

Abstract
The brain-derived neurotrophic factor (BDNF) Val66Met polymorphism is a common human single nucleotide polymorphism (SNP) that affects the regulated release of BDNF, and has been implicated in affective disorders and cognitive dysfunction. A decreased activation of the infralimbic medial prefrontal cortex (IL-mPFC), a brain region critical for the regulation of affective behaviors, has been described in BDNFMet carriers. However, it is unclear whether and how the Val66Met polymorphism affects the IL-mPFC synapses. Here, we report that spike timing-dependent plasticity (STDP) was absent in the IL-mPFC pyramidal neurons from BDNFMet/Met mice, a mouse that recapitulates the specific phenotypic properties of the human BDNF Val66Met polymorphism. Also, we observed a decrease in NMDA and GABA receptor-mediated synaptic transmission in the pyramidal neurons of BDNFMet/Met mice. While BDNF enhanced non-NMDA receptor transmission and depressed GABA receptor transmission in the wild-type mice, both effects were absent in BDNFMet/Met mice after BDNF treatment. Indeed, exogenous BDNF reversed the deficits in STDP and NMDA receptor transmission in BDNFMet/Met neurons. BDNF-mediated selective reversal of the deficit in plasticity and NMDA receptor transmission, but its lack of effect on GABA and non-NMDA receptor transmission in BDNFMet/Met mice, suggests separate mechanisms of Val66Met polymorphism upon synaptic transmission. The effect of the Val66Met polymorphism on synaptic transmission and plasticity in the IL-mPFC represents a mechanism to account for this impact of SNP on affective disorders and cognitive dysfunction.
Introduction
Both human and animal studies indicate that BDNF signaling is profoundly disrupted in affective disorders, particularly anxiety and depression. Postmortem studies have demonstrated abnormal expression of BDNF and TrkB, the receptor for BDNF, in patients with mental depression (Dwivedi et al., 2003; Thompson Ray et al., 2011). Consistently, serum BDNF was decreased in patients with depression (Gonul et al., 2005; Karege et al., 2005; Piccinni et al., 2008). Congruent with the decreased BDNF-TrkB signaling in affective disorders, antidepressants and agents that show antidepressant-like effects enhanced BDNF expression and TrkB activation in the brain (Nibuya et al., 1995; Russo-Neustadt et al., 1999; Koponen et al., 2005; Rantamäki et al., 2007; Dwivedi, 2009). These studies strongly suggest that BDNF signaling is involved in the pathophysiology of affective disorders. The BDNF Val66Met polymorphism (Met substitution for Val at codon 66 in the prodomain), a common human single nucleotide polymorphism in the BDNF gene, affects regulated release of BDNF, and this polymorphism has been implicated in anxiety, depression, neuroticism, and cognitive dysfunction (Egan et al., 2003; Jiang et al., 2005; Schumacher et al., 2005; Hwang et al., 2006; Terracciano et al., 2010). Despite strong evidence for the role of BDNF-TrkB signaling in affective disorders, the neural mechanisms involved in the effect of BDNF on affective behaviors are not well understood. The role of mPFC in anxiety and depression has been proposed (Drevets et al., 2008). Although the adult cortex expresses abundant levels of BDNF, the specific role of mPFC BDNF signaling in the regulation of affective behaviors is unclear.
Consistent with the increase in anxiety and depression in BDNFMet carriers (Jiang et al., 2005; Schumacher et al., 2005; Hwang et al., 2006; Terracciano et al., 2010), human Met allele carriers exhibit a fear extinction deficit, and decrease in the activation of the ventromedial PFC (the primate analog of the rodent IL-mPFC), a brain region critical for the regulation of affective behaviors (Rauch et al., 2006; Soliman et al., 2010). BDNFMet/Met mice exhibited impairments in extinction of conditioned aversive memory and cued fear (Yu et al., 2009; Soliman et al., 2010). However, the neuronal mechanism involved in the effect of the BDNF Val66Met polymorphism on the IL-mPFC is unknown. Both human and animal studies suggested that the ventromedial prefrontal cortex regulates the amygdala in a top-down fashion to modulate fear behavior and affective behaviors (Paré et al., 2004; Rauch et al., 2006; Amano et al., 2010). Given the elevated anxiety and fear extinction deficit in BDNFMet carriers, we examined synaptic transmission and plasticity in the IL-mPFC layer II/III and layer V pyramidal neurons, the major excitatory neurons, in BDNFMet/Met mice. We found a significant impairment of synaptic transmission and spike timing-dependent plasticity (STDP) in the IL-mPFC of BDNFMet/Met mice. In addition, acute activation of BDNF-TrkB signaling selectively modulated plasticity and NMDA receptor transmission, but not GABA or non-NMDA receptor transmission, in the IL-mPFC of BDNFMet/Met mice, suggesting that separate mechanisms may be involved in the effect of the Val66Met polymorphism on synaptic transmission.
