

Background: BST-2 inhibits virus release by tethering virions to the cell surface.
Results: The length of the BST-2 ectodomain can be increased or reduced without loss of function.
Conclusion: The positioning of ectodomain deletions rather than their size determines the impact on BST-2 function.
Significance: Understanding the importance of structural elements in BST-2 is critical for developing antiviral strategies.
Keywords: Protein Chimeras, Protein Domains, Retrovirus, Viral Replication, Virology, Virus Assembly, BST-2, Tetherin, Vpu
Abstract
BST-2/CD317/tetherin is a host factor that inhibits HIV-1 release and is counteracted by HIV-1 Vpu. Structural studies indicate that the BST-2 ectodomain assumes a coiled-coil conformation. Here we studied the role of the BST-2 ectodomain for tethering function. First, we addressed the importance of the length and structure of the ectodomain by adding or substituting heterologous coiled-coil or non-coiled-coil sequences. We found that extending or replacing the BST-2 ectodomain using non-coiled-coil sequences resulted in loss of BST-2 function. Doubling the size of the BST-2 ectodomain by insertion of a heterologous coiled-coil motif or substituting the BST-2 coiled-coil domain with a heterologous coiled-coil motif maintained tethering function. Reductions in the size of the BST-2 coiled-coil domain were tolerated as well. In fact, deletion of the C-terminal half of the BST-2 ectodomain, including a series of seven consecutive heptad motifs did not abolish tethering function. However, slight changes in the positioning of deletions affecting the relative placing of charged or hydrophobic residues on the helix severely impacted the functional properties of BST-2. Overall, we conclude that the size of the BST-2 ectodomain is highly flexible and can be reduced or extended as long as the positioning of residues important for the stability of the dimer interface is maintained.
Introduction
BST-2 is an interferon-inducible host factor responsible for the inhibition of HIV-1 release (1, 2). BST-2 can be neutralized by three different lentiviral proteins: HIV-1 employs Vpu, HIV-2 uses its Env glycoprotein, and SIV uses Nef. All three proteins are either integral membrane proteins (Vpu, Env) or are membrane-associated by means of a myristic acid modification (Nef) and are thought to interfere with BST-2 function via direct physical interaction (3–19).
BST-2 was originally identified as a membrane protein in terminally differentiated human B cells of patients with multiple myeloma (20, 21). BST-2 is a 30–36-kDa type II transmembrane glycoprotein consisting of 180 amino acids (22). The protein is predicted to have an N-terminal transmembrane (TM)2 region and a C-terminal glycosylphosphatidylinositol anchor (23). However, we recently found evidence to suggest that the C-terminal membrane anchor could be a second transmembrane domain rather than a glycosylphosphatidylinositol anchor (24). The two TM domains are separated by ∼120 residues that constitute the ectodomain of the protein, which is predicted to form a coiled-coil structure (25–28). The N-terminal half of the BST-2 ectodomain contains three cysteine residues, which are involved in the formation of covalent cysteine-linked dimers (20, 21, 29, 30). Interestingly, two of the three cysteines can be mutated without loss-of-function; in fact, any one of the three cysteines can independently contribute to the formation of cysteine-linked dimers (29, 30). In addition, BST-2 is modified by N-linked glycosylation at two sites in the BST-2 ectodomain (20, 23, 29, 30); however, the functional importance of BST-2 glycosylation for inhibition of virus release is under debate (29, 30). Finally, BST-2 protein associates with lipid rafts at the cell surface and on internal membranes (23, 31–34) and the C-terminal TM region has been implicated with raft targeting of BST-2 (23).
A current model suggests that BST-2 tethers mature virions to the cell surface by means of its N-terminal TM region and C-terminal domain. Structural studies suggest that the BST-2 ectodomain assumes a rod-like structure of about 16–17 nm (25–28), which constitutes the maximum distance between membranes that can be bridged by BST-2. Indeed, immune electron microscopy identified BST-2 on virions tethered to the cell surface and on particles tethered to each other (30, 35–38). Curiously, the distance between these structures frequently exceeded the 17-nm limit imposed by the BST-2 ectodomain, a phenomenon that has remained unexplained. Interestingly, Perez-Caballero et al. (30) demonstrated that an artificial tetherin consisting of the N-terminal TM region of transferrin receptor, a coiled-coil ectodomain of the cytoplasmic dimeric protein dystrophia myotonica protein kinase, and a glycosylphosphatidylinositol anchor signal derived from urokinase plasminogen activator receptor is capable of inhibiting the release of HIV-1 virions tethered to the cell surface. This suggests that the secondary structure rather than the primary amino acid sequence of BST-2 is important for tethering function. This conclusion is confirmed by a recent study demonstrating that replacement of four consecutive residues by alanines is well tolerated across much of the BST-2 ectodomain (39).
The goal of the current study was to gain additional insights into the role of the BST-2 ectodomain for tethering function. Our approach was two-pronged. First, we assessed the importance of the length and structure of the ectodomain by adding or substituting heterologous coiled-coil or non-coiled-coil sequences in the BST-2 ectodomain. Second, we introduced in-frame deletions at various positions and of various sizes to identify the minimum size of the BST-2 ectodomain. Results from our first approach revealed that extending or substituting the BST-2 ectodomain using non-coiled-coil sequences derived from the CD4 ectodomain inhibited BST-2 function. In contrast, substituting the BST-2 coiled-coil domain with a heterologous coiled-coil motif derived from vimentin maintained the tethering function, consistent with previous results involving artificial tetherin. Interestingly, addition of the vimentin coiled-coil to the existing BST-2 structure, which almost doubled the size of the BST-2 ectodomain, remained functional. These results suggest that there is significant tolerance with respect to increasing the length of the ectodomain. Results from our second approach revealed that much of the BST-2 ectodomain can be deleted without loss of function. Indeed, we found that approximately half of the ectodomain, including a series of seven consecutive heptad motifs, could be deleted without losing tethering function. Interestingly, the positioning of the deletions within the ectodomain was critical for BST-2 function. Utilizing molecular modeling, we found that the orientation of hydrophobic and hydrophilic residues along the dimer interface in the C-terminal part of BST-2 was critical for function. Overall, we conclude that the size of the BST-2 ectodomain can be reduced or enlarged with heterologous coiled-coil sequences, revealing a significant flexibility in the overall size of the protein, whereas changes to the heptad motifs or the register of the coil had variable effects depending on the positioning of the deletion.
