PAX2 is observed in a variety of cancers including leukemia, breast, prostate, kidney, and bladder carcinoma (1–4). As a result, PAX2 has become a hallmark of malignant cells, suggesting that it might be a potential target for cancer therapy. Heterozygous PAX2+/− mutations cause renal hypoplasia and increased apoptosis of the ureteric bud, suggesting an antiapoptotic function of PAX2 during nephrogenesis (5). The antiapoptotic function of PAX2 contributes to the resistance of renal carcinoma cells to chemotherapy (6, 7). Inhibition of PAX2 in renal cell carcinoma cells (8) and silencing of PAX2 in ovarian cancer and bladder cancer cells (3) trigger growth inhibition and cell death. Similarly, inhibition of PAX2 in Kaposi sarcoma cells led to cell death and reduced cell motility and invasiveness when challenged by serum deprivation or vincristine treatment (9). PAX2 inhibition also resulted in cell death of prostate cancer cells (10). Although a number of studies have demonstrated that PAX2 plays an important role in cancer, the underlying mechanisms are not well understood.
Colon cancer is the third commonest malignancy worldwide (11) and the second leading cause of cancer death in the United States (12). We demonstrated recently that the expression of PAX2 was up-regulated in human colon cancer (13). However, the functions of PAX2 in colon cancer cells remain unknown. Here, we report that knockdown of PAX2 repressed proliferation and xenograft growth of colon cancer cells. Mechanistic studies indicate that PAX2 regulated proliferation of colon cancer cells through AP-1. The role of PAX genes in cancer is emerging as an exciting research area. Abnormal PAX expression in cancer may offer new means to evaluate patient prognosis and provide target candidate for cancer treatments. We demonstrate here that PAX2 promotes proliferation of colon cancer cells via AP-1. These results suggest that PAX2 might be a potential therapeutic target of colon cancer.
MATERIALS AND METHODS
Cell Culture and Reagents
Human colon cancer HCT116 and RKO cells, human cervical cancer HeLa cells, human liver cancer HepG2 cells, human embryonic kidney 293T cells, and African green monkey SV40-transformed kidney fibroblast COS7 (PAX2-null) cells were maintained in DMEM. The human normal colon epithelial CCD841 cells, human colon cancer HT29, SW480 and SW620 cells, human lung cancer A549 cells, and gastric cancer SGC7901 cells were maintained in RPMI 1640 medium. All media were supplemented with 10% fetal bovine serum, 100 units/ml penicillin, and 100 μg/ml streptomycin. The cells were cultured at 37 °C in 5% CO2 incubator.
Construction of Vectors
The vector encoding PAX2 was constructed by PCR and cloned into pcDNA3.1-myc vector. The vector encoding GST-PAX2 fusion protein was constructed by inserting PCR-generated DNA fragment encoding PAX2 into pGEX4T1. NF-κB and AP-1 reporter plasmids were from Dr. C. Wang (The Institute for Biological Sciences, Shanghai, China). The TOP/FOP luciferase reporter plasmids were from Dr. D. Xie (The Institute for Nutritional Sciences, Shanghai, China). These two plasmids were used for determination of β-catenin transcriptional activity. The cyclin D1 promoter reporter plasmid (-964CD1LUC) that has one AP-1 binding site was constructed as described (14). The mutated cyclin D1 promoter at the AP-1 binding site (TGACTCA → TGCCGCA) was generated by site-directed mutagenesis (14). JunB(275–347), the fragment containing the leucine zipper domain of JunB, was cloned by PCR and inserted into pGEX4T1. To produce GST proteins, the Escherichia coli BL21-Gold(DE3)pLysS cells were transformed with pGEX4T1-PAX2 or pGEX4T1-JunB(275–347) vectors. The transformed cells were treated with 0.1 mmol/liter isopropyl-d-thiogalactoside for 4 h at 25 °C.
Western Blot and Immunoprecipitation
Western blot and immunoprecipitation were performed as described (15). Antibodies against PAX2, HA, Myc, cyclin D1, and p-c-Jun(Ser-63) 2 were products of Santa Cruz Biotechnology (Santa Cruz, CA). JNK and phospho-JNK antibodies were from Cell signaling Technology (Danvers, MA). C-Jun, ERK1/2, and p-ERK1/2 antibodies were from Cell Signaling (Beverly, MA). β-Actin antibody was from Sigma.
Transfection and Luciferase Assay
Transient transfection of the cells was performed using Lipofectamine 2000 (Invitrogen) as per the manufacturer's instructions. Stable transfection was performed as follows. The cells were transfected with plasmid and then selected in medium containing 600–800 μg/ml G418 (Sigma) for 2 weeks. A pool of cells that stably expressed the vector was selected and used in the following experiments. Relative luciferase activity was calculated as the ratio of luciferase/β-galactosidase activity.
Short Hairpin RNA (shRNA)
The PAX2-targeting recombinant adenovirus shPAX2 sequence was designed using BLOCK-iTTM RNAi designer (Invitrogen). ShPAX2 and control adenovirus were generated with the BLOCK-iTTM U6 RNAi entry vector kit and BLOCK-iTTM adenoviral RNAi expression system (Invitrogen). Each adenoviral vector was propagated in 293A cells according to the manufacturer's instructions. The LacZ sequence was used as control. In our experiments, HCT116 cells were infected at 30 multiplicity of infection, and RKO cells were infected at 100 multiplicity of infection. The virus designed for PAX2 knockdown contained the following PAX2 target sequence: shPAX2, 5′-CACCGCATCAGAGCACATCAAATCACGAATGATTTGATGTGCTCTGATGC-3′ (top); 5′AAAAGCATCAGAGCACATCAAATCATTCGTGATTTGATGTGCTCTGATGC-3′ (bottom); Control, 5′-CACCGCTACACAAATCAGCGATTTCGAAAAATCGCTGATTTGTGTAG-3′ (top); 5′-AAAACTACACAAATCAGCGATTTTTCGAAATCGCTGATTTGTGTAGC-3′ (bottom).
