

A fundamental challenge in plant biology is to discover the unknown connections within and between pathways and to associate previously unknown genes with specific biological processes. MORPH is a computational method for predicting genes that function in or regulate a biological pathway. MORPH fills pathway gaps and defines complex biological networks as shown for Arabidopsis and tomato.
Abstract
Closing gaps in our current knowledge about biological pathways is a fundamental challenge. The development of novel computational methods along with high-throughput experimental data carries the promise to help in the challenge. We present an algorithm called MORPH (for module-guided ranking of candidate pathway genes) for revealing unknown genes in biological pathways. The method receives as input a set of known genes from the target pathway, a collection of expression profiles, and interaction and metabolic networks. Using machine learning techniques, MORPH selects the best combination of data and analysis method and outputs a ranking of candidate genes predicted to belong to the target pathway. We tested MORPH on 230 known pathways in Arabidopsis thaliana and 93 known pathways in tomato (Solanum lycopersicum) and obtained high-quality cross-validation results. In the photosynthesis light reactions, homogalacturonan biosynthesis, and chlorophyll biosynthetic pathways of Arabidopsis, genes ranked highly by MORPH were recently verified to be associated with these pathways. MORPH candidates ranked for the carotenoid pathway from Arabidopsis and tomato are derived from pathways that compete for common precursors or from pathways that are coregulated with or regulate the carotenoid biosynthetic pathway.
INTRODUCTION
A biological pathway is the set of molecular entities involved in a given biological process and the interrelations among those entities. Pathways are to some extent the biologist’s simplification. Pathway boundaries are inherently fuzzy, but they are valuable for understanding biology and organizing biological knowledge (e.g., a metabolic or signaling pathway). Although current knowledge about some biological pathways may be substantial and useful for systems-level analyses, not all genes that participate in and/or affect function of these pathways are known. The challenge of identifying missing pathway members is a major challenge for biological research.
Prime examples of information gaps are biosynthetic pathways leading to secondary metabolites in plants. These pathways have been studied extensively (Saito et al., 2008), but little is known about pathway control mechanisms. We have limited understanding of the nature of interactions between metabolites and gene expression, and we have only a partial grasp of the relationship between transcriptional regulation and phenotype (Pigliucci, 2009). Even in the best-studied metabolic pathways, there remain information gaps where some participating genes are still unknown. Moreover, many plant genes remain to be annotated and have no known function (Gerdes et al., 2011).
In the postgenome era, the plethora of available high-throughput “omics” data (e.g., genomics, proteomics, metabolomics, and fluxomics) can assist in the task of linking genes to pathways. Several genome-wide computational methods attempt to close gaps in metabolic networks (Thimm et al., 2004; Yamanishi et al., 2004; Usadel et al., 2009; Orth and Palsson, 2010). Some methods use established knowledge about a pathway in a model that analyzes microarray expression data, since genes from the same pathway typically manifest coordinated expression under various conditions (Stuart et al., 2003; Allocco et al., 2004). One such tool is MapMan (Thimm et al., 2004), which displays large data sets (e.g., gene expression data from Arabidopsis thaliana Affymetrix arrays) onto diagrams of metabolic pathways or other processes.
Coexpression analysis can identify genes that tend to show similar expression profiles across many treatments (Usadel et al., 2009). By calculating coexpression for groups of genes, one can generate hypotheses about the function of unknown genes that show expression patterns similar to genes whose function is known, using the guilt by association paradigm. Several Web-based tools perform coexpression analysis in plants, including ACT (Manfield et al., 2006), GeneCAT (Mutwil et al., 2008), ATED-II (Obayashi et al., 2009), BAR Expression Angler (Toufighi et al., 2005), and CressExpress (Srinivasasainagendra et al., 2008). These tools use publicly available gene expression data and compute lists of genes coexpressed with bait genes given as input by the user. The PlaNet tool builds further on use of coexpression data by comparing coexpression networks across multiple plant species to predict functional homologs (Mutwil et al., 2011).
All of the plant tools mentioned above rely solely on coexpression. Methods developed for nonplant species analyze one or several types of high-throughput data, such as coexpression (Kharchenko et al., 2005), phylogeny similarity profiles (Pellegrini et al., 1999), and spatial clustering of genes on chromosomes (Lee and Sonnhammer, 2003). The ADOMETA method combines all of the data types above, together with a metabolic dependencies (MD) network, to predict enzymes of orphan reactions in metabolic pathways (Kharchenko et al., 2004; Chen and Vitkup, 2006; Kharchenko et al., 2006). In ADOMETA, genes are first partitioned into two classes: metabolic and nonmetabolic genes. Next, the genes are compared with their neighbors in the metabolic network using Adaboost, which combines different methods and association scores. The highest scoring genes are predicted as those encoding a catalyzer of the studied orphan reaction. These methods were designed for such organisms as Escherichia coli, Saccharomyces cerevisiae, or Bacillus subtilis, for which high-throughput information is much more abundant than for plants.
Closing information gaps in plant metabolic pathways is part of a larger community effort to predict gene function using heterogeneous data sources. Often, these data sources are represented as networks with nodes corresponding to genes and edges denoting functional similarity. Most network-based functional inference algorithms work under the assumption that the closer two nodes are in the network the more likely they are to share a common functionality (reviewed in Sharan et al., 2007). These approaches are generally useful in inferring broader functions, such as a biological process or a biological pathway, as opposed to the molecular/biochemical function, which are typically inferred by homology-based approaches. Thus, network- and homology-based approaches for annotating genomes are often complementary (reviewed in Janga et al., 2011).
Several methods were developed to detect associations using only protein–protein interaction (PPI) networks (Bader and Hogue, 2003; Deng et al., 2003; Letovsky and Kasif, 2003; Nabieva et al., 2005; Kourmpetis et al., 2010). Studies assumed that, given its neighbors in the graph, the function of a protein is independent of all other proteins. With this assumption, Markov random field (MRF) models were used for function prediction (Deng et al., 2003; Letovsky and Kasif, 2003). Other methods employed variations of the k-nearest neighbor (k-NN) classifier (Tan et al., 2005) for integrating gene expression profiles with interaction networks for functional prediction (Kuramochi and Karypis, 2005; Pandey et al., 2009). In k-NN, for each gene, the k genes most similar to it in terms of the input data are defined as its neighbors, and the gene is scored by the average similarity of its neighbors to genes of the target functionality (e.g., a biological process or a pathway). Typically k is 10 or 20. k-NN–based methods that integrate different data sources use them to calculate a combined similarity matrix. For example, Pandey et al. (2009) combined gene expression and network data by concatenating the gene expression matrices with the adjacency matrices of the networks and used cosine similarity to score candidate genes. Comparative studies reported that for protein function prediction, k-NN produces results comparable to other classifiers, such as support vector machines. Because of the simplicity of k-NN, its results are easier to interpret (Kuramochi and Karypis, 2005; Wang and Scott, 2005; Yao and Ruzzo, 2006).
In this study, we develop a new computational framework called MORPH (for module-guided ranking of candidate pathway genes) for high-confidence prediction of candidate genes that function in or regulate a given biological pathway. Our prediction method provides highly significant predictions despite the limited data available in plants compared with other model organisms. Our method is not limited to analyzing known enzymes (Popescu and Yona, 2005) or to discriminating metabolic from nonmetabolic genes as done in ADOMETA.
Like some other guilt by association coexpression methods, MORPH receives as input a list of genes known to participate in the specific target pathway, gene expression profiles from multiple studies, and an interaction network. In addition, and unlike other methods, MORPH uses a collection of gene partitions into functional modules. MORPH tests multiple combinations of expression data sets and partitions, identifies the best combination using cross-validation, and ranks genes in terms of strength of the evidence that they belong to the target pathway. Hence, MORPH builds upon coexpression analysis and finds the best combination of gene expression data and network information to assess the candidates of a specific pathway. Ranking is done by measuring expression similarity of each candidate gene to the target pathway genes that occupy the same module, with appropriate normalization to account for different module homogeneities.
MORPH was developed and tested using resources for two distinct plant species: Arabidopsis thaliana and tomato (Solanum lycopersicum). We tested our method using a cross-validation-based technique developed by Kharchenko et al. (2006) on 230 Arabidopsis biological pathways (downloaded from AraCyc and MapMan) and 93 tomato pathways (downloaded from MapMan). MORPH showed a consistently high prediction quality. Detailed analysis of the top-ranked candidates in three Arabidopsis pathways reveals accurate and valuable predictions. In tomato, MORPH uses gene networks that were generated by transforming Arabidopsis networks into tomato networks on the basis of homology. MORPH provides a twofold improvement upon standard coexpression analysis in our ability to predict tomato pathways. MORPH can be accessed online or downloaded and used independently on a PC from http://biocourse.weizmann.ac.il/morph/.
RESULTS
We developed a method for ranking candidate genes that are missing in a given target pathway and tested the method on data of Arabidopsis and tomato. The method, called MORPH, ranks genes using auxiliary data, including expression profiles and biological networks. See Figure 1 for an overview. Genes are partitioned into modules using several methods (described in detail below), and the best combination of grouping and further analysis is selected. We first describe the analyses performed in Arabidopsis and then in tomato and in each case demonstrate the ability of MORPH to provide high quality predictions and useful new candidates.
Figure 1.

Overview of the MORPH Algorithm.
Using several partitioning methods (see Partitioning Gene Modules and Networks), all candidate genes (black circles) are partitioned into modules (shaded boxes) together with a set of target genes (squares) known to belong to the target pathway. Modules represent distinct patterns of gene expression and/or protein interactions (candidate genes are now distinguished by the module to which they belong, with dark circles, triangles and light circles for the top, middle, and bottom modules, respectively). For each module, MORPH computes the average expression pattern of the known target genes that fall into that module (marked by a circle with an “x” in the center on the right) and then scores all other genes by their similarity to the average pattern in that module (depicted by the arrow length; numbers indicate ranking of the similarity for each module, where 1 is the most similar). Scores are normalized and united from all modules to obtain a single ranking of all candidate genes. A cross-validation procedure (data not shown) is used to score the resulting ranking.