EXPERIMENTAL PROCEDURES
Plasmids
The full-length infectious HIV-1 molecular clone pNL4-3 and the Vpu deletion mutant pNL4-3/Udel have been described (40, 41). Plasmid pcDNA-BST-2 is a vector for the expression of human BST-2 under control of the cytomegalovirus immediate-early promoter (29). HA-tagged BST-2 encoding a triple HA epitope tag in the ectodomain following BST-2 residue 148 (BST-2I) was constructed using PCR-based methodologies as described (24). BST-2I is referred to here as BST-2 wt and was used for construction of additional mutants. In pcDNA-BST-2-rCD4 residues, Lys106-Ile148 of BST-2I were removed and replaced by domain 3 of the ectodomain of human CD4 (residues Phe204-Met317). In pcDNA-BST-2-rCC1 and pcDNA-BST-2-rCC2, residues Lys106-Ile148 of BST-2I were replaced by vimentin residues Glu300-Asn388 or Arg122-Ser299, respectively, which were predicted to form coiled-coils using COILS predictor software (42). Insertions were made after residue Ile148 with the same portions of either CD4 (iCD4) or vimentin (iCC1 and iCC2). Deletions within BST-2I include Δ51–80 (residues Glu51-Gly80), Δ51–88 (residues Glu51-Ala88), Δ96–116 (residues Met96-Leu116), Δ96–127 (residues Met96-Leu127), Δ131–148 (residues Ser131-Ile148), Δ97–147 (residues Ala97-Arg147), Δ96–118 (residues Met96-Gly118), Δ55–87 (residues Asp55-Gln87), Δ81–94 (residues Phe81-Thr94), and Δ95–146 (residues Val95-Val146). All constructs were verified by sequence analysis and protein expression and dimerization was determined by immunoblotting.
Antisera
Rabbit polyclonal BST-2 antiserum, directed against the extracellular portion of BST-2, has been described (29, 43) and is available through the NIH AIDS Research and Reference Reagent Program (aidsreagent.org; catalog number 11721). HA-specific mouse monoclonal antibody was from Roche Diagnostics.
Tissue Culture and Transfections
HeLa and 293T cells were propagated in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum (FBS). HeLa TZM-bl cells were obtained from the NIH AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH (donated by Dr. John C. Kappes, Dr. Xiaoyun Wu, and Tranzyme Inc.) and propagated in DMEM. For transfection, cells were grown in 25-cm2 flasks to about 80% confluence. Cells were transfected using TransIT®-LT1 (Mirus, Madison, WI) or Lipofectamine PLUSTM (Invitrogen Corp.) following the manufacturer's recommendations. A total of 5 μg of plasmid DNA per 25-cm2 flask was used. Total amounts of transfected DNA was kept constant by adding empty vector DNA as appropriate. Cells were harvested 24 h post-transfection.
Viral Infectivity
Virus stocks were prepared by transfection of 293T cells with the indicated plasmid DNAs. Virus-containing supernatants were harvested 24 h after transfection. Cellular debris was removed by centrifugation (3 min, 1,500 × g) and clarified supernatants were filtered (0.45 μm) to remove residual cellular debris. 100 μl of viral stock was used to infect 5 × 104 TZM-bl cells in a 24-well plate in a total volume of 1.1 ml. Infection was allowed for 48 h at 37 °C. Media was removed and cells were lysed in 300 μl of Promega 1× reporter lysis buffer (Promega Corp., Madison, WI) and frozen at −80 °C for a minimum of 30 min. To determine the luciferase activity in the lysates, 5 μl of each lysate were combined with 20 μl of luciferase substrate (Steady-Glo; Promega Corp., Madison, WI) and light emission was measured using a Modulus ii microplate reader (Turner Biosystems Inc., Sunnyvale, CA).
Models of BST-2 Ectodomain
Models were built using the human BST-2 structure at 2.6 Å (PDB code 3NWH) (26) in FOLDX (44) within YASARA 12.4.1 (45). Residues were replaced using the swap function and no minimization of the structure was performed. Images were generated using the POV-Ray function within YASARA. For comparison of all the structures of BST-2, 3MQ7 (27), 3MQC (27), 3MQB (27), 3NWH (26), and 2XG7 (26) were aligned using the superpose function within YASARA 12.4.1.
Protein Structure Prediction
The sequence of untagged human BST-2 or human BST-2 with a 3× HA tag inserted following residue 148 was submitted for coiled-coil predictions to the online MARCOIL 1.0 server (101–140 (46)), the CCHMM_PROF server (68–151 (47)), the PAIRCOIL server (92–152 (48)), the PAIRCOIL2 server (99–148 (49)), Matcher (99–147 (50)), and JPRED3 (99–147 (51)). Residues shown in parentheses denote regions in the untagged protein predicted to participate in a coiled-coil interaction with 90% or greater certainty. The presence or absence of an HA tag had no effect on the coiled-coil predictions.
RESULTS
Replacing the C-terminal Coiled-coil Domain with a Non-coiled-coil Domain Results in Functional Inactivation of BST-2
A current working model suggests that the BST-2 ectodomain functions as a bridge between the terminal membrane anchors to tether virions to the plasma membranes or to each other (for review, see Ref. 52). A known structural requirement for virion tethering is the formation of cysteine-linked BST-2 dimers (29, 30). The cysteines involved in BST-2 dimerization are located in the N-terminal half of the ectodomain (see Fig. 1A). Structural analyses of BST-2 indicate that the BST-2 ectodomain forms a continuous α-helix that forms a parallel dimeric coiled-coil in its C-terminal region (25–28). However, the precise role of the coiled-coil and, in particular, possible constraints regarding the size of the coiled-coil remain unclear.
FIGURE 1.

BST-2 ectodomain requires a coiled-coil sequence at the C terminus. A, schematic structure of BST-2 variants rCC1, rCC2, and rCD4 compared with HA-tagged BST-2. Transmembrane domains (TM1, TM2) and the position of the HA tag are indicated as black and shaded boxes, respectively. Numbers indicate amino acid positions and the total number of residues for each of the proteins is indicated on the right. B, expression and dimerization of BST-2 variants. 293T cells were transfected with 1 μg each of the indicated constructs together with 4 μg of empty vector DNA. Mock-transfected 293T cells were included as negative control. Cells were harvested 24 h later, washed once with PBS, and resuspended in 400 μl of PBS. Cell suspensions were divided into two equal aliquots (200 μl each) and either mixed with an equal volume of reducing sample buffer (4% sodium dodecyl sulfate, 125 mm Tris-HCl, pH 6.8, 10% 2-mercaptoethanol, 10% glycerol, 0.002% bromphenol blue) or non-reducing sample buffer lacking 2-mercaptoethanol. Samples were heated for 10 to 15 min at 95 °C with occasional vortexing of the samples to shear cellular DNA. Residual insoluble material was removed by centrifugation (2 min, 13,500 × g in an Eppendorf Minifuge). Cell lysates were subjected to SDS-PAGE (12.5% for reduced samples; 10% for non-reduced samples); proteins were transferred to PVDF membranes and reacted with a BST-2-specific polyclonal rabbit antibody. Membranes were then incubated with horseradish peroxidase-conjugated secondary antibodies (GE Healthcare) and proteins were visualized by enhanced chemiluminescence (ECL, GE Healthcare). C, 293T cells were transfected with 5 μg of NL4–3/Udel together with increasing amounts (0, 0.1 μg (50:1), 0.25 μg (20:1), or 0.5 μg (10:1)) of DNA encoding BST-2 wt (solid circles), rCC1, rCC2, or rCD4 chimera (open circles) as indicated. Virus-containing supernatants were collected 24 h after transfection and used for the infection of HeLa TZM-bl indicator cells. Virus-induced luciferase activity was recorded 48 h later and was used as measure of virus release. Values are expressed as mean ± S.E. of three to six experiments. The infectivity of NL4–3/Udel virus produced in the absence of BST-2 was defined as 100%.