Real-time PCR
Real-time PCR was performed as described (15). β-Actin was used as internal control. The PAX2 primers are as follows: 5′-CCTCGCTCCAATGGTGAGAA-3′ (forward), 5′TGCTGCTGGGTGAAGGTGTC3′(reverse). The β-actin primers are as follows: 5′-GATCATTGCTCCTCCTGAGC-3′(forward), 5′-ACTCCTGCTTGCTGATCCAC-3′ (reverse).
Electrophoretic Mobility Shift Assay (EMSA)
The biotin 3′ end DNA labeling kit and the LightShift chemiluminescent EMSA kit from Pierce Biotechnology were used in the test. The cells were infected with shPAX2 or control adenovirus for 72 h followed by treatment with TNFα (10 ng/ml) for 1 h. The nuclear proteins were isolated, and EMSA was performed according to the manufacturer's instructions. For the binding assay, 10 μg of nuclear proteins and 20 fmol of probes were used. For competition with labeled probe for specificity, 4 pmol of unlabeled probes were employed. The probes for detecting AP-1 are as follows: 5′CGCTTGATGAGTCAGCCGGAA3′ (forward), 5′TTCCGGCTGACTCATCAAGCG3′ (reverse).
Chromatin Immunoprecipitation (ChIP) Assay
CHIP assay was performed using the ChIP-ITTM express enzymatic kit (Active Motif, catalog number 53009) as described previously (17). The primers used for quantitating the ChIP-enriched cyclin D1 promoter DNA are as follows: 5′-GGACGTCTACACCCCCAACA-3′ (forward), 5′AACACACCTCTGAATGGAAAGC-3′ (reverse)
Cell Proliferation, Cell Cycle, and Cell Apoptosis Analysis
Cell proliferation was measured by counting the number of the cells (16) and expressed as -fold change. The cell cycle was determined by staining the cells with propidium iodide followed by analysis in a BD Biosciences FACScan (16). Cell apoptosis was determined by staining the cells with propidium iodide, and the number of apoptotic cells was defined as the percentage of subdiploid cells (16).
Xenograft Tumor Growth Assay
In vivo xenograft tumor growth was performed as described (15). The 4-week-old male nude mice (BALB/cA-nu (nu/nu)) were injected subcutaneously at each flank with cells infected with control or shPAX2 virus. All animals were maintained and used in accordance with the guidelines of the Institutional Animal Care and Use Committee of the Institute of Nutritional Sciences.
Statistical Analysis
Statistical analysis was performed using the SAS statistical software package (V8.02). The data are presented as mean ± S.D., except where indicated. Student's t test was used for continuous variables. p value < 0.05 was considered significant.
RESULTS
PAX2 Promotes Proliferation and Xenograft Growth of Colon Cancer Cells
We determined the expression of PAX2 in colon cancer HCT116, RKO, HT29, and SW480 and SW620 cells and in normal colon epithelial CCD841 cells. All these cells produce PAX2 (Fig. 1A1), but CCD841 cells produce a low level of PAX2 when compared with these colon cancer cells. Expression of PAX2 in other human cancer cell lines including liver cancer HepG2, lung cancer A549, cervical cancer HeLa, and gastric cancer SGC7901 were determined (Fig. 1A2). SGC7901 and HepG2 cells produce high levels of PAX2, and A549 cells produce low level of PAX2 (Fig. 1A2). 293T and COS7 cells were also examined. When compared with RKO cells, 293T cells produce low level of PAX2. The PAX2-null COS7 cells do not produce any PAX2 (Fig. 1A2).
FIGURE 1.

PAX2 promotes proliferation of colon cancer cells. A, expression of PAX2 in colon cancer cells and normal colon epithelial cells (A1) and in other types of cells (A2). Expression of PAX2 was determined by Western blot as described under “Materials and Methods.” B, knockdown of PAX2 inhibited proliferation of colon cancer cells. HCT116 and RKO cells were infected with control (Con) or shPAX2 adenovirus. Cell proliferation was determined as described under “Materials and Methods.” The data are the mean ± S.E. (n = 3). *, p < 0.05 versus control. C, overexpression PAX2 increased cell proliferation. To determine the effect of PAX2 on proliferation of COS7 cell, a pool of COS7 cells that stably expressed PAX2 was prepared as described under “Materials and Methods.” CCD841 cells were transfected with the vector expressing Myc-tagged PAX2. *, p < 0.05 versus control (n = 3). D, knockdown of PAX2 led to cell G1 arrest. RKO cells were infected with control or shPAX2 adenovirus. The infected cells were subjected for cell cycle analysis as described under “Materials and Methods.” *, p < 0.05 versus control (n = 3). E, knockdown of PAX2 inhibited tumor growth of RKO cells. RKO cells were infected with control or shPAX2 adenovirus. 3 × 106 of the infected cells were injected subcutaneously into the flank of nude mice. The upper panel shows the knockdown efficiency of PAX2. The lower panel shows representative mice. F, the volume of the tumors. The tumor volumes were measured 5 days after injection. The data are mean ± S.E. (n = 13). *, p < 0.05 versus control.