[See online article for color version of this figure.]
Arabidopsis Expression Profiles and Pathways
We used 216 published microarray profiles of Arabidopsis gene expression responses to specific stimuli (see Methods; see Supplemental Data Set 1 online for array accession numbers). These data sets included 64 profiles of seedling tissues (the seedlings data set), 99 profiles of different tissues (the tissues data set), and 53 profiles of seed tissues at different developmental stages (the seeds data set). Each set was analyzed separately. In addition, we analyzed the combined seedlings and tissues data set, named DS1. A total of 66 pathways derived from AraCyc and 164 pathways from MapMan were used in the analysis (see Methods; see Supplemental Data Set 2 online for a complete list of pathways and associated genes).
Partitioning Gene Modules and Networks
A key step in MORPH is the partitioning of genes into modues or clusters. We used two different strategies to cluster Arabidopsis genes: gene expression–based clustering and modules defined using external information. First, we devised five different clustering solutions (see Methods): (1) clustering the genes by coexpression using two different algorithms, self-organizing map (SOM) and CLuster Identification via Connectivity Kernels (CLICK); (2) enzymes: a bipartition into the genes that encode enzymes and the rest; (3) orthologs: a bipartition of genes into those that have orthologs in rice (Oryza sativa), maize (Zea mays), and the rest; and (4) no clustering: the set of all genes as a single module. We also used two networks to construct additional modules: (i) MD network: the network comprises genes as nodes. An edge is added between two genes if their gene products share a metabolite (see Methods). (ii) PPI network: the network comprises genes as nodes. An edge is added between two genes if there is an evidence of interaction between their gene products (see Methods). In total, we devised eight clustering solutions.
To construct modules from gene expression and network data, we initially used the Matisse algorithm (Ulitsky and Shamir, 2007), which identifies sets of coexpressed genes that induce connected subnetworks. Since the MD network contained only ∼1200 genes, we modified Matisse to increase coverage of the underlying gene set. The modified version, called Matisse*, expands the modules by adding genes that show a high level of similarity to the average expression pattern of a given module even if these genes do not obey the connectivity constraints. The expansion step increased module size considerably –3.7-fold for the MD network. Therefore, we used Matisse* for the MD and PPI modules. For each network, we added an additional set containing all genes that were not included in other modules. We also constructed gene clusters in the PPI data using the Markov clustering (MCL) algorithm, which was reported to perform well in this task (Enright et al., 2002; Sharan et al., 2007; Vlasblom and Wodak, 2009).
Clustering Guided Scoring of Pathway Genes
Given a clustering solution (for a given data set, partitioning genes into modules) and a target pathway, our goal is to rank the remaining genes in terms of how plausible it is that a given gene is associated with the target pathway (Figure 1). For each module generated as described above, we identified genes from the target pathways and computed, for each other gene in the same module, its average coexpression similarity to the pathway genes in that module. The rationale is that while the modules reflect various broad functions, some of these functions may be related to the target pathway and, hence, on average, the pathway genes would show higher coexpression similarity than arbitrary genes in the same module. Since modules varied in size and homogeneity (i.e., average coexpression level), the gene pathway similarity scores within each module were standardized (see Methods). Pearson correlation was chosen for evaluating coexpression as it slightly outperformed Spearman correlation (see Supplemental Methods 1 online).
Assessment of Ranking Using the Self-Rank Curve
To assess the performance of the method, we used a technique developed by Kharchenko et al. (2006) based on leave-one-out cross-validation (LOOCV). The validation procedure repeatedly removes one gene from the target pathway (the test gene), generates the ranking based on the remaining genes (the training set), and calculates the rank of the test gene, denoted as the self-rank of that gene. Then, one can plot for every self-rank threshold in some predefined interval (e.g., 0 to 1000) the fraction of pathway genes that were detected at the threshold when acting as test genes. To obtain a score in the range 0 to 1, we rescaled the curve so that 1 denotes perfect ranking (see Methods). We call this score the relative area under the curve of the self-ranked genes (AUSR). Figure 2 shows an example of the self-rank plot using the carotenoid core pathway (see Supplemental Data Set 2 online for list of genes used). The plot was derived using MORPH with the SOM clustering algorithm. For reference, the expected self-rank derived by randomly choosing candidate genes is plotted as well.
Figure 2.

The Self-Rank Plot of the Carotenoid Biosynthetic Pathway That Contains 13 Genes.
For each value of the self-rank threshold on the x axis, the plot shows the fraction of genes in the pathway that were ranked below that threshold (black line) using the LOOCV method. The gray line shows the expected plot for a randomly selected gene set of size 13 (see Supplemental Data Set 2 online).
Matisse was shown to be effective in finding modules of functionally related genes, but it usually assigns only a small fraction of the genes into modules. We therefore first compared the quality of the rankings produced by Matisse and Matisse* using the metabolic dependency network and each gene expression data set on the 66 AraCyc pathways. Although Matisse* markedly improved the coverage, it produced slightly inferior results using all four examined data sets (difference of <0.05 average AUSR over all data sets; data not shown). On average, the seedlings data set provided the best scores. When we used the PPI network, both Matisse and Matisse* modules received very similar scores (data not shown). Using the MD network, we obtained higher quality predictions compared with using the PPI network. Since Matisse* was observed to perform much better in terms of coverage (i.e., the percentage of genes included in the modules), and only marginally worse in AUSR, it was used in all further analyses.
Customizing the Use of Gene Expression Data Sets
We compared the predictive power of using the tissues and seedlings profiles separately and of using the united data set DS1 on the 66 AraCyc pathways. We used Matisse*, combined with the MD network, to create modules. DS1 was significantly inferior, yielding an average AUSR of 0.34 compared with 0.43 provided by the seedlings data set (P = 0.016) and 0.37 provided by the tissues data set (P = 0.13). Figure 3 compares AUSR scores for each pathway using the DS1 and/or the seedlings data sets. Although more pathways attained better scores using the seedlings data set, some pathways had much higher scores using DS1. The differences in scores were often large. For example, the “ethylene biosynthesis from Met” pathway received a score of 0.115 using the seedlings data set and a score of 0.73 using DS1. These results inspired us to refine the MORPH algorithm by a model selection step to optimize analysis of a specific pathway, as will be explained in the next section.
Figure 3.

The Difference in AUSR Scores between the Seedlings Data Set and the Combined Seedlings and Tissues Data Sets (DS1).
Each column represents a difference in the AUSR score for one of the 66 tested pathways. The modules used here were created by Matisse* (see Methods) with the MD network.
[See online article for color version of this figure.]
Pathway-Specific Model Selection
The MORPH algorithm was applied using all combinations of (1) one out of four data sets (seeds, seedlings, tissues, and DS1) and (2) one clustering solution out of eight. In total, 32 different rankings were produced for each pathway. Our analysis showed that no single combination is best for most pathways; therefore, we wished to select the best one for each pathway. Optimizing predictive power, given a set of different possibilities to analyze data, has been addressed in the machine learning community and is generally referred to as “model selection” (Guyon et al., 2010).
We define a pathway’s learning configuration as a combination of gene expression data set and clustering method. Each learning configuration can be used to generate a ranking of candidates for a specific pathway. In order to match the optimal learning configuration to the pathway, we used LOOCV to estimate the predictive power of every configuration and selected the one that produced the highest AUSR score. We denote this ranking algorithm as selection. Importantly, for statistical validation of this method, the LOOCV procedure used by the selection algorithm is used internally, without taking into account the tested gene; therefore, we avoid overfitting.
To test the additional value of the selection process, we compared its prediction power to that of all possible configurations on the AraCyc pathways. Figure 4A shows the average AUSR scores for different learning configurations and of the selection algorithm. For every expression data set and clustering method, we compared the average AUSR score over 66 AraCyc pathways. Clustering by orthologs, CLICK and SOM produced inferior results, and these clustering methods are thus excluded. In general, the configurations based on enzymes or the MD network yielded better results than configurations that used the PPI network or no clustering of the genes. The enzymes and MD sources are derived from known metabolic information; therefore, we expect them to perform better and reflect more faithfully the signatures of metabolic pathways in expression data. Nevertheless, we observed some exceptional cases where higher scores were obtained by not using enzymes or the MD network. For example, the “homogalacturonan biosynthesis” pathway received a score <0.75 for all configurations except Matisse* with the PPI network, which yielded a score of 0.99 (see Supplemental Data Set 3 online for the AUSR and best configurations for each tested pathway).
Figure 4.

Performance of the Predictors on 230 Pathways in Arabidopsis.
(A) AUSR scores for different learning configurations. The average AUSR over 66 AraCyc pathways tested is displayed for each combination of gene expression data set and partitioning algorithm (white) and for the selection algorithm (black). Configurations denoted by a data set and PPI or MD network are a combination of a data set and a classifier.
(B) Average and median AUSR scores on 66 AraCyc pathways, using each of the seeds, tissues, and seedlings expression data sets together with PPI and MD networks as input.
(C) Average and median AUSR scores on 164 MapMan pathways, using the same input as in (B).
(D) The number of these pathways that had AUSR score above 0.8 when using MORPH, ACT, and the best performers among the other classifiers in (B).
(E) The number of those pathways that had AUSR score above 0.7 when using MORPH, ACT, and the best performers among the other classifiers in (D). CMRF ranks candidate genes by the number of pathway genes in their neighborhood. WMRF ranks candidates by their similarity with neighbor pathway genes. PPI, methods using the PPI network; MDN, methods using the MD network; Combined, methods using both networks.
Importantly, our model selection algorithm, which integrates all the data used in our framework, yielded the highest average score, 0.6, compared with individual configurations. We therefore use the model selection methodology to rank candidates in the next phase.