To assess the functional importance of the coiled-coil sequence in BST-2, we constructed a series of BST-2 variants where the coiled-coil domain was replaced by two different segments of the vimentin coiled-coil or by a non-coiled-coil element derived from the ectodomain of human CD4. For consistency with immune recognition, we utilized a BST-2 construct containing a triple HA tag downstream of residue 148 near the C terminus of the ectodomain as described previously (24). We refer to this construct as BST-2 wt for the remainder of the study. We used coiled-coil prediction software to predict the coiled-coil region in BST-2. Five of the six predictors suggested the coiled-coil in the ectodomain starts downstream of residue 90, whereas one predicted residue 101 as start of the coiled-coil region (see “Experimental Procedures”). Based on these predictions, we created replacement variants rCD4, rCC1, and rCC2 by replacing residues 106 to 148 as described under “Experimental Procedures” and schematically depicted in Fig. 1A. Expression and dimerization of the resulting variants was confirmed by immunoblotting (Fig. 1B). The functional properties of the BST-2 variants were tested by determining their effects on HIV-1 particle release. For that purpose, 293T cells, lacking endogenous BST-2 expression, were transfected with the vpu-defective pNL4–3/Udel plasmid DNA either alone or in combination with BST-2 wt, rCC1, rCC2, or rCD4 at virus:BST-2 ratios of 50:1, 20:1, and 10:1. Viral supernatants were collected 24 h later and used for the infection of TZM-bl indicator cells. Relative virus titers were determined by measuring the virus-induced expression of luciferase in the TZM-bl cells 48 h later. The signal produced by the NL4–3/Udel virus in the absence of BST-2 was defined as 100% (Fig. 1C, 0.0 μg of BST-2). As expected, BST-2 wt inhibited the release of NL4–3/Udel virions in a dose-dependent manner (Fig. 1C, solid circles). Similarly, BST-2 rCC1, encoding a 89-residue coiled-coil segment of vimentin, inhibited the release of vpu-defective HIV-1 (Fig. 1C, left panel, open circles). BST-2 rCC2 contains a 178-residue coiled-coil segment of vimentin, which more than doubled the size of the ectodomain. This variant retained partial activity (Fig. 1C, middle panel, open circles). Finally, replacing the C-terminal coiled-coil region of BST-2 by a 114-amino acid non-coiled-coil segment of CD4 resulted in loss of function (Fig. 1C, right panel, open circles). These results therefore suggest that whereas a coiled-coil domain at the C-terminal region of the BST-2 ectodomain is important for tethering function, there may be limitations to the overall size of the ectodomain.
The Length of the BST-2 Ectodomain Is Flexible
To further address the importance of the size of the coiled-coil region, we tested whether the length of the BST-2 ectodomain could be extended by adding coiled-coil regions of vimentin to the existing BST-2 coiled-coil domain. As a control, a non-coiled-coil portion of the ectodomain of CD4 was added as well (Fig. 2A). Regions of CD4 or vimentin were inserted just upstream of the HA tag to create iCD4, iCC1, and iCC2, respectively, as detailed under “Experimental Procedures.” Thus, this series of BST-2 variants contains the full BST-2 ectodomain in addition to heterologous sequences inserted upstream of the HA tag. The resulting proteins encode ectodomains of 239 to 328 residues compared with 150 residues (i.e. residues 45–194) of the HA-tagged wild type protein (Fig. 2A). As before, protein expression and dimerization properties of the resulting variants were analyzed by immunoblotting (Fig. 2B).
FIGURE 2.

The length of the ectodomain of BST-2 is variable. A, schematic diagram of constructs used in this experiment. Transmembrane domains (TM1, TM2) and the position of the HA tag are indicated as black and shaded boxes, respectively. Numbers refer to amino acid positions and the total number of residues for each of the proteins is indicated on the right. B, expression and dimerization of BST-2 variants was tested as described for Fig. 1B. C, 293T cells were transfected with 5 μg of NL4–3/Udel together with varying amounts of BST-2 wt (closed circles), or BST-2 variants iCC1, iCC2, or iCD4 (open circles) as indicated. Virus-containing supernatants were collected 24 h later and used to infect HeLa TZM-bl indicator cells. Data analysis was performed as described in the legend to Fig. 1C. The signal produced by NL4–3/Udel virus in the absence of BST-2 was each defined as 100% infectivity.
We tested the ability of the iCD4, iCC1, and iCC2 variants to inhibit virus particle release. 293T cells were transfected with pNL4–3/Udel plasmid DNA either alone or in combination with BST-2 wt, iCD4, iCC1, or iCC2 at virus:BST-2 ratios of 50:1, 20:1, and 10:1, and virus release was determined as in Fig. 1C by measuring the virus-induced expression of luciferase in infected TZM-bl cells (Fig. 2C). As expected, BST-2 wt efficiently inhibited the release of NL4–3/Udel virions (Fig. 2C, solid circles). BST-2 iCD4 containing an insertion of 114 residues encompassing domain 3 of the CD4 ectodomain had only a very modest effect on the release of NL4–3/Udel (Fig. 2C, right panel, open circles). Similarly, BST-2 iCC2 containing a 178-residue coiled-coil element of vimentin (Fig. 2C, middle panel, open circles) was virtually inactive. In contrast, BST-2 iCC1 containing an 89-residue coiled-coil element of vimentin was capable of inhibiting virus release with almost wild type efficiency when compared with BST-2 wt (Fig. 2B, left panel, open circles). The lack of function of iCD4 could be explained by the inability of this sequence to form a coiled-coil because the vimentin coiled-coil element of similar size (iCC1) was functional. It is unclear why insertion of the 178-residue coiled-coil element of vimentin in iCC2 led to functional inactivation of BST-2. Immunoblot analysis under reducing conditions revealed reduced expression of iCC2 (Fig. 2B, left panel). Nevertheless, iCC2 appeared to efficiently dimerize as indicated by the complete absence of the 52-kDa monomer in the non-reducing gel (Fig. 2B, right panel). Therefore, an inability to form cysteine-linked dimers is unlikely to explain the functional defect. Instead, reduced protein stability could at least partially explain the functional defect. It is also possible, however, that the inability of iCC2 to restrict virus release reflects an upper limit to the size of the BST-2 ectodomain even if the coiled-coil structure is maintained. Finally, because the same vimentin fragment could not rescue BST-2 function in the context of the replacement variant (rCC2) in Fig. 1, the functional defect could indicate a general incompatibility of this vimentin segment with the BST-2 structure. Nevertheless, together with the data from Fig. 1, we conclude that changes to both the length and the primary sequence of the BST-2 ectodomain are tolerated provided a coiled-coil structure is maintained.