We determined the possible role of PAX2 on proliferation of colon cancer cells. Knockdown of PAX2 attenuated the proliferation of HCT116 and RKO cells (Fig. 1B). Then, we determined the effect of overexpression of PAX2 on proliferation of CCD841 cells that produced low level of PAX2. As expected, overexpression of PAX2 enhanced the proliferation of the cells (Fig. 1C). Similarly, reintroduction of PAX2 into COS7 cells promoted proliferation of the cells (Fig. 1C). These data suggest that PAX2 promotes cell proliferation. We performed cell cycle analysis in RKO cells and found that knockdown of PAX2 led to G1 arrest (Fig. 1D). A few studies showed that PAX2 inhibition resulted in apoptosis of cancer cells (3, 10). We examined RKO cells and found that, under our experimental conditions, knockdown of PAX2 did not significantly induce cell apoptosis (supplemental Fig. S1).
We next determined the effect of PAX2 on xenograft growth of RKO cells. RKO cells that were infected with control or shPAX2 virus were injected into nude mice, and the xenograft growth was determined. We found that knockdown of PAX2 suppressed the xenograft growth of RKO cells (Fig. 1, E and F). Taken together, our results suggest that PAX2 promotes proliferation and tumor growth of colon cancer cells.
PAX2 Induces Expression of Cyclin D1
We found that knockdown of PAX2 decreased the protein levels of cyclin D1 in both HCT116 and RKO cells (Fig. 2A). To exclude the possibility that the altered levels of cyclin D1 in cells with or without shPAX2 was due to difference in cell cycle distribution, RKO cells were synchronized at G1 (24 h of serum starvation) followed by release from cell cycle arrest by the addition of serum and subsequent inspection of cyclin D1 protein levels. The results showed that knockdown of PAX2 repressed expression of cyclin D1 at various time points after release (Fig. 2B). Overexpression of PAX2 in CCD841 cells and reintroduction of PAX2 into COS7 cells enhanced expression of cyclin D1 (Fig. 2C). As expected, PAX2 knockdown inhibited the phosphorylation of Rb, a downstream target of cyclin D1 (supplemental Fig. S2).
FIGURE 2.

PAX2 induces expression of cyclin D1. A, knockdown of PAX2 decreased expression of cyclin D1. HCT116 and RKO cells infected with control or shPAX2 adenovirus were harvested for immunoblotting. B, knockdown of PAX2 decreased expression of cyclin D1. RKO cells were infected as described above. In 48 h, the cells were switched to serum-free medium and incubated for 24 h. Serum was added, and the cells were harvested at different times as indicated. The cells incubated in serum-containing medium were used as a control. C, overexpression of PAX2 increased expression of cyclin D1. The CCD841 and COS7 cells described in the legend for Fig. 1 (panel C) were used for determination of cyclin D1 by immunoblotting. D, expression of PAX2 and cyclin D1 in xenografts of RKO cells. E, expression of cyclin D1 is associated with that of PAX2 in human colon tumors. Sixty pairs of human primary colon tumors were analyzed. The immunoblotting of the tissue extracts was described previously (13). The upper panel indicates a representative Western blot. N, normal; T, tumor. A Fisher's exact test was performed to analyze the data.
We determined the protein levels of cyclin D1 in xenografts of RKO cells from Fig. 1E. Three control and three shPAX2 tumors were randomly selected. The results show that the tumors infected with shPAX2 virus have lower protein levels of cyclin D1 (Fig. 2D). Our previous study showed that expression of PAX2 was up-regulated in human colon tumors (13). We determined the protein levels of cyclin D1 in these tumors paired with adjacent normal tissues. We found that expression of cyclin D1 was up-regulated in colon tumors (Fig. 2E). Statistical analysis indicated that the increased expression of cyclin D1 was associated with increased expression of PAX2 (Fig. 2E). Altogether, these results suggest that PAX2 induces expression of cyclin D1.
PAX2 Induces Expression of Cyclin D1 through AP-1
We found that knockdown of PAX2 decreased cyclin D1 mRNA levels (Fig. 3A), suggesting that PAX2 regulates expression of cyclin D1 at a transcriptional level. We checked cyclin D1 promoter for up to −10 kb and did not find any PAX2 binding motif (TCA(C/T)GC(G/A)TGAC). We presumed that PAX2 might regulate transcription of cyclin D1 indirectly. It is known that NF-κB, β-catenin/TCF4, and AP-1 are regulators of transcription of cyclin D1 (14, 18–20). We asked whether PAX2 induced expression of cyclin D1 through these factors. To know this, we tested the effects of PAX2 on the transcriptional activities of β-catenin/TCF4, NF-κB, and AP-1. Our results showed that knockdown of PAX2 had little effect on the transcriptional activities of either β-catenin/TCF4 (Fig. 3B) or NF-κB (Fig. 3C). Interestingly, we found that knockdown of PAX2 repressed the transcriptional activity of AP-1 (Fig. 3D). Thus, PAX2 might regulate expression of cyclin D1 through AP-1. We performed an EMSA experiment, and the results showed that knockdown of PAX2 reduced the DNA binding capacity of AP-1 (Fig. 3E). Our ChIP assay demonstrated that knockdown of PAX2 decreased the binding of AP-1 to cyclin D1 promoter (Fig. 3F). These results suggest that PAX2 is a positive regulator of AP-1 and that it may regulate expression of cyclin D1 via AP-1. To confirm this, we employed the cyclin D1 promoter reporter plasmid that has the AP-1 binding motif. We found that overexpression of PAX2 increased the cyclin D1 promoter reporter activities (Fig. 3G). If a mutant cyclin D1 reporter plasmid (at AP-1 binding site) was used, the PAX2-induced cyclin D1 reporter activity was reduced significantly (Fig. 3G). Taken together, these results suggest that PAX2 induces expression of cyclin D1 through AP-1.
FIGURE 3.