Robustness
We tested the robustness of our method by comparing AUSR scores obtained from randomly selected metabolic gene sets and using the 66 AraCyc metabolic pathways. We used only metabolic genes since we observed that these genes tend to show higher coexpression level than all genes (data not shown). We ran the selection algorithm with target pathways comprised of randomly selected genes from all genes that appeared in AraCyc. The sizes of the sets were 10 to 44, the same range of the known pathways. We repeated the process 100 times for each set size. For each set size, Figure 5 shows the distribution of AUSR scores on the randomly generated gene sets as compared with scores of known pathways. Overall, no random gene set received an AUSR score >0.75. By contrast, 15 of the 66 real pathways tested received scores >0.75 and as high as 0.99. Moreover, 29 pathways scored higher than the maximal scoring random pathway of the same size. The test gives additional support for the robustness of our ranking algorithms.
Figure 5.

AUSR Scores of Real and Random Pathways.
For each pathway size, we generated 100 random gene sets (using only the pathway genes in Supplemental Data Set 2 online as background) and used our algorithm to compute an AUSR score. Each box plot depicts the average and the range of 25 to 75% of the AUSR scores obtained for the random gene sets. The scores of all real biological pathways are marked with an “x,” except those that received a score above 0.75, which are individually noted with symbols shown in the legend box at the top.
Comparison with Other Function Prediction Algorithms on Arabidopsis Data
We compared MORPH to a k-NN based classifier, two MRF predictors, and two coexpression schemes that do not use any network information. The comparison included 66 AraCyc and 164 MapMan pathways. The coexpression schemes rank genes by their average similarity to the pathway genes, where similarity is measured by coexpression in a reference gene expression data set. The ACT data set (see Methods) and our data set DS1 were used as such references. We tested one pathway at a time and treated all genes outside the pathway as candidates. In particular, we did not use negative samples of genes that are known to belong to other pathways so that all methods use the same annotation as input. Thus, MRF approaches can be reduced to ranking gene candidates according to the number of neighbors that participate in the tested pathway (see Supplemental Methods 1 online). Since this approach may result in ranking with many ties, we used two MRF configurations: ranking candidate genes according to the number of pathway gene neighbors, denoted as CMRF, and a weighted version that ranks candidates by their total similarity to their annotated neighbors, denoted as WMRF. To integrate network information and gene expression, we used the combined network based on cosine similarity (Pandey et al., 2009). For MRF-based methods, in order to maintain graph sparseness, we used a threshold of 0.4 to define the neighborhood of a gene; higher thresholds resulted in graphs with low average gene degree (i.e., the average number of edges per gene). All classifiers in this analysis were given as input the seeds, tissues, and seedlings gene expression data sets, together with network information. We defined three types of classifiers according to the additional network information and gene expression data: (1) the PPI network, (2) the MD network, and (3) the combined PPI and MD network.
Figure 4B summarizes the performance of the different predictors for 66 AraCyc pathways. Figure 4C shows the results for 164 MapMan pathways. The coexpression scheme that used our DS1 data set achieved better scores than that using the ACT data in both cases, suggesting that our preprocessing is better than that used in assembling the ACT data. In all cases, the k-NN–based predictors performed better than MRF-based predictors. Of the k-NN classifiers, the one that uses both networks outperformed the other two. Notably, MORPH performed better than all other classifiers. In particular, in terms of prediction, MORPH improved significantly over all other classifiers in both sets of pathways: (1) on the AraCyc pathways the second-place classifier, k-NN combined, achieved an average AUSR of 0.53 versus 0.603 achieved by MORPH (P = 0.03); (2) on the MapMan pathways, the coexpression scheme based on DS1 was second, achieving an average AUSR of 0.24, compared with 0.27 of MORPH (P = 0.009). Figure 4D compares MORPH, k-NN–based predictors, and ACT in terms of the number of AraCyc pathways that received an AUSR score above 0.8. The k-NN–based classifiers had one to seven such pathways, while MORPH had 12 (binomial test P = 0.043). Figure 4E compares MORPH and the top five predictors in terms of the number of MapMan pathways that received an AUSR score above 0.7. The other classifiers had two to five pathways, while MORPH had 10 (binomial test P = 0.012).
While MORPH overall outperforms the k-NN classifiers, the two methods appear complementary, as they show large difference in the AUSR on some pathways (see Supplemental Data Set 3A online). For example, for the homogalacturonan degradation pathway, MORPH and k-NN combined obtained AUSR of 0.27 and 0.99, respectively. Only one AraCyc pathway, photosynthesis light reaction, received a score above 0.8 by both methods.
Literature Consistent with the Predictions of Several Top-Ranking Genes in Arabidopsis
In order to demonstrate the relatedness of candidate genes generated by MORPH, we briefly review the top candidates obtained for three of the 66 biological pathways tested (genes listed in Supplemental Data Set 2 online).
The Homogalacturonan Biosynthesis Pathway for Primary Cell Wall Construction
This is a classic example of a pathway that, according to our configuration selection (machine-learning) results, with an AUSR of 0.99, works best with the PPI network as a clustering solution together with seedling gene expression data. MORPH suggested only nine candidates, and all of them are labeled as being putatively involved in homogalacturonan synthesis or in the synthesis of other structurally similar compounds that are involved in cell wall biosynthesis (see Supplemental Data Set 4 online). These proteins are mostly annotated via homology. When the homogalacturonan biosynthesis gene set is used as an input in other coexpression analysis programs, such as GeneCAT, we see a much larger set of predictions consisting of a range of protein encoding genes with vague descriptions. While vague descriptions do not rule out these genes as homogalacturonan pathway candidates, it should be noted that none of the MORPH candidates (with their strong homogalacturonan related descriptions) appear in the GeneCAT candidate list. With the exception of known homogalacturonan pathway genes (input genes that also show up in the GeneCAT output), only three genes (At3g53520, At5g07720, and At1g13860) in the GeneCAT list have descriptions that strongly suggest homogalacturonan pathway function. One other gene (At5g15490) has a description strongly suggesting a function in cell wall biosynthesis. This comparison shows the unique analysis provided by MORPH and how it can be used to provide strong pathway candidate genes.
The Chlorophyll Biosynthesis Pathway
Coordinated expression of genes involved in the Calvin cycle, chlorophyll biosynthesis, and photosystem subunit synthesis has been observed in previous studies (Ghassemian et al., 2006). Supplemental Data Set 5 online shows the input pathway genes and output candidate genes with AUSR score of 0.88. The top-ranked candidate gene (AT2G46820) for the MORPH input pathway (chlorophyll biosynthesis) was validated to function as the P subunit of photosystem I (Khrouchtchova et al., 2005). Out of the top 25 genes ranked by MORPH, eight are annotated as being involved in assembly and function of photosystem I and five are annotated as being involved in assembly and function of photosystem II (PSII). The photosystem I and PSII complexes are the ultimate site of chlorophyll accumulation and function, explaining why these complexes are synthesized and assembled in coordination with chlorophyll biosynthesis. Six of the top 25 ranked genes encode proteins involved in the Calvin cycle and are therefore required for utilization of energy generated from photosynthesis, for carbon fixation, and the generation of metabolites for the cell. The Calvin cycle intermediate glyceraldehyde 3-phosphate is a precursor of isoprenoid biosynthesis, and isoprenoids are required for the synthesis of chlorophylls. Considering the Calvin cycle provides precursors for chlorophyll biosynthesis and uses the energy harvested by chlorophylls, it is expected to see enzymes of the Calvin cycle ranked high in our list. The few remaining genes had roles in electron transport and protection against reactive oxygen species (ROS). Inhibition of electron transport is a cause of photoinhibition and, therefore, ROS production. ROS can damage and degrade many compounds, including chlorophylls. The prevention of chlorophyll degradation likely consumes less energy than the synthesis of new chlorophylls. It is therefore not surprising to find these enzymes ranked as related to the chlorophyll biosynthesis pathway. In general, the candidate genes reflect a strong and not unexpected connection between chlorophyll biosynthesis and assembly, function, and maintenance of the photosynthetic apparatus.