The Structure of the Ectodomain of BST-2 at Its N Terminus Is Flexible
Recent studies revealed the crystallographic structure of the human and mouse BST-2 ectodomain (25–28). We compiled the full-length structures of human BST-2 deposited into the RCSB PDB protein database using YASARA (Fig. 3A). Partial sequences and the murine structure were not included in this alignment. We found that whereas the structures of the C-terminal halves of the BST-2 ectodomain dimer are a near perfect match, the structures identified for the N-terminal portion appear to be more flexible with a hinge region around residue 80 (Fig. 3A). Cys53 is displaced 12 Å between PDB codes 3NWH and 3MQC, whereas Cys91 is only displaced 1.4 Å demonstrating the hinge region allows at least 12° of motion. The N-terminal region of the BST-2 ectodomain includes three cysteine residues (positions 53, 63, and 91), each of which can independently contribute to the formation of covalent cysteine-linked dimers. It also includes two N-linked carbohydrates at residues 65 and 92, which we previously reported to be dispensable for tetherin function (29).
FIGURE 3.

The N-terminal region of the ectodomain of BST-2 is flexible. A, an alignment of human BST-2 crystal structures was generated using the superpose function within YASARA 12.4.1. Structures aligned were 3MQ7 (27) (white), 3MQC (27) (magenta), 3MQB (27) (yellow), 3NWH (26) (red), and 2XG7 (26) (blue). B, schematic diagram of constructs used in this experiment. Transmembrane domains (TM1, TM2) and the position of the HA tag are indicated. Dashed boxes indicate deletions. Numbers refer to amino acid positions. C, expression and dimerization of BST-2 variants was tested as described in the legend to Fig. 1B. D, 293T cells were transfected with 5 μg each of NL4–3/Udel together with varying amounts of BST-2 wt (closed circles) or deletion mutants (open circles) as indicated. Virus-containing supernatants were collected 24 h later and used to infect HeLa TZM-bl indicator cells. Analysis of the data were performed as described in the legend to Fig. 1C. The signal produced by NL4–3/Udel virus in the absence of BST-2 was each defined as 100% infectivity.
In an attempt to characterize the functional importance of the N-terminal region of the BST-2 ectodomain, we introduced deletions within this region such that at least one cysteine residue remained (Fig. 3B). Mutants Δ51–80 and Δ51–88 retain cysteine 91, whereas mutant Δ55–87 retains cysteines 53 and 91. Finally, mutant Δ81–94 contains cysteines 53 and 63. All mutants were expressed and formed cysteine-linked dimers (Fig. 3C). The functional properties of these BST-2 deletion mutants were tested in a virus release assay as described for the previous figures. Interestingly, deletion of residues 51–80 did not affect tethering activity of BST-2 (Fig. 3D). However, deletion of eight additional residues (Δ51–88) that extended into the central hinge region resulted in functional inactivation of BST-2 (Fig. 3D).
Crystallography data indicate that the BST-2 ectodomain forms a continuous α-helix of 3.6 residues per turn. Hydrophobic residues are spaced along this helix about every fourth position so that they form an interface along one surface of the helix with another molecule of BST-2. The intervening positions consist of aliphatic or hydrophilic residues, which interact with the membrane or the extracellular fluid. One possible explanation for the lack of function of BST-2 Δ51–88 could be a change in the residue spacing on the α-helix relative to wild type BST-2 that might induce misplacement of hydrophobic interactions throughout the α-helices. To test this hypothesis, we created an additional mutant, BST-2 Δ55–87. This construct was designed to maintain the spacing of hydrophobic and hydrophilic residues in the α-helix, in-frame within the region we previously deleted. Analysis of this BST-2 variant in our virus release assay revealed that this mutant was unable to efficiently inhibit virus release (Fig. 3D) suggesting that our hypothesis was incorrect. Of note, a deletion affecting the hinge region (Δ81–94) was also poorly functional (Fig. 3D). Because the deletion in BST-2 Δ51–88 also affects the hinge region, it is possible that the hinge region in BST-2 is critical for tetherin function. Taken together our data suggest a critical function for the central hinge region in BST-2, whereas upstream sequences encoding two of the three cysteine residues and one of the two glycosylation sites are expendable for the tethering function of BST-2. Of note, the functional importance of the central hinge region in the BST-2 ectodomain was not recognized in a recent study employing alanine scanning mutagenesis (39).
Maintaining the Register of the α-Helix in the C-terminal Region of the BST-2 Ectodomain May Be Important for Tethering Function
To test the minimum sequence requirements in the C-terminal region of BST-2 we created a series of in-frame deletions as depicted in Fig. 4A. Expression and dimerization of the resulting constructs was assessed by immunoblot analysis (Fig. 4B). Initially, mutants Δ96–116, Δ96–127, and Δ131–148 were tested for their ability to inhibit virion release of NL4–3/Udel. Although alanine scanning mutagenesis of the region encompassed by residues 96 to 127 was previously reported to be without phenotype (39), neither mutant Δ96–116 nor Δ96–127 was able to inhibit virus release (Fig. 4C). In contrast, deletion of residues 131 to 148 in mutant Δ131–148, previously found to be sensitive to alanine scanning mutagenesis (39), was tolerated and did not affect BST-2 function (Fig. 4C). We reasoned that structural elements such as the register of the helix encoded by residues 96 to 127 might be important for BST-2 function. To test this hypothesis we designed an additional mutant, BST-2 Δ96–118, that extended the deletion present in the inactive Δ96–116 variant by 2 amino acids to bring the register of the α-helix back in sync with the wild type helix. Deletion of two additional residues did indeed restore the tethering activity of BST-2 (Fig. 4C, Δ96–118). These results suggest that deletions in the C-terminal coiled-coil domain of BST-2 are tolerated as long as the proper register of the BST-2 encoded α-helix is maintained.
FIGURE 4.