PAX2 induces cyclin D1 through AP-1. A, knockdown of PAX2 decreased cyclin D1 mRNA level. HCT116 and RKO cells were infected with control or shPAX2 adenovirus. In 48 h, the cells were harvested for real-time PCR analysis. B, PAX2 did not affect the transcriptional activity of β-catenin/TCF4. HCT116 and RKO cells were transfected with TOP/FOP reporter and β-galactosidase plasmids. The next day, the cells were infected with control (Con) or shPAX2 adenovirus. In 48 h, the luciferase activity was determined. C, PAX2 did not affect NF-κB activity. HCT116 and RKO cells were transfected with NF-κB reporter and β-galactosidase plasmids. The cells were incubated overnight followed by infection with control or shPAX2 adenovirus. In 48 h, the relative NF-κB activity was determined by measuring luciferase activity. D, knockdown of PAX2 repressed AP-1 activity. HCT116 and RKO cells were transfected with AP-1 reporter and β-galactosidase plasmids. The transfected cells were incubated overnight and then infected with control or shPAX2 adenovirus. In 48 h, the cells were harvested for luciferase assay. E, knockdown of PAX2 inhibited AP-1 DNA binding activity. HCT116 and RKO cells were infected with control or shPAX2 adenovirus. In 72 h, the infected cells were harvested for EMSA assay. F, ChIP assay was performed as described under “Materials and Methods.” RKO cells were used in the experiment. G, PAX2 regulated expression of cyclin D1 via AP-1. The cyclin D1 promoter reporter and mutated cyclin D1 promoter reporter plasmids were prepared as described under “Materials and Methods.” RKO and HCT116 cells were transfected with cyclin D1 promoter reporter, PAX2, and β-galactosidase plasmids. In 24 h, the cells were harvested for luciferase activity assay. *, p < 0.05 versus control (n = 3).
PAX2 Inhibits the Interaction between c-Jun and JunB
We determined the possible mechanisms that PAX2 enhanced AP-1 activity. Knockdown of PAX2 had little effect on expression of AP-1 members including c-Jun, c-Fos, and JunB (Fig. 4A). We found that knockdown of PAX2 increased the interaction between JunB and c-Jun (Fig. 4, B and C). Consistent with the results, overexpression of PAX2 prevented JunB from binding c-Jun (Fig. 4D). The data indicate that PAX2 blocks the interaction between JunB and c-Jun. It is known that JunB binds c-Jun and inhibits AP-1 activity (21–23). We found that PAX2 bound JunB (Fig. 4E and supplemental Fig. S3) but not c-Jun (supplemental Fig. S4). Taken together, the results suggest that PAX2 binds JunB and blocks the interaction of JunB and c-Jun, leading to AP-1 activation.
FIGURE 4.

PAX2 inhibits the interaction between JunB and c-Jun. A, PAX2 had little effect on expression of c-Jun, c-Fos, and JunB. HCT116 and RKO cells were infected with control or shPAX2 adenovirus. Forty-eight hours after infection, the cells were harvested for immunoblotting. B, knockdown of PAX2 enhanced the c-Jun-JunB interaction. RKO cells were infected as above. Forty-eight hours after infection, the cells were harvested for immunoprecipitation (IP). The c-Jun/JunB ratio (c-Jun/JunB) was determined by measuring the density of co-immunoprecipitated c-Jun protein band and normalized to that of immunoprecipitated JunB. The c-Jun/JunB ratio in control (Con) cells was designated as 1. The data are mean ± S.E. (n = 3). *, p < 0.05 versus control. C, knockdown of PAX2 enhanced the c-Jun-JunB interaction in a dose-dependent manner. RKO cells were infected with different amounts of shPAX2 virus. Forty-eight hours after infection, the cells were harvested for immunoprecipitation. D, PAX2 inhibited the interaction between c-Jun and JunB. HeLa cells were transfected as indicated. In 24 h, the cells were harvested for immunoprecipitation. The c-Jun/JunB ratio was determined as above. E, the endogenous PAX2 and JunB were co-immunoprecipitated. HCT116 cell lysates were used for immunoprecipitation. F, c-Jun bound to JunB(275–347). 293T cells were transfected with HA-c-Jun. Equal amounts of cell lysates containing HA-c-Jun were incubated with the glutathione-Sepharose beads that already captured GST or GST-JunB(275–347). The beads were washed, and HA-c-Jun retained on beads was determined. G, PAX2 bound to GST-JunB(275–347). 293T cells were transfected with Myc-PAX2. Equal amounts of cell lysates containing Myc-PAX2 were incubated with the glutathione-Sepharose beads that captured GST or GST-JunB(275–347). The beads were washed, and Myc-PAX2 retained on beads was determined. H, PAX2 competed with c-Jun for binding JunB(275–347). 293T cells were transfected with HA-c-Jun or different amounts of Myc-PAX2 plasmids. Equal amounts of lysates containing HA-c-Jun were incubated with glutathione-Sepharose beads containing GST-JunB(275–347) at 4 °C for 3 h. The beads were washed, and equal amounts of lysates containing different amounts of Myc-PAX2 were added. The beads were incubated at 4 °C for 3 h. The beads were washed and incubated in SDS-PAGE loading buffer, and the resolved proteins were analyzed by immunoblotting.
C-Jun binds JunB at the leucine zipper motif (24, 25). We constructed a vector encoding GST-JunB(275–347) that contains the binding motif. We found that both c-Jun and PAX2 bound to GST-JunB(275–347) (Fig. 4, F and G), indicating that PAX2 and c-Jun binds JunB at the same domain. Further experiments indicated that PAX2 repressed the interaction of c-Jun and JunB(275–347) dose-dependently (Fig. 4H). These results provide more evidence that PAX2 binds JunB and prevents the JunB-c-Jun interaction.