The Carotenoid Biosynthesis Pathway
For the carotenoid biosynthetic pathway of 13 genes, MORPH achieved an AUSR score of 0.86. Input pathway genes and output candidate genes are shown in Figure 6, Supplemental Data Set 2, and Supplemental Data Set 6A online. Carotenoids play a multitude of roles in plant cells, including as antioxidants for protection against ROS, as essential components of the light-harvesting apparatus, and as precursors to the hormones abscisic acid and strigolactones. The enzymes of the carotenoid biosynthetic pathway are well known (Cuttriss et al., 2011). Therefore, we expect to identify genes that control ancillary functions related to carotenoid biosynthesis. Not surprisingly, the carotenoid cleavage enzyme CCD1, responsible for the degradation of carotenoids into apocarotenoids (Vogel et al., 2008), was highly ranked at number two in the list. The list also includes AT2G31750, ranked at number 25, encoding a UDP-glucosyl transferase, capable of glucosylating the carotenoid derived hormone abscisic acid (Lim et al., 2005). Other genes identified in this list encode enzymes for biosynthetic pathways intricately linked with carotenoid biosynthesis. Three genes, AT3G48730, AT4G27600, and AT3G51820, are involved in chlorophyll biosynthesis, a pathway known to show coregulation with carotenoid biosynthesis (Ghassemian et al., 2006; Meier et al., 2011). Ranked at number one is the gene SQE3 encoding squalene monooxygenase (AT4G37760), which is responsible for catalyzing the conversion of squalene into 2,3-oxidosqualene (Phillips et al., 2006), a precursor of the plant hormone, brassinosteroid. It was recently shown that genes in the carotenoid pathway are coordinately expressed in response to brassinosteroids (Meier et al., 2011), explaining the presence of this gene on this list. Ranked at number four, the gene encoding SPS2 (AT1G17050) has been shown to have solanesyl diphosphate synthase activity (Jun et al., 2004). Solanesyl diphosphate is required for the final step of plastoquinone synthesis in Arabidopsis. Plastoquinones serve an essential role as electron acceptors during the desaturation of the carotenoid intermediates phytoene and ζ-carotene, catalyzed by phytoene desaturase and ζ-carotene desaturase, respectively (Mayer et al., 1990; Norris et al., 1995). Chemical or genetic disruption of plastoquinone function causes an early termination of the carotenoid biosynthetic pathway at the phytoene desaturase step (Josse et al., 2000; Breitenbach et al., 2001; Matthews et al., 2003), showing the essential nature of plastoquinones in carotenoid biosynthesis. Reflecting the antioxidant nature of carotenoids, we see many genes in the list encoding enzymes involved in synthesizing antioxidant compounds. Ranked at numbers 21 and 11 are genes encoding two enzymes involved in the production of the antioxidant pigments anthocyanin (Hanumappa et al., 2007) and anthocyanidin (Xu et al., 2008), respectively. Thioredoxin reductase (ranked at number six) is involved in an oxidative stress response (Serrato et al., 2004), and tocopherol cyclase (ranked at number three) is involved in synthesis of the antioxidant tocopherol (Mène-Saffrané et al., 2010). These four enzymes are likely found in this list because they perform similar antioxidant actions as carotenoids and are therefore expressed in a similar manner (i.e., in response to oxidative stress). It should be noted, however, that tocopherols are partially derived from isoprenoids, which are also precursors of carotenoid biosynthesis. Blocking isoprenoid incorporation into tocopherols may provide more substrates for carotenoid biosynthesis. Another stress-related gene, AT2G26800 (ranked at number five), encodes hydroxymethylglutaryl-CoA lyase involved in catabolism of amino acids to provide substrates to the citric acid cycle during severe stress conditions (Taylor et al., 2004). This enzyme may be present in our list simply because it plays a role in response to stress, as do carotenoids. It should be noted, however, that in the absence of photosynthesis, the citric acid cycle ultimately provides precursors for isoprenoids, which are channeled into biosynthesis of carotenoids. This relationship of AT2G26800 and carotenoid biosynthesis, suggested by our work, has not been previously considered and may be useful for increasing carotenoid biosynthetic flux in organisms under stress conditions. At least eight genes within the top 25 of this list only have predicted functions, with very little details available on their role in plants. These unstudied genes are of great interest for further studies on their effects on carotenogenesis.
Figure 6.

Candidate Genes (Numbered Octagons) Generated by MORPH for the Carotenoid Biosynthetic Pathway (Starting from Phytoene).
The MEP pathway generates substrates for GGPPS (geranylgeranyl pyrophosphate synthase), which serves as a metabolic hub to feed metabolites to several pathways, including the carotenoid pathway. For simplicity, CCD1 is shown at the pathway end, but the enzyme is known to degrade multiple carotenoid substrates (Vogel et al., 2008). Input genes for MORPH analysis included phytoene synthase (PSY), phytoene desaturase (PDS), zeta-carotene isomerase (Z-ISO), zeta-carotene desaturase (ZDS), carotenoid isomerase (CrtISO), lycopene epsilon cyclase (LCYE), lycopene beta cyclase (LCYB), beta-carotene hydroxylase 1 and 2 (HYD 1 and 2), cytochrome P450 hydroxylase 97A (CYP97A), cytochrome P450 hydroxylase 97C (CYP97C), violaxanthin de-epoxidase (VDE), and zeaxanthin epoxidase (ZEP).
Extending MORPH to Tomato
To test the utility of MORPH beyond Arabidopsis, we gathered a compendium of gene expression data from tomato. We used 220 published microarray expression profiles of tomato gene responses to specific stimuli. These data sets included 115 profiles of fruit tissues (the fruit data set) and 105 profiles of root and leaf tissues (the root and leaf data set). We did not split the root and leaf data set further since the resulting sets would be too small after merging biological replicates. We generated networks computationally using homology because no large-scale MD and protein interaction networks are available for tomato. Beginning with the Arabidopsis (AT) network, we used the tomato genome from the Sol Genomics Network (http://solgenomics.net/; see Methods) to transform each edge onto one or more new tomato (SL) network edges connecting the tomato orthologs of Arabidopsis genes (see Methods). For each gene expression data set, we created the same eight learning configurations as in Arabidopsis. Thus, to analyze our tomato data, MORPH uses two gene expression matrices and eight learning configurations.
As in the Arabidopsis tests, the selection process used by MORPH achieved on average better AUSR scores as compared with individual configurations (see Supplemental Figure 1 online). Supplemental Data Set 3B online shows AUSR scores of all tomato pathways tested. Remarkably, except for the MCL-based configurations, all configurations achieved an average AUSR score below 0.18, whereas the MCL-based configurations achieved an average AUSR of 0.36. Specifically, with using only coexpression to rank candidate genes, the average AUSR was 0.167 and 0.14 for the fruit and root plus leaf data sets, respectively. Hence, in spite of the inherent noise, homology-based networks allow dramatic improvement in prediction quality. The selection process slightly improved upon the MCL configurations, achieving an average AUSR of 0.375.
Compared with other methods, MORPH achieved the best AUSR scores across all 93 tested pathways. The results are shown in Figure 7A. In particular, the second place predictor, k-NN, which combines all data sources, achieved an average AUSR of 0.345 compared with 0.375 by MORPH (P = 0.08). Figure 7B shows the number of pathways with an AUSR score above 0.7 for the five best predictors. MORPH achieved an AUSR above 0.7 in 17 pathways (P = 0.15).
Figure 7.

Performance of the Predictors on 93 Pathways in Tomato.
Predictors include MORPH, k-NN, MRF-based, and coexpression based classifiers.
(A) Average and median AUSR scores.
(B) The number of pathways that had AUSR score above 0.7 when using MORPH and the best performers among the other classifiers. All classifiers were given the fruit and root and leaf (RL) gene expression data set, together with network information. CMRF ranks candidate genes by the number of pathway genes in their neighborhood. WMRF ranks candidates by their similarity with neighbor pathway genes. PPI, methods using the PPI network; MDN, methods using the MD network; Combined, methods using both networks.
We applied MORPH to rank genes associated with the test pathway “carotenoid biosynthesis” (see Supplemental Data Set 6 online). Our analysis of the tomato carotenoid pathway produced an AUSR of 0.306 (P value= 0.00104) based on a fruit data set. It is known that carotenoid biosynthesis and carotenoid function in plants are linked with chlorophyll biosynthesis, photosynthesis, and protection against photooxidative damage (Ghassemian et al., 2006; Li et al., 2008; Meier et al., 2011). Genes for the tomato carotenoid biosynthetic pathway (Tomato Genome Consortium, 2012) are linked to fruit ripening (Lee et al., 2012) and are light regulated in leaf and fruit tissues (Toledo-Ortiz et al., 2010; Powell et al., 2012). The candidate genes identified are consistent with these pathway connections. Furthermore, for the top 10 candidates in tomato, 50% matched similar functions in the Arabidopsis candidate list (top 123 genes) drawn from a seedling data set.
We observed tomato gene candidates consistent with the known coordinate transcriptional regulation of carotenoid and chlorophyll pathways. Tomato candidate gene #1 encodes magnesium chelatase (Solyc04g015750), the committed enzyme for chlorophyll biosynthesis. Magnesium chelatase is involved in the retrograde signaling between the chloroplast and nucleus that modulates carotenoid gene transcript levels (Mochizuki et al., 2001; Koussevitzky et al., 2007; Pogson et al., 2008; Huang and Li, 2009). A magnesium chelatase candidate was also identified in the Arabidopsis list (AT5G45930). Other top-ranked tomato genes function in chlorophyll biosynthesis (#5, Solyc10g007320; #25, Solyc10g005110). The gene encoding NYC3, a protein involved in chlorophyll degradation (Morita et al., 2009), was found in both the tomato (Solyc12g098660) and Arabidopsis lists (AT5G19850).
Carotenoids play roles in protecting against damage from ROS (Havaux et al., 2007; Johnson et al., 2007; Li et al., 2008; Zhu et al., 2010; Li et al., 2012; Bradbury et al., 2012). Related to this role, MORPH ranked as number 2 (Solyc06g007350) a gene encoding a chloroplast PP2C phosphatase that is required for dephosphorylation of PSII proteins (Samol et al., 2012). Reversible phosphorylation-dephosphorylation of PSII regulates light acclimation and signals repair of damaged PSII. Candidate number 6 (Solyc06g071960) encodes nucleoside diphosphate kinase-2 (NDK-2), a regulator of cellular redox state and mediator of tolerance to ROS (Moon et al., 2003). NDK-2 has been shown to affect levels of catalase (Solyc02g082760, candidate number 4), an enzyme that reduces levels of ROS (Queval et al., 2007; Kim et al., 2011).
Several top-ranked tomato candidates encode proteins required for carbon fixation (e.g., Solyc12g094640, Solyc08g076220, Solyc02g020940, and Solyc01g106010), three of which form part of the glyceraldehyde-3-phosphate dehydrogenase complex. Glyceraldehyde-3-phosphate is an intermediate in the Calvin cycle and a precursor of isoprenoid biosynthesis, from which carotenoids are ultimately derived. MORPH ranking of the glyceraldehyde-3-phosphate dehydrogenase candidates exposes competing pathways: Isoprenoid biosynthetic enzymes required for carotenoid biosynthesis compete with Calvin cycle enzymes for access to glyceraldehyde-3-phosphate.
Thus, we demonstrate that MORPH can reliably predict candidate genes to be associated with a pathway in two evolutionarily distinct plant species. Despite the wide genetic separation, application of MORPH to a conserved pathway reveals candidates shared in both species. MORPH also identified species-specific candidates as would be expected from the unique physiology and structure of the individual species or specific data sets used in the selection process.
DISCUSSION
The results presented here demonstrate the power of MORPH, a novel methodology for ranking candidate (annotated and unannotated) genes according to their predicted association with well-studied pathways. MORPH was initially developed for analysis of Arabidopsis data, but we demonstrated that it can be extended to other model systems by showing its utility in tomato. The results of such a tool can contribute to extending gene annotations as well as generate hypotheses to fuel efforts for enhancing metabolic engineering in plants. Thus, MORPH expands the plant gene annotation toolkit and can suggest many new targets for investigation.