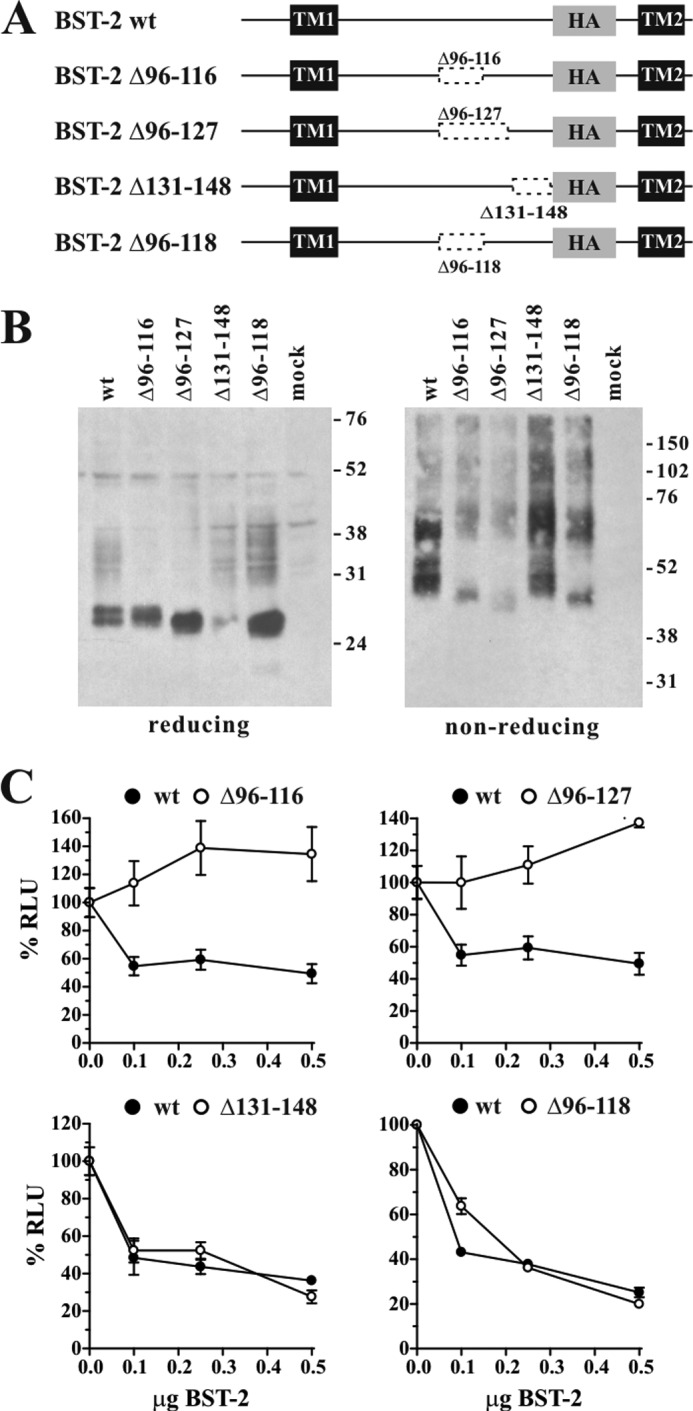
The functional properties of BST-2 deletions do not correlate with the size of the deletions. A, schematic diagram of constructs used in this experiment. Transmembrane domains (TM1, TM2) and the position of the HA tag are indicated as black and shaded boxes, respectively. Dashed boxes indicate deletions. Numbers refer to amino acid positions. B, expression and dimerization of BST-2 variants was tested as described in the legend to Fig. 1B. C, 293T cells were transfected with 5 μg each of NL4–3/Udel together with varying amounts of BST-2 wt (closed circles) or mutant DNA (open circles) as indicated. Virus-containing supernatants were collected 24 h later and used to infect HeLa TZM-bl indicator cells. Analysis of the data were performed as described in the legend to Fig. 1C. The signal produced by NL4–3/Udel virus in the absence of BST-2 was each defined as 100% infectivity.
Heptad Motifs in the BST-2 Ectodomain Are Dispensable for BST-2 Function
An important structural feature of a coiled-coil is the presence of heptad motifs. In classical coiled-coils, the 7-amino acid heptad motif places hydrophobic residues in the a and d positions and charged residues in the e and g positions to form stabilizing salt bridges resulting in “knobs into holes” packing of the helices (53). Heptad repeats in the BST-2 ectodomain were reported for the C-terminal half of the BST-2 ectodomain (25, 26) and are indicated as green and yellow boxes in Fig. 5A. The deletion mutants shown in Fig. 4 affected many of the heptad repeats, which could have contributed to their functional properties. In an effort to determine whether heptad repeats in the BST-2 ectodomain play a critical role, we created two deletion mutants, BST-2 Δ97–147 and Δ95–146. Both variants lack all seven heptad motifs but they differ slightly in the positioning and the size of the deletions (Fig. 5A). Both constructs were expressed with similar efficiency as wild type BST-2 and they formed cysteine-linked dimers even though dimerization appeared to be less efficient for both constructs (Fig. 5B). Analysis of the functional properties of these mutants revealed that despite their close similarity, BST-2 Δ97–147 was able to inhibit virus release, albeit somewhat less efficiently than the wild type protein, whereas BST-2 Δ95–146 was completely non-functional (Fig. 5C).
FIGURE 5.

Heptad motifs in the C-terminal coiled-coil region of the BST-2 ectodomain are dispensable for tetherin function. A, amino acid alignment of BST-2 deletions Δ97–147 and Δ95–146 as compared with BST-2 wt. Heptad motifs are highlighted. Green highlights indicate motifs that perfectly match the HXXHEXE consensus motif where H is hydrophobic or uncharged and E is a charged residue; yellow highlights depict motifs in which one residue in the heptad sequence does not match the canonical motif. Black and shaded areas represent the transmembrane domains and the HA tag, respectively. B, expression and dimerization of BST-2 variants was tested as described in the legend to Fig. 1B. C, 293T cells were transfected with 5 μg each of NL4–3/Udel together with varying amounts of BST-2 wt (closed circles) or mutant DNA (open circles) as indicated. Virus-containing supernatants were collected 24 h later and used to infect HeLa TZM-bl indicator cells. Analysis of the data were performed as described in the legend to Fig. 1C. Values are expressed as mean ± S.E. of nine experiments. The signal produced by NL4–3/Udel virus in the absence of BST-2 was defined as 100%.
To better understand the basis for the functional differences between BST-2 Δ96–116 and Δ96–118 as well as Δ97–147 and Δ95–146, we modeled these mutants on PDB structure 2XG7 (26) using FOLDX within YASARA (Fig. 6B). The amino acid alignment is shown in Fig. 6A. Interestingly, the structures derived for the non-functional BST-2 Δ96–116 and Δ95–146 show more hydrophobic residues on the outside of the helix (indicated in orange color in Fig. 6B) compared with the active BST-2 Δ96–118 and Δ97–147. In addition, charged residues (indicated in red and blue color in Fig. 6B) are located on the inside of the coil in BST-2 Δ96–116 and Δ95–146, inconsistent with the typical location outside the interface for mediating interactions with the extracellular fluid. These data suggest that changes to charge distributions that are unrelated to alterations of the coil register can affect the stability of the coiled-coil and determine the functional properties of BST-2. Overall, our data suggest that the formation of a cysteine-linked coiled-coil dimer of BST-2 is critical for tethering activity. Importantly, however, the presence of C-terminal heptad motifs is not critical for proper assembly of BST-2 dimers and inhibition of virus release.
FIGURE 6.

The positioning of residues along the helix interface may determine the functional properties of BST-2 variants. A, sequence alignment of BST-2 deletion mutants Δ96–116, Δ96–118, Δ97–147, and Δ95–146 against wild type BST-2. Only the region encompassing positions 81–159 in wild type BST-2 is shown. Amino acids labeled in panel B are shown in red and marked as raised fonts. B, models of BST-2 Δ96–118 and Δ96–116 as well as Δ97–147 and Δ95–146 were created using the human BST-2 structure (PDB code 2XG7) (26) as a template using FOLDX within YASARA 12.4.1. Thr98 in BST-2 Δ96–118 and the corresponding Thr100 in Δ96–116 are indicated in orange. Similarly, Leu98 in BST-2 Δ97–147 and the corresponding Leu97 in Δ95–146 are indicated in orange. Charged residues Glu98 and Arg95 are indicated in red and blue, respectively. Cys91 in Δ97–147 and Δ95–146 is highlighted in purple.