PAX2 Promotes Phosphorylation of c-Jun
Blockade of the interaction between c-Jun and JunB might enhance the association of c-Jun and c-Fos. We presumed that PAX2 might influence the interaction of c-Jun and c-Fos. As expected, PAX2 knockdown decreased the interaction of c-Jun and c-Fos (Fig. 5A). We asked whether this would affect phosphorylation of c-Jun. Phosphorylation of c-Jun at Ser-63/73 is important for AP-1 activation (25). We therefore determined the effects of PAX2 on c-Jun phosphorylation using p-c-Jun(Ser-63) antibody. We found that knockdown of PAX2 did decrease c-Jun phosphorylation (Fig. 5A). To confirm this, we examined RKO and HCT116 cells. As shown in Fig. 5B, inhibition of PAX2 suppressed the c-Jun phosphorylation in both cells. We found that overexpression of PAX2 enhanced the phosphorylation of c-Jun (Fig. 5C). These results suggest that c-Jun is prone to be phosphorylated when it is not associated with JunB. The c-Jun N-terminal kinase (JNK) is the kinase that phosphorylates c-Jun at Ser-63/73 (25). Our results showed that overexpression of PAX2 did enhance the binding of JNK to c-Jun (Fig. 5D). We found that neither knockdown nor overexpression of PAX2 affected JNK phosphorylation (supplemental Fig. S5). ERK also phosphorylates c-Jun at Ser-63/73 (26). PAX2 had little effect on ERK phosphorylation either (supplemental Fig. S5). Thus, PAX2 regulates c-Jun phosphorylation without influencing either JNK or ERK activities.
FIGURE 5.
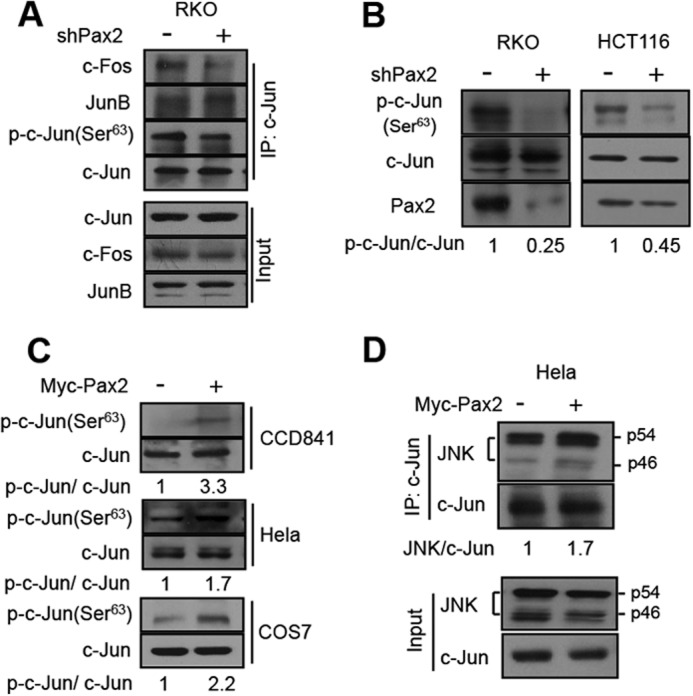
PAX2 enhances phosphorylation of c-Jun. A, knockdown of PAX2 decreased the c-Jun-c-Fos interaction and c-Jun phosphorylation. RKO cells were infected with control or shPAX2 virus. In 48 h, the cells were harvested, and cell lysates were prepared for immunoprecipitation (IP) and immunoblotting. B, knockdown of PAX2 suppressed the phosphorylation of c-Jun. HCT116 and RKO cells were infected with control or shPAX2 adenovirus. In 48 h, the cells were harvested for immunoblotting. The antibody against phospho-c-Jun at Ser-63 was used. The p-c-Jun(Ser-63) to c-Jun ratio (p-c-Jun/c-Jun) was determined by measuring the density of the p-c-Jun(Ser-63) band and normalized to that of c-Jun. C, overexpression of PAX2 increased phosphorylation of c-Jun. CCD841, HeLa, and COS7 cells were transfected with Myc-PAX2 plasmid. The lysates of these cells were used to determine the level of p-c-Jun(Ser-63) by Western blot. p-c-Jun/c-Jun ratio was determined as described above. D, PAX2 enhanced the interaction of JNK and c-Jun. HeLa cells were transfected with control or Myc-PAX2 plasmid. In 24 h, the cells were harvested for determination of the interaction of JNK and c-Jun by means of immunoprecipitation.
DISCUSSION
Aberrant expression of PAX2 has been observed in a variety of cancers. Although PAX2 exhibits oncogenic properties when aberrantly re-expressed in adult tissues, the underlying molecular mechanisms are not understood. We found that expression of PAX2 was also up-regulated in human colon cancer (13). The function of PAX2 in colon cancer has yet to be determined. In this work, we investigated the possible function of PAX2 in human colon cancer cells and studied the possible mechanisms that underlie it. We found that inhibition of PAX2 suppressed proliferation and xenograft growth of colon cancer cells. Mechanistic studies indicate that PAX2 promotes proliferation of colon cancer cells via AP-1.