Arabidopsis was the first plant model system used in genomics. Consequently, among plants, it has the best gene annotations, the largest publicly available microarray data, and the most detailed known PPI and metabolic networks. For these reasons, we chose to develop MORPH using Arabidopsis data. The ability of MORPH to predict candidates for each model system requires only a large collection of microarray data and gene annotations and information that ties gene IDs to specific biological pathways or biological groupings. Since tomato is an important crop and model plant subject of many metabolic studies, we chose to adjust MORPH to support tomato data in addition to Arabidopsis.
From a computational point of view, we addressed the question of learning the characteristics (mathematical features of the genes given the data) of a class of genes that reside within a much larger group of genes, without availability of a set of genes that are known to not belong to that set (negative examples). This situation arises in studying organisms with low annotation coverage: To perform well in gene function prediction, an algorithm cannot rely on broad evidence based on functionality of other genes. To evaluate a predictor, we used the self-rank curve suggested by Kharchenko et al. (2004) which summarizes the leave-one-out validation. We then calculate the area under this curve and call the final score AUSR. The AUSR score quantifies the prediction power quite effectively by concentrating on a predefined number of top-ranking genes, but the score depends on that number (i.e., using 5000 gives higher AUSR scores than using 1000 although the significance might not change). In our data, since there were many genes in the tested organism, we selected this number to be 1000.
MORPH and the other predictors tested here combine two main goals for learning the set of candidates associated with a given pathway: (1) filtering genes that are unrelated to the pathway and (2) ranking candidate genes. In MORPH, the filtering effect is achieved by disregarding genes in clusters that do not contain pathway genes, whereas in k-NN–based methods, genes that do not have pathway genes among their nearest neighbors are not considered.
We observed that many predictors, especially those that do not integrate diverse data sources, lack robustness, in that they can perform well on one type of pathway and poorly on others. For example, AraCyc pathways typically form a low diameter subnetwork in the MD network (i.e., the shortest path between every pair of pathway genes has only a few edges). Consequently, k-NN–based approaches that only consider gene distances in the MD network yielded high AUSR scores on AraCyc metabolic pathways, but almost random results on MapMan signaling pathways, which have higher diameters. In addition, such predictors consider only genes in the network, and since the MD network coverage is low, k-NN–based approaches cannot suggest new candidate genes that were not previously annotated. By contrast, MORPH outperformed all other predictors in all types of pathways and also provided new valuable candidate genes, as demonstrated on specific pathways.
When integrating different data sources, a possible alternative to the approach MORPH uses is to rank each candidate gene according to one data source at a time and aggregate the scores into a ranking of the candidates. For example, for each gene, we can average its ranks in the different data sources or first standardize the scores in each data source and then sum the scores of the candidate. Such methodology is well established in machine learning and is sometimes called an ensemble approach in problems of feature selection (Abeel et al., 2010). We tested a simple ensemble method that integrates ranking of candidates based on the networks, without considering the gene expression data (see Supplemental Methods 1 online). On average this predictor achieved higher AUSR scores than MORPH. For example, on the 164 Arabidopsis MapMan pathways, it achieved an average of 0.375, whereas MORPH achieved an average score of 0.28. However, the ensemble predictor does not consider genes that are not present in the networks and therefore is more suitable to model organisms for which network information is abundant (e.g., S. cerevisiae). Hence, in plant model organisms, due to the low coverage of networks, MORPH is more suitable as a predictor, as it was specifically designed to integrate the network information without penalizing candidate genes that are not present in the networks. This effect is reached mainly by the Matisse* process, which expands the network clustering solutions to contain many highly coexpressed genes. This problem of data coverage was observed in bioinformatics also in the context of disease gene learning (Piro and Di Cunto, 2012). Future improvement of MORPH should aim to improve the AUSR scores without automatically penalizing genes that are not present in the networks.
METHODS
Microarray Data Sets and Preprocessing
Arabidopsis thaliana Data
We collected 216 published microarray expression profiles of Arabidopsis thaliana reflecting responses to specific stimuli, developmental stages, and selected mutants (summarized in Supplemental Data Set 1 online). Only data sets that passed quality control test by their original authors were chosen. Normalized matrices were retrieved from NASCArrays (http://affymetrix.Arabidopsis.info/narrays/experimentbrowse.pl) (Craigon et al., 2004), TAIR-ATGenExpress (http://www.ebi.ac.uk/microarray-as/ae/), and the Gene Expression Omnibus (the National Center for Biotechnology Information) (http://www.ncbi.nlm.nih.gov/geo/) (Barrett et al., 2009). Altogether, we formed three data sets: (1) 64 experiments conducted on seedling tissues generated by 13 different labs, (2) 99 experiments conducted on different tissues (leaves, roots, seeds, and flowers) generated by 10 different labs, and (3) 53 experiments of laser dissected seed tissues from different developmental stages (Le et al., 2010).We also analyzed the combined seedlings and tissues data sets, which we called DS1.
We removed probes that displayed consistently low detection calls (raw detection call <100 in at least 80% of the experiments) as done by others (Scherzer et al., 2007). We also removed probes that exhibited low variation (sd <120) in either the seeds data set or DS1, so that only genes that were expressed in both data sets remained for further analysis. A total of 12,459 genes (out of 21,751) survived this filtering step. Since DS1 profiles were collected from 23 different sources, for each source we averaged replicates, divided treatments by control, and standardized each experiment. Dividing by the respective controls was applied in each of the seedlings and the tissues data sets but not in the seeds time series data set.
Tomato Data
We collected 53 tomato (Solanum lycopersicum) microarray expression profiles reflecting responses to specific stimuli, developmental stages, and selected mutants (see Supplemental Data Set 1 online). Only data sets that passed quality control test by their original authors were chosen. Normalized matrices were retrieved from Mintz-Oron et al. (2008), Adato et al. (2009), and Fukushima et al. (2012). These matrices included 32 profiles (after uniting replicates and dividing by the control) from fruit tissues (the fruit data set) and 21 profiles (after uniting replicates and dividing by control) of root and leaf tissues (the root and leaf data set). We used BLAST to match probes on the tomato Affymetrix chips to tomato genome Solyc genes (version ITAG2.3) by screening for hits that showed 100% identity. Probes that were not mapped to Solyc genes were not removed from the data matrices since such genes can be used as novel candidates if they are ranked high in the output list of MORPH. Replicates were averaged, and since the microarray profiles were collected from different sources, we divided the treatments by control values. We did not standardize each experiment separately (i.e., standardize the genes in each submatrix of an experiment) since most experiments were too small.
Pathways
Arabidopsis AraCyc pathways were downloaded from the PMN database (ftp://ftp.plantcyc.org/Pathways/) and MapMan pathways were downloaded from http://mapman.gabipd.org/web/guest/mapmanstore;jsessionid=97B79F4C0839F79FA214EF6AF31638B4.ajp13_mapman_gabipd_org. We excluded pathways with <10 genes in the gene expression data sets, leaving 64 pathways. We manually added the carotenoid biosynthetic pathway (13 genes in the carotenoid biosynthetic pathway) and the carotenoid pathway combined with the upstream MEP pathway (23 genes in CarotenoidsMEP), as they were the initial focus of the study. Supplemental Data Set 2 online lists for each pathway the Arabidopsis gene members used.
Tomato pathways were downloaded from MapMan. We excluded pathways with <10 genes in our gene expression data sets, resulting in 164 pathways (http://mapman.gabipd.org/web/guest/mapmanstore; see Supplemental Data Set 2 online for a list of pathway genes).
Additional Information Sources
We used four types of additional information for pathway ranking, as follows.
Gene Expression–Based Clustering Method
We used the SOM clustering algorithm (Kohonen, 1990) with 5 × 5 grid layout settings and the CLICK algorithm (Sharan et al., 2003). For both algorithms, we used the EXPANDER platform (Ulitsky et al., 2010). SOM partitions all genes into modules, while CLICK can leave some unclustered genes. In addition, we defined one cluster (denoted as “no-clustering”) containing all the genes.
Enzymes
Enzymatic gene annotations were downloaded from PlantCyc (ftp://ftp.plantcyc.org/Pathways/). The genes were divided into two sets: 2933 genes annotated as enzymes and the remaining 9526 genes.
MD Network
Arabidopsis metabolic interactions were downloaded from AraCyc (ftp://ftp.plantcyc.org/Pathways/) and were used to construct the MD network, as described (Kharchenko et al., 2004, 2005). The MD network is an unweighted and undirected graph in which nodes denote metabolic genes and edges connect genes whose corresponding enzymes share a common metabolite among their reactants or products. As described by Kharchenko et al. (2006), in the process of building the network, we excluded the most common metabolites. We iteratively removed the most common metabolite and recalculated the percentage of genes that were annotated in the AraCyc reactions covered by the network. We repeated this process until coverage dropped below 95%. Overall, 20 metabolites were removed. The final network contained 1987 genes and 56,244 interactions.
PPI Network
Our PPI network was constructed from the PAIR database (Lin et al., 2011; http://www.cls.zju.edu.cn/pair/) and the newly published Arabidopsis Interactome Map (Arabidopsis Interactome Mapping Consortium, 2011). A total of 145,404 predicted interactions and 5990 experimentally reported interactions were extracted from PAIR. A total of 11,374 experimentally verified interactions were obtained from the Arabidopsis Interactome Map. The united PPI network contained 149,229 interactions.
Clustering the PPI network
We clustered the PPI network using the MCL algorithm (Van Dongen, 2000). This method was selected because it was shown to outperform other clustering algorithms on PPI data (Enright et al., 2002; Sharan et al., 2007; Vlasblom and Wodak, 2009).