DISCUSSION
BST-2 is thought to provide a physical tether between otherwise fully detached virions and the cell surface. One of the structural prerequisites for virus tethering is the formation of cysteine-linked dimers (29, 30). Indeed, structural models of the BST-2 ectodomain suggest that BST-2 forms a parallel dimer that forms a 16–17-nm long rod-like coiled-coil (25–28), which must be anchored at both ends in a membrane. Exactly how a cell-virus tether is formed or how many BST-2 dimers are required to effectively tether a virion to the plasma membrane is currently unclear. It is also not clear exactly how Vpu prevents BST-2 from tethering nascent virions to the cell surface. Our previous studies on antibody-mediated neutralization of BST-2 indicate that inhibition of virus release by BST-2 involves a very early step of virus assembly prior to Gag proteins reaching the cell surface (54). However, much of the Vpu-BST-2 interaction remains to be investigated.
Previous work by Hammonds and co-workers (39) used alanine scanning mutagenesis to identify regions of functional importance in the BST2 ectodomain. In that study, alanine mutagenesis was well tolerated across much of the BST-2 ectodomain and only changes near the N and C termini of the ectodomain significantly affected BST-2 function (39). These results may not be surprising if one takes into consideration that alanine is the least destabilizing residue to add to the interaction surface of an oligomeric coiled-coil (55–57) and that replacing four consecutive residues at a time by alanine would not be expected to greatly disrupt the structure and interaction of the BST2 coiled-coil (58, 59). Because of these limitations, previous alanine scanning mutagenesis did not recognize the importance of the central hinge region nor the relative unimportance of the size of the BST-2 ectodomain.
Our current study focused on the characterization of functionally critical elements in the BST-2 ectodomain using insertion, substitution, and deletion mutagenesis. The fact that in a previous study the BST-2 ectodomain could be replaced by a ∼120 residue heterologous sequence consisting of 32 residues of the transferrin receptor extracellular stalk plus 75 residues of the cytoplasmic dimeric protein dystrophia myotonica protein kinase coiled-coil, already demonstrated that the primary sequence of the BST-2 ectodomain was not strictly required for tethering function (30). Our own observation showing that the coiled-coil region in the C-terminal region of the BST-2 ectodomain could be substituted with a coiled-coil region of vimentin but not with a region from the CD4 ectodomain (Fig. 1) is consistent with this conclusion and, in addition, highlights the importance of a C-terminal coiled-coil structure. Interestingly, we found that in addition to flexibility in the primary sequence of the coiled-coil, there is also significant flexibility in its size. Indeed, we were able to almost double the size of the BST-2 ectodomain by adding a segment of the vimentin coiled-coil without loss of function (Fig. 2). Surprisingly, whereas those data point to a critical role of a coiled-coil structure of the BST-2 ectodomain, we were able to significantly reduce the size of the BST-2 ectodomain without loss of tethering function. In fact, we were able to delete more than 50 residues of the C-terminal region of the BST-2 ectodomain (Fig. 5, Δ97–147). This result is particularly striking because the deletion in BST-2 Δ97–147 removes most of the coiled-coil domain including all seven heptad motifs thought to be critical for the formation of a coiled-coil (see Fig. 5A). We were also able to delete up to 30 residues from the N-terminal portion of the BST-2 ectodomain, including two of the three cysteines critical for dimerization (Fig. 3, Δ51–80), again without loss of function. In all cases where functional inactivation was observed, differences in protein expression or the ability to form cysteine-linked dimers did not account for the observed effects on virus release with the possible exception of variant iCC2 (Fig. 2B).
Structural data indicate that the BST-2 ectodomain forms a continuous α-helix in the crystal lattice (26–28). It was therefore conceivable that loss of function in at least some of our deletion mutants was caused by structural disturbances due to changes in the register of the helix. In an attempt to fit our data into a functional model, we compared the functional properties of mutations that differed by only two or three amino acids. We have two sets of mutants (Δ96–116/Δ96–118 and Δ97–147/Δ95–146) that fit this description (Fig. 6). Two of these mutants (Δ96–118 and Δ97–147) are functional, whereas the other two (Δ96–116 and Δ95–146) are not (Figs. 4 and 5). The amino acid alignment of these mutants against BST-2 wt (Fig. 6A) indicates that the functional properties of these BST-2 variants are not determined by the size of the deletions because the deletion in BST-2 Δ97–147 (functional) is much larger (51 amino acids) than in the non-functional BST-2 Δ96–116 (21 residues). Interestingly, however, we found that in both cases, the non-functional variant includes a charged residue in the dimer interface that was not present in the functional variant. Molecular modeling of the mutants based on published x-ray crystallographic data of the BST-2 ectodomain suggests differential positioning of these charged residues on the helices that could explain the functional properties of the variants. In both cases, the charged residue, Glu98 in Δ96–116 (Fig. 6B, red) and Arg95 in Δ95–146 (Fig. 6B, blue), points into the dimer interface, whereas adjacent hydrophobic residues (Fig. 6B, orange) point toward the water phase of the helix. In contrast, in the functional variants the hydrophobic residues point toward the dimer interface and could thus stabilize the structure of the BST-2 dimer. Taken together our data demonstrate significant flexibility in the size of the BST-2 ectodomain as long as the positioning of residues critical for the formation of the dimer interface is conserved.
Acknowledgments
We thank Drs. Eri Miyagi, Sarah Welbourn, Takeshi Yoshida, and Haruka Yoshii for helpful suggestions and critical comments on the manuscript. We also thank Dr. Alicia Buckler-White and Ronald Plishka for conducting nucleotide sequence analyses.
This work was supported by the Intramural Research Program of the NIAID, National Institutes of Health.
- TM
- transmembrane
- PDB
- Protein Data Bank.