Overexpression of cyclin D1 occurs in one-third or more of colorectal cancers (27–29). It has been suggested that cyclin D1 trans-activation secondary to Adenomatous polyposis coli or β-catenin mutations participates in colonic cancer initiation (18, 30). Molecular correlates with cyclin D1 activation are important in understanding carcinogenic mechanisms of colorectal cancer. We found in colon cancer cells that PAX2 induced expression of cyclin D1 at a transcriptional level (Fig. 3). No PAX2 binding consensus was found in cyclin D1 promoter, suggesting that PAX2 regulates transcription of cyclin D1 indirectly. AP-1 is a canonical regulator of transcription of cyclin D1 (31). Knockdown of PAX2 repressed AP-1 transcriptional activity and DNA binding capacity (Fig. 3, D–F), suggesting that PAX2 is a positive regulator of AP-1. Further, our results show that PAX2 regulated cyclin D1 transcriptional activation through AP-1 (Fig. 3G). Thus, PAX2 may function to activate AP-1 and induce expression of cyclin D1. Expression of cyclin D1 can also be regulated by other transcriptional factors including NF-κB and β-catenin/TCF4 (18, 19). PAX2 had little effects on activities of β-catenin/TCF4 (Fig. 3B) or NF-κB (Fig. 3C), indicating that these two factors are not involved in PAX2-induced transcription of cyclin D1. SP-1 is also known to regulate transcription of cyclin D1 (32). Therefore, we could not exclude the possibility that PAX2 may also regulate cyclin D1 via SP-1.
AP-1 is a transcription factor that consists of either a Jun-Fos heterodimer or a Jun-Jun homodimer. It regulates the expression of multiple genes essential for cell proliferation, differentiation, and apoptosis (31). AP-1 has been implicated in the pathogenesis of cancer in various tissues and plays a critical role in tumor promotion (25, 33–35). Increased expression of c-Jun has been reported in human colorectal carcinoma (36). A previous study revealed the role of AP-1 in the proliferation of the colon cancer HT29 cells (37). Increased AP-1 activity resulted in enhanced tumorigenic properties of colorectal cancer HCT15 cells (38). Because AP-1 is a crucial regulator of intestinal progenitor proliferation and tumorigenesis (39, 40), therapeutic inhibition of AP-1 activity has attracted considerable interest (41). However, the regulation of AP-1 activity in colon cancer is not well understood. Our previous study indicates that PAX2 was up-regulated in colon cancer (13). We demonstrate here that PAX2 prevents the interaction of c-Jun and JunB (Fig. 4), which leads to AP-1 activation.
In contrast to Jun-Fos heterodimers, the stability of c-Jun homodimers as well as c-Jun-JunB is fairly low. To validate that PAX2 promotes AP-1 activity via inhibiting the c-Jun-JunB interaction, we knocked down PAX2 and determined the c-Jun-JunB interaction. We found that knockdown of PAX2 did enhance the interaction of c-Jun and JunB (Fig. 4, B and C). Thus, PAX2 activates AP-1, probably through blocking the c-Jun-JunB interaction. We found that knockdown of PAX2 blocked the interaction of c-Jun and c-Fos (Fig. 5A). This might be due to the increased c-Jun-JunB interaction. In addition, interestingly, the phosphorylation of c-Jun was also reduced (Fig. 5B). PAX2 promoted the JNK-c-Jun interaction (Fig. 5D). It is most likely that PAX2 blocks the interaction of c-Jun and JunB, leading to increased interaction of c-Jun and JNK. We found that PAX2 had no effect on phosphorylation of JNK and ERK (supplemental Fig. S5), excluding the possibility that PAX2 regulates c-Jun phosphorylation through influencing either JNK or ERK activities.
Cell proliferation is regulated by many factors. Thus, we also determined the effects of PAX2 on expression of other cell cycle regulators. We showed that knockdown of PAX2 led to G1 arrest in RKO cells (Fig. 1D). It is known that p21 is a cyclin-dependent kinase inhibitor and regulates cell cycle progression at G1. Cyclin E1 is a G1/S-specific protein and influences cell cycle G1/S transition. We determined the effects of PAX2 knockdown on expression of these two factors. Inhibition of PAX2 had little effect on expression of p21 and cyclin E1 (supplemental Fig. S6), suggesting that PAX2 does not regulate cell proliferation through p21 and cyclin E1. Knockdown of PAX2 did not influence the expression of G2/mitotic-specific protein cyclin B1 (supplemental Fig. S6). This is consistent with our results that PAX2 influences cell cycle at G1 (Fig. 1D). It was reported that inhibition of PAX2 led to cancer cell apoptosis (3, 10). However, in our work, knockdown of PAX2 did not significantly induce apoptosis of RKO cells (supplemental Fig. S1). This might be due to the different types of cancer cells that were examined. These results may also suggest that PAX2 functions differently in different cells.
Significant advances have been made in understanding the biology of colon cancer. Many signaling pathways play key roles in maintaining the growth and proliferation of colon cancer, such as Wnt/β-catenin, TGF-β/SMAD, epidermal growth factor receptor, PI3K/Akt, Notch, Hedgehog, and Ras. The enhanced understanding of cancer biology provides important insights for developing novel therapies that target specific molecules and/or certain critical signal pathways. These targeted therapies can be used alone or in combination with cytotoxic chemotherapy. Despite many advances in treatment, colon cancer is still a major health problem in the world. We report here that PAX2 promotes proliferation of colon cancer cells through AP-1 and that inhibition of PAX2 attenuated xenograft of colon cancer cells. These results suggest that PAX2 plays an important role in colon cancer development and that it may serve as a potential target for colon cancer therapy.
Acknowledgments
We thank Dr. Dong Xie (The Institute for Nutritional Sciences, Shanghai, China) and Dr. Chen Wang (The Institute for Biological Sciences, Shanghai, China) for kindly providing the plasmids.
This work was supported by the Natural Science Foundation of China (Grant 30970586) and the Innovation Program of the Chinese Academy of Sciences (Grant KSCX2-EW-R-09).

This article contains supplemental Figs. S1–S6.