Module-Guided Ranking Algorithm
We developed a new algorithm for prioritizing novel candidate genes in a given pathway (Figure 1). The algorithm receives as input a set S of genes that are known to participate in the pathway, a set of gene expression profiles, a similarity function D, and a partitioning of all genes in the gene expression data into k modules: M1, …, Mk. As a first step, we filter out modules that do not contain any pathway genes. For a module Mi that contains a set of pathway genes s1, …, sl and for every gene g within Mi that is not a part of the pathway, we first calculate its average expression similarity with s1, …, sl:
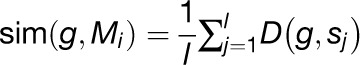 |
where D(g, sj) is the similarity between the expression patterns of g and sj. This calculation provides a ranking of the candidates within the same module. However, we seek a common ranking of the candidates from all modules, each of which may differ considerably in size and homogeneity. Hence, we standardize the similarity scores within each module. Formally, let sim1, …, simCi be the average similarity scores of all candidate genes in module Ki. Let μ be the average of sim1, …, simCi and let σ be the sd of these scores. For every candidate gene g in module Mi we calculate its z-score:
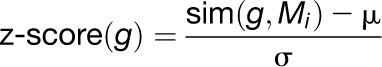 |
The final ranking of candidate genes is as follows: All genes that were not clustered with any pathway gene are placed at the bottom of the ranking. All the other genes are sorted in descending order of their z-scores. For the similarity function D, we used Pearson correlation as it performed better than Spearman rank correlation on each gene expression data set (see Supplemental Figure 2 online).
Statistical Validation Procedure
For each tested pathway S, we run a leave-one-out cross validation (LOOCV) procedure as follows (Figure 2). One of the genes v in S is removed from the list and the algorithm is applied using the reduced set S\{v} of the pathway genes. The performance of the ranking procedure is evaluated using the self-rank measure of Kharchenko et al. (2006). The self-rank of v is defined as the place of v in the ranking produced by the algorithm when v is left out. A perfect prediction would give v a self-rank of 1 (top candidate), and a completely noninformative method would result in a uniform distribution of ranks. The process is repeated for every gene v in S. The results are summarized by the self-rank plot (Figure 2), which shows for every rank threshold k (k = 1 to 1000), the percentage of pathway genes with self rank ≤ k. We calculate the area under the self-rank curve and divide it by the area under the line y = 1. This ratio is defined as the AUSR score, which ranges between 0 and 1.
Learning a Pathway-Specific Configuration
The choice of expression data and a specific clustering algorithm (possibly employing a network) affect the results of the analysis. For each data set, six clustering options were considered: no clustering, CLICK, MCL with the PPI, MATISSE* with the PPI or the MD networks, and enzyme/nonenzyme partition. In Arabidopsis, we used four data sets (a total of 24 combinations), and in tomato, we used two data sets (a total of 12 combinations). We tested all possible combinations for each examined pathway. The SOM and orthologs clustering solutions were excluded since their prediction power was inferior. The selection procedure chooses the combination producing the highest AUSR score. This scheme was statistically evaluated by the same LOOCV procedure. Specifically, we repeat the following process for every gene in the pathway: We remove (hide) the gene from the pathway and for each possible learning configuration apply the LOOCV procedure on the remaining pathway genes and select the configuration that obtains the maximum AUSR score. That configuration is then used to evaluate the rank of the hidden gene. The self-rank curve and the overall AUSR score are obtained using the ranks of every single gene as the hidden gene. Note that different test sets may use different configurations.
Utilizing Network Information: Matisse Modules
Matisse is a method for detection of functional modules using an interaction network and expression data (Ulitsky and Shamir, 2009). A Matisse module is a gene set composed of coexpressed genes that are connected in the network, possibly through inclusion of additional genes for which expression data are not available (back nodes). These modules are used in the MORPH algorithm.
Overcoming the Low Coverage of Networks in Plants
The coverage provided by the Matisse modules using the MD network was very low (1204 genes on average). We therefore sought an improvement to the coverage of the algorithm. The modified version, called Matisse*, starts with the Matisse set of modules and repeatedly inserts the gene with the highest correlation to a module into that module, possibly violating the connectivity constraints, and updates gene-module correlations. The process was repeated until the correlation of expression dropped below 0.4. This step improved the coverage to an average of 4446 genes for the MD network. For the PPI network, the improvement was milder, from 4642 to 4923 genes on average. The candidate genes generated in the ranking process comprised all available genes in the gene expression profiles.
In the tomato data, since the clusters provided by MCL covered only 2273 genes, we applied the expansion step of Matisse* using the initial solution of the MCL algorithm. This step improved the coverage of the clustering solution to 8933 genes in the fruit data set and 9069 genes in the root and leaf data set.
Transforming Arabidopsis Networks to Tomato Networks
We computationally generated tomato (SL) PPI and MDN networks using homology gene mapping from Arabidopsis (AT). This mapping was based on TBLASTX (Altschul et al., 1997) analysis of the Arabidopsis genome (version TAIR10) against the latest version of the tomato genome (ITAG2.3; SGN, http://solgenomics.net/organism/Solanum_lycopersicum/genome). For each edge (u,v) in the AT network, we added a set of edges to the corresponding SL network. If an AT gene u was mapped to SL gene w and AT gene v was mapped to SL gene y, then we added the edge (w,y) in the SL network. Note that this process may produce several edges in the SL network. Isolated nodes were removed from the final SL network. The resulting PPI network contained 12,895 genes and 274,212 edges. The resulting MD network contained 2689 genes and 140,409 edges.
ACT Data
The ACT data set was assembled by transforming a collection of 12,459 text files (one for each gene in our data sets) into a correlation matrix. Each file contained Pearson coexpression correlation between one gene and all the AT genes. The retrieval of the ACT text files was done by crawling the ACT website (http://www.Arabidopsis.leeds.ac.uk/act/coexpanalyser.php) with a Ruby script.
MORPH Tool
MORPH can be accessed online or downloaded and used independently on a PC from http://biocourse.weizmann.ac.il/morph/.
Accession Numbers
See Supplemental Data Set 1 Online for a list of microarray accession numbers.
Supplemental Data
The following materials are available in the online version of this article.
Supplemental Figure 1. AUSR Scores for Different Learning Configurations on the Tomato Data.
Supplemental Figure 2. A Comparison between the Quality of Results Obtained Using Pearson and Spearman Correlation.
Supplemental Methods 1. Comparing the Pearson and Spearman Correlation Measures, Markov Field Algorithm, and Ensemble Method.
Supplemental Data Set 1. List of Microarray Experiments Used in This Study.
Supplemental Data Set 2. Biological Pathways from AraCyc and MapMan.
Supplemental Data Set 3. Arabidopsis and Tomato AUSR Scores of the Selection Algorithm and Selected Learning Configurations for All Pathways Tested.
Supplemental Data Set 4. Top-Ranked Genes for the Arabidopsis Homogalacturonan Biosynthesis Pathway.
Supplemental Data Set 5. Top-Ranked Genes for the Chlorophyll Biosynthesis Pathway.
Supplemental Data Set 6. Top-Ranked Candidate Genes for the Arabidopsis Carotenoid Biosynthetic Pathway.
Acknowledgments
We thank Erich Grotewold (Ohio State University) and Avi Ma’ayan (Mount Sinai School of Medicine) for their valuable suggestions. We thank Veronika Berman (Weizmann Institute of Science) for help in tomato data collection and Roee Hayut (Tel Aviv University) for help in developing the MORPH interface. This research was supported by grants (to E.T.W.) from the U.S. National Institutes of Health (GM081160) and by funding from Lehman College and the Graduate Center of The City University of New York and a grant (to R.S.) from the Israel Science Foundation (802/08). D.A. was supported in part by fellowships from the Edmond J. Safra Center for Bioinformatics at Tel Aviv University and from the Israeli Centers of Research Excellence (I-CORE), Gene Regulation in Complex Human Disease, Center No 41/11.
AUTHOR CONTRIBUTIONS
O.T. and D.A. designed the research, performed research, contributed new computational tools, analyzed data, and wrote the article. L.M.T.B. and E.T.W. analyzed data and wrote the article. R.S. designed the research, analyzed data, and wrote the article.