REFERENCES
- 1. Van Damme N., Goff D., Katsura C., Jorgenson R. L., Mitchell R., Johnson M. C., Stephens E. B., Guatelli J. (2008) The interferon-induced protein BST-2 restricts HIV-1 release and is down-regulated from the cell surface by the viral Vpu protein. Cell. Host. Microbe. 3, 245–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Neil S. J., Zang T., Bieniasz P. D. (2008) Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature 451, 425–430 [DOI] [PubMed] [Google Scholar]
- 3. Gupta R. K., Hué S., Schaller T., Verschoor E., Pillay D., Towers G. J. (2009) Mutation of a single residue renders human tetherin resistant to HIV-1 Vpu-mediated depletion. PLoS Pathog. 5, e1000443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jia B., Serra-Moreno R., Neidermyer W., Rahmberg A., Mackey J., Fofana I. B., Johnson W. E., Westmoreland S., Evans D. T. (2009) Species-specific activity of SIV Nef and HIV-1 Vpu in overcoming restriction by tetherin/BST2. PLoS Pathog. 5, e1000429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hauser H., Lopez L. A., Yang S. J., Oldenburg J. E., Exline C. M., Guatelli J. C., Cannon P. M. (2010) HIV-1 Vpu and HIV-2 Env counteract BST-2/tetherin by sequestration in a perinuclear compartment. Retrovirology 7, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Le Tortorec A., Neil S. J. (2009) Antagonism to and intracellular sequestration of human tetherin by the human immunodeficiency virus type 2 envelope glycoprotein. J. Virol. 83, 11966–11978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sauter D., Schindler M., Specht A., Landford W. N., Münch J., Kim K. A., Votteler J., Schubert U., Bibollet-Ruche F., Keele B. F., Takehisa J., Ogando Y., Ochsenbauer C., Kappes J. C., Ayouba A., Peeters M., Learn G. H., Shaw G., Sharp P. M., Bieniasz P., Hahn B. H., Hatziioannou T., Kirchhoff F. (2009) Tetherin-driven adaptation of Vpu and Nef function and the evolution of pandemic and nonpandemic HIV-1 strains. Cell Host Microbe 6, 409–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yang S. J., Lopez L. A., Hauser H., Exline C. M., Haworth K. G., Cannon P. M. (2010) Anti-tetherin activities in Vpu-expressing primate lentiviruses. Retrovirology 7, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhang F., Wilson S. J., Landford W. C., Virgen B., Gregory D., Johnson M. C., Munch J., Kirchhoff F., Bieniasz P. D., Hatziioannou T. (2009) Nef proteins from simian immunodeficiency viruses are tetherin antagonists. Cell Host Microbe 6, 54–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kobayashi T., Ode H., Yoshida T., Sato K., Gee P., Yamamoto S. P., Ebina H., Strebel K., Sato H., Koyanagi Y. (2011) Identification of amino acids in the human tetherin transmembrane domain responsible for HIV-1 Vpu interaction and susceptibility. J. Virol. 85, 932–945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yoshida T., Kao S., Strebel K. (2011) Identification of residues in the BST-2 TM domain important for antagonism by HIV-1 Vpu using a gain-of-function approach. Front. Microbiol. 2, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rong L., Zhang J., Lu J., Pan Q., Lorgeoux R. P., Aloysius C., Guo F., Liu S. L., Wainberg M. A., Liang C. (2009) The transmembrane domain of BST-2 determines its sensitivity to down-modulation by HIV-1 Vpu. J. Virol. 83, 7536–7546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mangeat B., Gers-Huber G., Lehmann M., Zufferey M., Luban J., Piguet V. (2009) HIV-1 Vpu neutralizes the antiviral factor Tetherin/BST-2 by binding it and directing its beta-TrCP2-dependent degradation. PLoS Pathog. 5, e1000574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dubé M., Roy B. B., Guiot-Guillain P., Binette J., Mercier J., Chiasson A., Cohen E. A. (2010) Antagonism of tetherin restriction of HIV-1 release by Vpu involves binding and sequestration of the restriction factor in a perinuclear compartment. PLoS Pathog. 6, e1000856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Douglas J. L., Viswanathan K., McCarroll M. N., Gustin J. K., Früh K., Moses A. V. (2009) Vpu directs the degradation of the human immunodeficiency virus restriction factor BST-2/Tetherin via a βTrCP-dependent mechanism. J. Virol. 83, 7931–7947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Vigan R., Neil S. J. (2011) Separable determinants of subcellular localization and interaction account for the inability of group O HIV-1 Vpu to counteract tetherin. J. Virol. 85, 9737–9748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Skasko M., Wang Y., Tian Y., Tokarev A., Munguia J., Ruiz A., Stephens E. B., Opella S. J., Guatelli J. (2012) HIV-1 Vpu protein antagonizes innate restriction factor BST-2 via lipid-embedded helix-helix interactions. J. Biol. Chem. 287, 58–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhou J., Zhang Z., Mi Z., Wang X., Zhang Q., Li X., Liang C., Cen S. (2012) Characterization of the interface of the bone marrow stromal cell antigen 2-Vpu protein complex via computational chemistry. Biochemistry 51, 1288–1296 [DOI] [PubMed] [Google Scholar]
- 19. Kueck T., Neil S. J. (2012) A cytoplasmic tail determinant in HIV-1 Vpu mediates targeting of tetherin for endosomal degradation and counteracts interferon-induced restriction. PLoS Pathog. 8, e1002609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ohtomo T., Sugamata Y., Ozaki Y., Ono K., Yoshimura Y., Kawai S., Koishihara Y., Ozaki S., Kosaka M., Hirano T., Tsuchiya M. (1999) Molecular cloning and characterization of a surface antigen preferentially overexpressed on multiple myeloma cells. Biochem. Biophys. Res. Commun. 258, 583–591 [DOI] [PubMed] [Google Scholar]
- 21. Goto T., Kennel S. J., Abe M., Takishita M., Kosaka M., Solomon A., Saito S. (1994) A novel membrane antigen selectively expressed on terminally differentiated human B cells. Blood 84, 1922–1930 [PubMed] [Google Scholar]
- 22. Ishikawa J., Kaisho T., Tomizawa H., Lee B. O., Kobune Y., Inazawa J., Oritani K., Itoh M., Ochi T., Ishihara K. (1995) Molecular cloning and chromosomal mapping of a bone marrow stromal cell surface gene, BST2, that may be involved in pre-B-cell growth. Genomics 26, 527–534 [DOI] [PubMed] [Google Scholar]
- 23. Kupzig S., Korolchuk V., Rollason R., Sugden A., Wilde A., Banting G. (2003) Bst-2/HM1.24 is a raft-associated apical membrane protein with an unusual topology. Traffic 4, 694–709 [DOI] [PubMed] [Google Scholar]
- 24. Andrew A. J., Kao S., Strebel K. (2011) C-terminal hydrophobic region in human bone marrow stromal cell antigen 2 (BST-2)/tetherin protein functions as second transmembrane motif. J. Biol. Chem. 286, 39967–39981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hinz A., Miguet N., Natrajan G., Usami Y., Yamanaka H., Renesto P., Hartlieb B., McCarthy A. A., Simorre J. P., Göttlinger H., Weissenhorn W. (2010) Structural basis of HIV-1 tethering to membranes by the BST-2/tetherin ectodomain. Cell Host Microbe 7, 314–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Schubert H. L., Zhai Q., Sandrin V., Eckert D. M., Garcia-Maya M., Saul L., Sundquist W. I., Steiner R. A., Hill C. P. (2010) Structural and functional studies on the extracellular domain of BST2/tetherin in reduced and oxidized conformations. Proc. Natl. Acad. Sci. U.S.A. 107, 17951–17956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yang H., Wang J., Jia X., McNatt M. W., Zang T., Pan B., Meng W., Wang H. W., Bieniasz P. D., Xiong Y. (2010) Structural insight into the mechanisms of enveloped virus tethering by tetherin. Proc. Natl. Acad. Sci. U.S.A. 107, 