- p-c-Jun
- phospho-c-Jun
- TCF4
- transcription factor 4.
REFERENCES
- 1. Robson E. J., He S. J., Eccles M. R. (2006) A PANorama of PAX genes in cancer and development. Nat. Rev. Cancer 6, 52–62 [DOI] [PubMed] [Google Scholar]
- 2. Lang D., Powell S. K., Plummer R. S., Young K. P., Ruggeri B. A. (2007) PAX genes: roles in development, pathophysiology, and cancer. Biochem. Pharmacol. 73, 1–14 [DOI] [PubMed] [Google Scholar]
- 3. Muratovska A., Zhou C., He S., Goodyer P., Eccles M. R. (2003) Paired-box genes are frequently expressed in cancer and often required for cancer cell survival. Oncogene 22, 7989–7997 [DOI] [PubMed] [Google Scholar]
- 4. Luu V. D., Boysen G., Struckmann K., Casagrande S., von Teichman A., Wild P. J., Sulser T., Schraml P., Moch H. (2009) Loss of VHL and hypoxia provokes PAX2 up-regulation in clear cell renal cell carcinoma. Clin. Cancer Res. 15, 3297–3304 [DOI] [PubMed] [Google Scholar]
- 5. Torban E., Eccles M. R., Favor J., Goodyer P. R. (2000) PAX2 suppresses apoptosis in renal collecting duct cells. Am. J. Pathol. 157, 833–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hueber P. A., Waters P., Clark P., Eccles M., Goodyer P. (2006) PAX2 inactivation enhances cisplatin-induced apoptosis in renal carcinoma cells. Kidney Int. 69, 1139–1145 [DOI] [PubMed] [Google Scholar]
- 7. Hueber P. A., Iglesias D., Chu L. L., Eccles M., Goodyer P. (2008) In vivo validation of PAX2 as a target for renal cancer therapy. Cancer Lett. 265, 148–155 [DOI] [PubMed] [Google Scholar]
- 8. Gnarra J. R., Dressler G. R. (1995) Expression of Pax2 in human renal cell carcinoma and growth inhibition by antisense oligonucleotides. Cancer Res. 55, 4092–4098 [PubMed] [Google Scholar]
- 9. Buttiglieri S., Deregibus M. C., Bravo S., Cassoni P., Chiarle R., Bussolati B., Camussi G. (2004) Role of PAX2 in apoptosis resistance and proinvasive phenotype of Kaposi's sarcoma cells. J. Biol. Chem. 279, 4136–4143 [DOI] [PubMed] [Google Scholar]
- 10. Gibson W., Green A., Bullard R. S., Eaddy A. C., Donald C. D. (2007) Inhibition of PAX2 expression results in alternate cell death pathways in prostate cancer cells differing in p53 status. Cancer Lett. 248, 251–261 [DOI] [PubMed] [Google Scholar]
- 11. Davies R. J., Miller R., Coleman N. (2005) Colorectal cancer screening: prospects for molecular stool analysis. Nat. Rev. Cancer 5, 199–209 [DOI] [PubMed] [Google Scholar]
- 12. Jemal A., Siegel R., Ward E., Hao Y., Xu J. Q., Murray T., Thun M. J. (2008) Cancer statistics, 2008. CA Cancer J. Clin. 58, 71–96 [DOI] [PubMed] [Google Scholar]
- 13. Yan B., Jiao S., Zhang H. S., Lv D. D., Xue J., Fan L., Wu G. H., Fang J. (2011) Prolyl hydroxylase domain protein 3 targets Pax2 for destruction. Biochem. Biophys. Res. Commun. 409, 315–320 [DOI] [PubMed] [Google Scholar]
- 14. Albanese C., Johnson J., Watanabe G., Eklund N., Vu D., Arnold A., Pestell R. G. (1995) Transforming p21ras mutants and c-Ets-2 activate the cyclin D1 promoter through distinguishable regions. J. Biol. Chem. 270, 23589–23597 [DOI] [PubMed] [Google Scholar]
- 15. Xue J., Li X., Jiao S., Wei Y., Wu G., Fang J. (2010) Prolyl hydroxylase-3 is downregulated in colorectal cancer cells and inhibits IKKβ, independent of hydroxylase activity. Gastroenterology 138, 606–615 [DOI] [PubMed] [Google Scholar]
- 16. Fang J., Zhou Q., Shi X. L., Jiang B. H. (2007) Luteolin inhibits insulin-like growth factor 1 receptor signaling in prostate cancer cells. Carcinogenesis 28, 713–723 [DOI] [PubMed] [Google Scholar]
- 17. Li Z., Zhang H., Chen Y., Fan L., Fang J. (2012) Forkhead transcription factor FOXO3a protein activates nuclear factor κB through B-cell lymphoma/leukemia 10 (BCL10) protein and promotes tumor cell survival in serum deprivation. J. Biol. Chem. 287, 17737–17745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tetsu O., McCormick F. (1999) β-Catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature 398, 422–426 [DOI] [PubMed] [Google Scholar]
- 19. Hinz M., Krappmann D., Eichten A., Heder A., Scheidereit C., Strauss M. (1999) NF-κB function in growth control: regulation of cyclin D1 expression and G0/G1-to-S-phase transition. Mol. Cell. Biol. 19, 2690–2698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bakiri L., Lallemand D., Bossy-Wetzel E., Yaniv M. (2000) Cell cycle-dependent variations in c-Jun and JunB phosphorylation: a role in the control of cyclin D1 expression. EMBO J. 19, 2056–2068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Deng T., Karin M. (1993) JunB differs from c-Jun in its DNA-binding and dimerization domains, and represses c-Jun by formation of inactive heterodimers. Genes Dev. 7, 479–490 [DOI] [PubMed] [Google Scholar]