References
- Abeel T., Helleputte T., Van de Peer Y., Dupont P., Saeys Y. (2010). Robust biomarker identification for cancer diagnosis with ensemble feature selection methods. Bioinformatics 26: 392–398 [DOI] [PubMed] [Google Scholar]
- Adato A., et al. (2009). Fruit-surface flavonoid accumulation in tomato is controlled by a SlMYB12-regulated transcriptional network. PLoS Genet. 5: e1000777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allocco D.J., Kohane I.S., Butte A.J. (2004). Quantifying the relationship between co-expression, co-regulation and gene function. BMC Bioinformatics 5: 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul S.F., Madden T.L., Schäffer A.A., Zhang J., Zhang Z., Miller W., Lipman D.J. (1997). Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 25: 3389–3402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arabidopsis Interactome Mapping Consortium (2011). Evidence for network evolution in an Arabidopsis interactome map. Science 333: 601–607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bader G.D., Hogue C.W. (2003). An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinformatics 4: 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett T., et al. (2009). NCBI GEO: Archive for high-throughput functional genomic data. Nucleic Acids Res. 37(Database issue): D885–D890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradbury L.M.T., Shumskaya M., Tzfadia O., Wu S.-B., Kennelly E.J., Wurtzel E.T. (2012). Lycopene cyclase paralog CruP protects against reactive oxygen species in oxygenic photosynthetic organisms. Proc. Natl. Acad. Sci. USA 109: E1888–E1897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitenbach J., Zhu C., Sandmann G. (2001). Bleaching herbicide norflurazon inhibits phytoene desaturase by competition with the cofactors. J. Agric. Food Chem. 49: 5270–5272 [DOI] [PubMed] [Google Scholar]
- Chen L., Vitkup D. (2006). Predicting genes for orphan metabolic activities using phylogenetic profiles. Genome Biol. 7: R17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craigon D.J., James N., Okyere J., Higgins J., Jotham J., May S. (2004). NASCArrays: A repository for microarray data generated by NASC’s transcriptomics service. Nucleic Acids Res. 32(Database issue): D575–D577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuttriss A.J., Cazzonelli C.I., Wurtzel E.T., Pogson B.J. (2011). Carotenoids. In Biosynthesis of Vitamins in Plants (Advances in Botanical Research, Part A), F. Rébeillé and R. Douce, eds (Amsterdam: Elsevier), pp. 1–36
- Deng M.H., Zhang K., Mehta S., Chen T., Sun F.Z. (2003). Prediction of protein function using protein-protein interaction data. J. Comput. Biol. 10: 947–960 [DOI] [PubMed] [Google Scholar]
- Enright A.J., Van Dongen S., Ouzounis C.A. (2002). An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 30: 1575–1584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukushima A., Nishizawa T., Hayakumo M., Hikosaka S., Saito K., Goto E., Kusano M. (2012). Exploring tomato gene functions based on coexpression modules using graph clustering and differential coexpression approaches. Plant Physiol. 158: 1487–1502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerdes S., et al. (2011). Synergistic use of plant-prokaryote comparative genomics for functional annotations. BMC Genomics 12(suppl. 1): S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghassemian M., Lutes J., Tepperman J.M., Chang H.-S., Zhu T., Wang X., Quail P.H., Lange B.M. (2006). Integrative analysis of transcript and metabolite profiling data sets to evaluate the regulation of biochemical pathways during photomorphogenesis. Arch. Biochem. Biophys. 448: 45–59 [DOI] [PubMed] [Google Scholar]
- Guyon I., Saffari A., Dror G., Cawley G. (2010). Model selection: Beyond the Bayesian/frequentist divide. J. Mach. Learn. Res. 11: 61–87 [Google Scholar]
- Hanumappa M., Choi G., Ryu S., Choi G. (2007). Modulation of flower colour by rationally designed dominant-negative chalcone synthase. J. Exp. Bot. 58: 2471–2478 [DOI] [PubMed] [Google Scholar]
- Havaux M., Dall’osto L., Bassi R. (2007). Zeaxanthin has enhanced antioxidant capacity with respect to all other xanthophylls in Arabidopsis leaves and functions independent of binding to PSII antennae. Plant Physiol. 145: 1506–1520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y.-S., Li H.M. (2009). Arabidopsis CHLI2 can substitute for CHLI1. Plant Physiol. 150: 636–645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janga S.C., Díaz-Mejía J.J., Moreno-Hagelsieb G. (2011). Network-based function prediction and interactomics: The case for metabolic enzymes. Metab. Eng. 13: 1–10 [DOI] [PubMed] [Google Scholar]
- Johnson M.P., Havaux M., Triantaphylidès C., Ksas B., Pascal A.A., Robert B., Davison P.A., Ruban A.V., Horton P. (2007). Elevated zeaxanthin bound to oligomeric LHCII enhances the resistance of Arabidopsis to photooxidative stress by a lipid-protective, antioxidant mechanism. J. Biol. Chem. 282: 22605–22618 [DOI] [PubMed] [Google Scholar]
- Josse E.M., Simkin A.J., Gaffé J., Labouré A.M., Kuntz M., Carol P. (2000). A plastid terminal oxidase associated with carotenoid desaturation during chromoplast differentiation. Plant Physiol. 123: 1427–1436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jun L., Saiki R., Tatsumi K., Nakagawa T., Kawamukai M. (2004). Identification and subcellular localization of two solanesyl diphosphate synthases from Arabidopsis thaliana. Plant Cell Physiol. 45: 1882–1888 [DOI] [PubMed] [Google Scholar]
- Kharchenko P., Chen L., Freund Y., Vitkup D., Church G.M. (2006). Identifying metabolic enzymes with multiple types of association evidence. BMC Bioinformatics 7: 177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharchenko P., Church G.M., Vitkup D. (2005). Expression dynamics of a cellular metabolic network. Mol. Syst. Biol. 1: 2005.0016
- Kharchenko P., Vitkup D., Church G.M. (2004). Filling gaps in a metabolic network using expression information. Bioinformatics 20(suppl. 1): i178–i185 [DOI] [PubMed] [Google Scholar]
- Khrouchtchova A., Hansson M., Paakkarinen V., Vainonen J.P., Zhang S., Jensen P.E., Scheller H.V., Vener A.V., Aro E.M., Haldrup A. (2005). A previously found thylakoid membrane protein of 14kDa (TMP14) is a novel subunit of plant photosystem I and is designated PSI-P. FEBS Lett. 579: 4808–4812 [DOI] [PubMed] [Google Scholar]
- Kim Y.-H., Kim M.D., Choi Y.I., Park S.-C., Yun D.-J., Noh E.W., Lee H.-S., Kwak S.-S. (2011). Transgenic poplar expressing Arabidopsis NDPK2 enhances growth as well as oxidative stress tolerance. Plant Biotechnol. J. 9: 334–347 [DOI] [PubMed] [Google Scholar]
- Kohonen T. (1990). Cortical maps. Nature 346: 24. [DOI] [PubMed] [Google Scholar]
- Kourmpetis Y.A.I., van Dijk A.D.J., Bink M.C.A.M., van Ham R.C.H.J., ter Braak C.J.F. (2010). Bayesian Markov Random Field analysis for protein function prediction based on network data. PLoS ONE 5: e9293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koussevitzky S., Nott A., Mockler T.C., Hong F., Sachetto-Martins G., Surpin M., Lim J., Mittler R., Chory J. (2007). Signals from chloroplasts converge to regulate nuclear gene expression. Science 316: 715–719 [PubMed] [Google Scholar]
- Kuramochi M., Karypis G. (2005). Gene classification using expression profiles: A feasibility study. Int. J. Artif. Intell. Tools 14: 641–660 [Google Scholar]
- Le B.H., et al. (2010). Global analysis of gene activity during Arabidopsis seed development and identification of seed-specific transcription factors. Proc. Natl. Acad. Sci. USA 107: 8063–8070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.M., Joung J.-G., McQuinn R., Chung M.-Y., Fei Z., Tieman D., Klee H., Giovannoni J. (2012). Combined transcriptome, genetic diversity and metabolite profiling in tomato fruit reveals that the ethylene response factor SlERF6 plays an important role in ripening and carotenoid accumulation. Plant J. 70: 191–204 [DOI] [PubMed] [Google Scholar]
- Lee J.M., Sonnhammer E.L. (2003). Genomic gene clustering analysis of pathways in eukaryotes. Genome Res. 13: 875–882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letovsky S., Kasif S. (2003). Predicting protein function from protein/protein interaction data: A probabilistic approach. Bioinformatics 19(suppl. 1): i197–i204 [DOI] [PubMed] [Google Scholar]
- Li F., Vallabhaneni R., Yu J., Rocheford T., Wurtzel E.T. (2008). The maize phytoene synthase gene family: Overlapping roles for carotenogenesis in endosperm, photomorphogenesis, and thermal stress tolerance. Plant Physiol. 147: 1334–1346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z., Keasling J.D., Niyogi K.K. (2012). Overlapping photoprotective function of vitamin E and carotenoids in Chlamydomonas. Plant Physiol. 158: 313–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim E.K., Doucet C.J., Hou B., Jackson R.G., Abrams S.R., Bowles D.J. (2005). Resolution of (+)-abscisic acid using an Arabidopsis glycosyltransferase. Tetrahedron Asymmetry 16: 143–147 [Google Scholar]
- Lin M, Shen X, Chen X. (2011). PAIR: the predicted Arabidopsis interactome resource. Nucl. Acids Res. 39 (suppl 1): D1134–D1140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manfield I.W., Jen C.H., Pinney J.W., Michalopoulos I., Bradford J.R., Gilmartin P.M., Westhead D.R. (2006). Arabidopsis Co-expression Tool (ACT): Web server tools for microarray-based gene expression analysis. Nucleic Acids Res. 34(Web Server issue): W504–W509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews P.D., Luo R., Wurtzel E.T. (2003). Maize phytoene desaturase and zeta-carotene desaturase catalyse a poly-Z desaturation pathway: Implications for genetic engineering of carotenoid content among cereal crops. J. Exp. Bot. 54: 2215–2230 [DOI] [PubMed] [Google Scholar]
- Mayer M.P., Beyer P., Kleinig H. (1990). Quinone compounds are able to replace molecular oxygen as terminal electron acceptor in phytoene desaturation in chromoplasts of Narcissus pseudonarcissus L. Eur. J. Biochem. 191: 359–363 [DOI] [PubMed] [Google Scholar]