18428–18432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Swiecki M., Scheaffer S. M., Allaire M., Fremont D. H., Colonna M., Brett T. J. (2011) Structural and biophysical analysis of BST-2/tetherin ectodomains reveals an evolutionary conserved design to inhibit virus release. J. Biol. Chem. 286, 2987–2997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Andrew A. J., Miyagi E., Kao S., Strebel K. (2009) The formation of cysteine-linked dimers of BST-2/tetherin is important for inhibition of HIV-1 virus release but not for sensitivity to Vpu. Retrovirology 6, 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Perez-Caballero D., Zang T., Ebrahimi A., McNatt M. W., Gregory D. A., Johnson M. C., Bieniasz P. D. (2009) Tetherin inhibits HIV-1 release by directly tethering virions to cells. Cell 139, 499–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Masuyama N., Kuronita T., Tanaka R., Muto T., Hirota Y., Takigawa A., Fujita H., Aso Y., Amano J., Tanaka Y. (2009) HM1.24 is internalized from lipid rafts by clathrin-mediated endocytosis through interaction with α-adaptin. J. Biol. Chem. 284, 15927–15941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rollason R., Korolchuk V., Hamilton C., Schu P., Banting G. (2007) Clathrin-mediated endocytosis of a lipid-raft-associated protein is mediated through a dual tyrosine motif. J. Cell. Sci. 120, 3850–3858 [DOI] [PubMed] [Google Scholar]
- 33. Fritz J. V., Tibroni N., Keppler O. T., Fackler O. T. (2012) HIV-1 Vpu's lipid raft association is dispensable for counteraction of the particle release restriction imposed by CD317/tetherin. Virology 424, 33–44 [DOI] [PubMed] [Google Scholar]
- 34. Lopez L. A., Yang S. J., Exline C. M., Rengarajan S., Haworth K. G., Cannon P. M. (2012) Anti-tetherin activities of HIV-1 Vpu and Ebola virus glycoprotein do not involve removal of tetherin from lipid rafts. J. Virol. 86, 5467–5480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Fitzpatrick K., Skasko M., Deerinck T. J., Crum J., Ellisman M. H., Guatelli J. (2010) Direct restriction of virus release and incorporation of the interferon-induced protein BST-2 into HIV-1 particles. PLoS Pathog. 6, e1000701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Habermann A., Krijnse-Locker J., Oberwinkler H., Eckhardt M., Homann S., Andrew A., Strebel K., Kräusslich H. G. (2010) CD317/tetherin is enriched in the HIV-1 envelope and down-regulated from the plasma membrane upon virus infection. J. Virol. 84, 4646–4658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hammonds J., Wang J. J., Yi H., Spearman P. (2010) Immunoelectron microscopic evidence for tetherin/BST2 as the physical bridge between HIV-1 virions and the plasma membrane. PLoS Pathog. 6, e1000749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jones P. H., Mehta H. V., Maric M., Roller R. J., Okeoma C. M. (2012) Bone marrow stromal cell antigen 2 (BST-2) restricts mouse mammary tumor virus (MMTV) replication in vivo. Retrovirology 9, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hammonds J., Ding L., Chu H., Geller K., Robbins A., Wang J. J., Yi H., Spearman P. (2012) The tetherin/BST-2 coiled-coil ectodomain mediates plasma membrane microdomain localization and restriction of particle release. J. Virol. 86, 2259–2272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Adachi A., Gendelman H. E., Koenig S., Folks T., Willey R., Rabson A., Martin M. A. (1986) Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J. Virol. 59, 284–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Klimkait T., Strebel K., Hoggan M. D., Martin M. A., Orenstein J. M. (1990) The human immunodeficiency virus type 1-specific protein vpu is required for efficient virus maturation and release. J. Virol. 64, 621–629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lupas A., Van Dyke M., Stock J. (1991) Predicting coiled coils from protein sequences. Science 252, 1162–1164 [DOI] [PubMed] [Google Scholar]
- 43. Miyagi E., Andrew A. J., Kao S., Strebel K. (2009) Vpu enhances HIV-1 virus release in the absence of Bst-2 cell surface down-modulation and intracellular depletion. Proc. Natl. Acad. Sci. U.S.A. 106, 2868–2873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Van Durme J., Delgado J., Stricher F., Serrano L., Schymkowitz J., Rousseau F. (2011) A graphical interface for the FoldX forcefield. Bioinformatics 27, 1711–1712 [DOI] [PubMed] [Google Scholar]
- 45. Krieger E., Koraimann G., Vriend G. (2002) Increasing the precision of comparative models with YASARA NOVA. A self-parameterizing force field. Proteins 47, 393–402 [DOI] [PubMed] [Google Scholar]
- 46. Delorenzi M., Speed T. (2002) An HMM model for coiled-coil domains and a comparison with PSSM-based predictions. Bioinformatics 18, 617–625 [DOI] [PubMed] [Google Scholar]
- 47. Bartoli L., Fariselli P., Krogh A., Casadio R. (2009) CCHMM_PROF. A HMM-based coiled-coil predictor with evolutionary information. Bioinformatics 25, 2757–2763 [DOI] [PubMed] [Google Scholar]
- 48. Berger B., Wilson D. B., Wolf E., Tonchev T., Milla M., Kim P. S. (1995) Predicting coiled coils by use of pairwise residue correlations. Proc. Natl. Acad. Sci. U.S.A. 92, 8259–8263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. McDonnell A. V., Jiang T., Keating A. E., Berger B. (2006) Paircoil2. Improved prediction of coiled-coils from sequence. Bioinformatics 22, 356–358 [DOI] [PubMed] [Google Scholar]
- 50. Fischetti V. A., Landau G. M., Sellers P. H., Schmidt J. P. (1993) Identifying periodic occurences of a template with applications to protein structure. Inf. Process. Lett. 45, 11–18 [Google Scholar]
- 51. Cole C., Barber J. D., Barton G. J. (2008) The Jpred 3 secondary structure prediction server. Nucleic Acids Res. 36, W197-W201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Andrew A., Strebel K. (2011) The interferon-inducible host factor bone marrow stromal antigen 2/tetherin restricts virion release, but is it actually a viral restriction factor? J. Interferon. Cytokine Res. 31, 137–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Crick F. H. (1953) The packing of α-helices. Simple coiled-coils. Acta Crystallogr. 6, 689–697 [Google Scholar]
- 54. Miyagi E., Andrew A., Kao S., Yoshida T., Strebel K. (2011) Antibody-mediated enhancement of HIV-1 and HIV-2 production from BST-2/tetherin+ cells. J. Virol. 85, 11981–11994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wagschal K., Tripet B., Lavigne P., Mant C., Hodges R. S. (1999) The role of position a in determining the stability and oligomerization state of α-helical coiled coils. 20 amino acid stability coefficients in the hydrophobic core of proteins. Protein Sci. 8, 2312–2329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Tripet B., Wagschal K., Lavigne P., Mant C. T., Hodges R. S. (2000) Effects of side-chain characteristics on stability and oligomerization state of a de novo-designed model coiled-coil. 20 amino acid substitutions in position “d.” J. Mol. Biol. 300, 377–402 [DOI] [PubMed] [Google Scholar]
- 57. Pace C. N., Scholtz J. M. (1998) A helix propensity scale based on experimental studies of peptides and proteins. Biophys. J. 75, 422–427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Lu S. M., Hodges R. S. (2004) Defining the minimum size of a hydrophobic cluster in two-stranded α-helical coiled-coils. Effects on protein stability. Protein Sci. 13, 714–726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kwok S. C., Hodges R. S. (2004) Effect of chain length on coiled-coil stability. Decreasing stability with increasing chain length. Biopolymers 76, 378–390 [DOI] [PubMed] [Google Scholar]