- 22. Schütte J., Viallet J., Nau M., Segal S., Fedorko J., Minna J. (1989) jun-B inhibits and c-fos stimulates the transforming and trans-activating activities of c-jun. Cell 59, 987–997 [DOI] [PubMed] [Google Scholar]
- 23. Chiu R., Angel P., Karin M. (1989) Jun-B differs in its biological properties from, and is a negative regulator of, c-Jun. Cell 59, 979–986 [DOI] [PubMed] [Google Scholar]
- 24. Reddy S. P., Mossman B. T. (2002) Role and regulation of activator protein-1 in toxicant-induced responses of the lung. Am. J. Physiol. Lung Cell. Mol. Physiol. 283, L1161–L1178 [DOI] [PubMed] [Google Scholar]
- 25. Eferl R., Wagner E. F. (2003) AP-1: a double-edged sword in tumorigenesis. Nat. Rev. Cancer 3, 859–868 [DOI] [PubMed] [Google Scholar]
- 26. Pulverer B. J., Kyriakis J. M., Avruch J., Nikolakaki E., Woodgett J. R. (1991) Phosphorylation of c-jun mediated by MAP kinases. Nature 353, 670–674 [DOI] [PubMed] [Google Scholar]
- 27. Palmqvist R., Stenling R., Oberg A., Landberg G. (1998) Expression of cyclin D1 and retinoblastoma protein in colorectal cancer. Eur. J. Cancer 34, 1575–1581 [DOI] [PubMed] [Google Scholar]
- 28. Arber N., Hibshoosh H., Moss S. F., Sutter T., Zhang Y., Begg M., Wang S., Weinstein I. B., Holt P. R. (1996) Increased expression of cyclin D1 is an early event in multistage colorectal carcinogenesis. Gastroenterology 110, 669–674 [DOI] [PubMed] [Google Scholar]
- 29. Palmqvist R., Rutegârd J. N., Bozoky B., Landberg G., Stenling R. (2000) Human colorectal cancers with an intact p16/cyclin D1/pRb pathway have up-regulated p16 expression and decreased proliferation in small invasive tumor clusters. Am. J. Pathol 157, 1947–1953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shtutman M., Zhurinsky J., Simcha I., Albanese C., D'Amico M., Pestell R., Ben-Ze'ev A. (1999) The cyclin D1 gene is a target of the β-catenin/LEF-1 pathway. Proc. Natl. Acad. Sci. U.S.A. 96, 5522–5527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shaulian E., Karin M. (2002) AP-1 as a regulator of cell life and death. Nat. Cell Biol. 4, E131–E136 [DOI] [PubMed] [Google Scholar]
- 32. Nagata D., Suzuki E., Nishimatsu H., Satonaka H., Goto A., Omata M., Hirata Y. (2001) Transcriptional activation of the cyclin D1 gene is mediated by multiple cis-elements, including SP1 sites and a cAMP-responsive element in vascular endothelial cells. J. Biol. Chem. 276, 662–669 [DOI] [PubMed] [Google Scholar]
- 33. Maeda S., Karin M. (2003) Oncogene at last–c-Jun promotes liver cancer in mice. Cancer Cell 3, 102–104 [DOI] [PubMed] [Google Scholar]
- 34. Jochum W., Passegué E., Wagner E. F. (2001) AP-1 in mouse development and tumorigenesis. Oncogene 20, 2401–2412 [DOI] [PubMed] [Google Scholar]
- 35. Matthews C. P., Colburn N. H., Young M. R. (2007) AP-1 a target for cancer prevention. Curr. Cancer Drug Targets 7, 317–324 [DOI] [PubMed] [Google Scholar]
- 36. Wang H. L., Wang J., Xiao S. Y., Haydon R., Stoiber D., He T. C., Bissonnette M., Hart J. (2002) Elevated protein expression of cyclin D1 and Fra-1 but decreased expression of c-Myc in human colorectal adenocarcinomas overexpressing β-catenin. Int. J. Cancer 101, 301–310 [DOI] [PubMed] [Google Scholar]
- 37. Suto R., Tominaga K., Mizuguchi H., Sasaki E., Higuchi K., Kim S., Iwao H., Arakawa T. (2004) Dominant-negative mutant of c-Jun gene transfer: a novel therapeutic strategy for colorectal cancer. Gene. Ther. 11, 187–193 [DOI] [PubMed] [Google Scholar]
- 38. Xu Y. M., Zhu F., Cho Y. Y., Carper A., Peng C., Zheng D., Yao K., Lau A. T., Zykova T. A., Kim H. G., Bode A. M., Dong Z. (2010) Extracellular signal-regulated kinase 8-mediated c-Jun phosphorylation increases tumorigenesis of human colon cancer. Cancer Res. 70, 3218–3227 [DOI] [PubMed] [Google Scholar]
- 39. Nateri A. S., Spencer-Dene B., Behrens A. (2005) Interaction of phosphorylated c-Jun with TCF4 regulates intestinal cancer development. Nature 437, 281–285 [DOI] [PubMed] [Google Scholar]
- 40. Sancho R., Nateri A. S., de Vinuesa A. G., Aguilera C., Nye E., Spencer-Dene B., Behrens A. (2009) JNK signalling modulates intestinal homeostasis and tumourigenesis in mice. EMBO J. 28, 1843–1854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ashida R., Tominaga K., Sasaki E., Watanabe T., Fujiwara Y., Oshitani N., Higuchi K., Mitsuyama S., Iwao H., Arakawa T. (2005) AP-1 and colorectal cancer. Inflammopharmacology 13, 113–125 [DOI] [PubMed] [Google Scholar]