- Meier S., Tzfadia O., Vallabhaneni R., Gehring C., Wurtzel E.T. (2011). A transcriptional analysis of carotenoid, chlorophyll and plastidial isoprenoid biosynthesis genes during development and osmotic stress responses in Arabidopsis thaliana. BMC Syst. Biol. 5: 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mène-Saffrané L., Jones A.D., DellaPenna D. (2010). Plastochromanol-8 and tocopherols are essential lipid-soluble antioxidants during seed desiccation and quiescence in Arabidopsis. Proc. Natl. Acad. Sci. USA 107: 17815–17820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mintz-Oron S., Mandel T., Rogachev I., Feldberg L., Lotan O., Yativ M., Wang Z., Jetter R., Venger I., Adato A., Aharoni A. (2008). Gene expression and metabolism in tomato fruit surface tissues. Plant Physiol. 147: 823–851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochizuki N., Brusslan J.A., Larkin R., Nagatani A., Chory J. (2001). Arabidopsis genomes uncoupled 5 (GUN5) mutant reveals the involvement of Mg-chelatase H subunit in plastid-to-nucleus signal transduction. Proc. Natl. Acad. Sci. USA 98: 2053–2058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon H., et al. (2003). NDP kinase 2 interacts with two oxidative stress-activated MAPKs to regulate cellular redox state and enhances multiple stress tolerance in transgenic plants. Proc. Natl. Acad. Sci. USA 100: 358–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita R., Sato Y., Masuda Y., Nishimura M., Kusaba M. (2009). Defect in non-yellow coloring 3, an α/β hydrolase-fold family protein, causes a stay-green phenotype during leaf senescence in rice. Plant J. 59: 940–952 [DOI] [PubMed] [Google Scholar]
- Mutwil M., Klie S., Tohge T., Giorgi F.M., Wilkins O., Campbell M.M., Fernie A.R., Usadel B., Nikoloski Z., Persson S. (2011). PlaNet: Combined sequence and expression comparisons across plant networks derived from seven species. Plant Cell 23: 895–910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutwil M., Obro J., Willats W.G., Persson S. (2008). GeneCAT–Novel webtools that combine BLAST and co-expression analyses. Nucleic Acids Res. 36: W320–W326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabieva E., Jim K., Agarwal A., Chazelle B., Singh M. (2005). Whole-proteome prediction of protein function via graph-theoretic analysis of interaction maps. Bioinformatics 21(suppl. 1): i302–i310 [DOI] [PubMed] [Google Scholar]
- Norris S.R., Barrette T.R., DellaPenna D. (1995). Genetic dissection of carotenoid synthesis in Arabidopsis defines plastoquinone as an essential component of phytoene desaturation. Plant Cell 7: 2139–2149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obayashi T., Hayashi S., Saeki M., Ohta H., Kinoshita K. (2009). ATTED-II provides coexpressed gene networks for Arabidopsis. Nucleic Acids Res. 37(Database issue): D987–D991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orth J.D., Palsson B.O. (2010). Systematizing the generation of missing metabolic knowledge. Biotechnol. Bioeng. 107: 403–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey G., Myers C.L., Kumar V. (2009). Incorporating functional inter-relationships into protein function prediction algorithms. BMC Bioinformatics 10: 142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellegrini M., Marcotte E.M., Thompson M.J., Eisenberg D., Yeates T.O. (1999). Assigning protein functions by comparative genome analysis: Protein phylogenetic profiles. Proc. Natl. Acad. Sci. USA 96: 4285–4288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips D.R., Rasbery J.M., Bartel B., Matsuda S.P. (2006). Biosynthetic diversity in plant triterpene cyclization. Curr. Opin. Plant Biol. 9: 305–314 [DOI] [PubMed] [Google Scholar]
- Pigliucci M. (2009). Genotype-phenotype mapping and the end of the 'genes as blueprint' metaphor. Philos. Trans. R. Soc. Lond. B Biol. Sci. 365: 557–566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piro R.M., Di Cunto F. (2012). Computational approaches to disease-gene prediction: Rationale, classification and successes. FEBS J. 279: 678–696 [DOI] [PubMed] [Google Scholar]
- Pogson B.J., Woo N.S., Förster B., Small I.D. (2008). Plastid signalling to the nucleus and beyond. Trends Plant Sci. 13: 602–609 [DOI] [PubMed] [Google Scholar]
- Popescu L., Yona G. (2005). Automation of gene assignments to metabolic pathways using high-throughput expression data. BMC Bioinformatics 6: 217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell A.L.T., et al. (2012). Uniform ripening encodes a Golden 2-like transcription factor regulating tomato fruit chloroplast development. Science 336: 1711–1715 [DOI] [PubMed] [Google Scholar]
- Queval G., Issakidis-Bourguet E., Hoeberichts F.A., Vandorpe M., Gakière B., Vanacker H., Miginiac-Maslow M., Van Breusegem F., Noctor G. (2007). Conditional oxidative stress responses in the Arabidopsis photorespiratory mutant cat2 demonstrate that redox state is a key modulator of daylength-dependent gene expression, and define photoperiod as a crucial factor in the regulation of H2O2-induced cell death. Plant J. 52: 640–657 [DOI] [PubMed] [Google Scholar]
- Saito K., Hirai M.Y., Yonekura-Sakakibara K. (2008). Decoding genes with coexpression networks and metabolomics - ‘Majority report by precogs’. Trends Plant Sci. 13: 36–43 [DOI] [PubMed] [Google Scholar]
- Samol I., Shapiguzov A., Ingelsson B., Fucile G., Crèvecoeur M., Vener A.V., Rochaix J.-D., Goldschmidt-Clermont M. (2012). Identification of a photosystem II phosphatase involved in light acclimation in Arabidopsis. Plant Cell 24: 2596–2609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherzer C.R., et al. (2007). Molecular markers of early Parkinson’s disease based on gene expression in blood. Proc. Natl. Acad. Sci. USA 104: 955–960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrato A.J., Pérez-Ruiz J.M., Spínola M.C., Cejudo F.J. (2004). A novel NADPH thioredoxin reductase, localized in the chloroplast, which deficiency causes hypersensitivity to abiotic stress in Arabidopsis thaliana. J. Biol. Chem. 279: 43821–43827 [DOI] [PubMed] [Google Scholar]
- Sharan R., Maron-Katz A., Shamir R. (2003). CLICK and EXPANDER: A system for clustering and visualizing gene expression data. Bioinformatics 19: 1787–1799 [DOI] [PubMed] [Google Scholar]
- Sharan R., Ulitsky I., Shamir R. (2007). Network-based prediction of protein function. Mol. Syst. Biol. 3: 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasasainagendra V., Page G.P., Mehta T., Coulibaly I., Loraine A.E. (2008). CressExpress: A tool for large-scale mining of expression data from Arabidopsis. Plant Physiol. 147: 1004–1016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart J.M., Segal E., Koller D., Kim S.K. (2003). A gene-coexpression network for global discovery of conserved genetic modules. Science 302: 249–255 [DOI] [PubMed] [Google Scholar]
- Tan P.N., Steinbach M., Kumar V. (2005). Introduction to Data Mining. (New York: Pearson Addison Wesley)
- Taylor N.L., Heazlewood J.L., Day D.A., Millar A.H. (2004). Lipoic acid-dependent oxidative catabolism of alpha-keto acids in mitochondria provides evidence for branched-chain amino acid catabolism in Arabidopsis. Plant Physiol. 134: 838–848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thimm O., Blasing O., Gibon Y., Nagel A., Meyer S., Kruger P., Selbig J., Muller L.A., Rhee S.Y., Stitt M. (2004). MAPMAN: A user-driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. Plant J. 37: 914–939 [DOI] [PubMed]
- Toledo-Ortiz G., Huq E., Rodríguez-Concepción M. (2010). Direct regulation of phytoene synthase gene expression and carotenoid biosynthesis by phytochrome-interacting factors. Proc. Natl. Acad. Sci. USA 107: 11626–11631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomato Genome Consortium (2012). The tomato genome sequence provides insights into fleshy fruit evolution. Nature 485: 635–641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toufighi K., Brady S.M., Austin R., Ly E., Provart N.J. (2005). The botany array resource: e-Northerns, expression angling, and promoter analyses. Plant J. 43: 153–163 [DOI] [PubMed]
- Ulitsky I., Maron-Katz A., Shavit S., Sagir D., Linhart C., Elkon R., Tanay A., Sharan R., Shiloh Y., Shamir R. (2010). Expander: From expression microarrays to networks and functions. Nat. Protoc. 5: 303–322 [DOI] [PubMed] [Google Scholar]
- Ulitsky I., Shamir R. (2007). Identification of functional modules using network topology and high-throughput data. BMC Syst. Biol. 1: 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulitsky I., Shamir R. (2009). Identifying functional modules using expression profiles and confidence-scored protein interactions. Bioinformatics 25: 1158–1164 [DOI] [PubMed] [Google Scholar]
- Usadel B., Obayashi T., Mutwil M., Giorgi F.M., Bassel G.W., Tanimoto M., Chow A., Steinhauser D., Persson S., Provart N.J. (2009). Co-expression tools for plant biology: Opportunities for hypothesis generation and caveats. Plant Cell Environ. 32: 1633–1651 [DOI] [PubMed] [Google Scholar]
- Van Dongen S. (2000). Graph Clustering by Flow Simulation. PhD dissertation (Utrecht, The Netherlands: University of Utrecht)
- Vlasblom J., Wodak S.J. (2009). Markov clustering versus affinity propagation for the partitioning of protein interaction graphs. BMC Bioinformatics 10: 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel J.T., Tan B.-C., McCarty D.R., Klee H.J. (2008). The carotenoid cleavage dioxygenase 1 enzyme has broad substrate specificity, cleaving multiple carotenoids at two different bond positions. J. Biol. Chem. 283: 11364–11373 [DOI] [PubMed] [Google Scholar]
- Wang C., Scott S. (2005). New kernels for protein structural motif discovery and function classification. In Proceedings of the 22nd International Conference on Machine Learning (ICML), ACM, New York pp. 940–947
- Xu F., Cheng H., Cai R., Li L.L., Chang J., Zhu J., Zhang F.X., Chen L.J., Wang Y., Cheng S.H., Cheng S.Y. (2008). Molecular cloning and function analysis of an anthocyanidin synthase gene from Ginkgo biloba, and its expression in abiotic stress responses. Mol. Cells 26: 536–547 [PubMed] [Google Scholar]
- Yamanishi Y., Vert J.P., Kanehisa M. (2004). Protein network inference from multiple genomic data: A supervised approach. Bioinformatics 20(suppl. 1): i363–i370 [DOI] [PubMed] [Google Scholar]
- Yao Z.Z., Ruzzo W.L. (2006). A regression-based K nearest neighbor algorithm for gene function prediction from heterogeneous data. BMC Bioinformatics 7: 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y., Graham J.E., Ludwig M., Xiong W., Alvey R.M., Shen G., Bryant D.A. (2010). Roles of xanthophyll carotenoids in protection against photoinhibition and oxidative stress in the cyanobacterium Synechococcus sp strain PCC 7002. Arch. Biochem. Biophys. 504: 86–99 [DOI] [PubMed] [Google Scholar]