Materials and Methods
Animals.
The generation of BDNFMet/Met mice was described previously (Chen et al., 2006). Two month-old male BDNFMet/Met mice and wild-type littermates derived from heterozygous BDNF+/Met parents were used for all experiments. All animals were kept on a 12/12 light–dark cycle at 22°C with food and water available ad libitum. All experiments were performed in accordance with institutional guidelines. Mice were genotyped as described previously (Chen et al., 2006).
Fear extinction.
The fear conditioning apparatus consisted of a mouse shock-chamber (Coulbourn Instruments) placed in a sound-attenuated box. After a 2 min acclimation period to the conditioning chamber (scented with 0.1% peppermint in 70% EtOH), mice were fear conditioned with three tone-shock pairings, consisting of a 30 s presentation of a (5 kHz, 70 dB) tone (CS) that coterminated with a 0.7 mA foot shock (US) during the last 1.0 s of the tone with an intertrial interval (ITI) of 30 s. After the final tone-shock pairing, mice remained in the conditioning chamber for 1 min before being returned to their home cages. Twenty-four hours after fear conditioning, the extinction procedure began in which mice were exposed to 5 presentations of the CS in the absence of the US. To eliminate any confounding interactions of contextual fear, tones were presented in a novel context, consisting of a green cylindrical arena (scented with 0.1% lemon in 70% EtOH). Tone presentations lasted for 30 s with an ITI of 30 s. After the final tone presentation, mice remained in the conditioning chamber for 1 min before being returned to their home cages. Fear extinction trials were repeated daily for a total of 4 d of extinction training. Experiments were controlled by a computer using Graphic State software. Mice were videotaped for subsequent analysis by raters blind to mouse genotype. Freezing was defined as the absence of visible movement except that required for respiration (Fanselow, 1980). The freezing during the initial acclimation period was measured and used as an assay for unconditioned effects on general locomotor activity. The percentage time spent freezing was calculated by dividing the amount of time spent freezing during the 30 s tone presentations by the duration of the tone. Extinction trials were binned into early and late trials with the early trials representing the average of the trials on day 1 of extinction (24 h after conditioning), while late trials represent the average of the trials on day 4 of extinction (96 h after conditioning).
Electrophysiology.
After pentobarbital anesthesia, mice were intracardially perfused with ice-cold artificial CSF (ACSF) containing the following (in mm): NaCl (118), KCl (2.5), CaCl2 (1), MgSO4 (1.5), NaHCO3 (26), NaH2PO4 (1), and d-glucose (10), osmolarity adjusted to 325 mOsm with sucrose, and aerated by 95%O2/5%CO2, pH 7.4 for 2 min. Mouse brains were removed and placed in ice-cold ACSF immediately following decapitation. Coronal brain slices (300 μm) were prepared using a vibratome (Campden Instruments) and kept submerged in ACSF in a brain slice keeper (Scientific Systems Design) at room temperature for at least 1–1.5 h to allow recovery. A single slice was then transferred to a recording chamber, and held submerged in ACSF by a nylon net. The recording chamber was continuously perfused by ACSF containing the following (in mm): NaCl (118), KCl (2.5), CaCl2 (2.5), MgSO4 (1.5), NaHCO3 (26), NaH2PO4 (1), and d-glucose (10), osmolarity adjusted to 325 mOsm with sucrose, and aerated by 95%O2/5%CO2, pH 7.4, at a constant rate of 2 ml/min and held at a temperature of 32°C, controlled with a TC324B in-line solution heater (Warner Instruments). The IL-mPFC layer II/III and layer V pyramidal neurons were visualized using video-enhanced infrared differential interference contrast microscopy (Hamamatsu C5405), with an Olympus BX50WI upright microscope fitted with a 40× long working distance water-immersion objective. For measuring EPSCs, patch electrodes (4–6 MΩ) were filled with an intracellular pipette solution consisting of the following (in mm): CsCl (145), HEPES (10), EGTA (0.5), QX-314 (5), GTP (0.2), and MgATP (5). The osmolarity was adjusted to 290 mOsm with sucrose, and pH was adjusted to 7.4 with CsOH. EPSCs were recorded in IL-mPFC layer II/III pyramidal neurons by electrical stimulation using an extracellular electrode placed in layer IV. EPSCs were recorded in IL-mPFC layer V pyramidal neurons by electrical stimulation of layer II/III (Huang et al., 2004; Couey et al., 2007). Non-NMDA EPSCs were recorded at −60 mV in the presence of GABAA receptor blocker bicuculline (10 μm), and NMDA EPSCs were recorded at +40 mV in the presence of bicuculline and the non-NMDA receptor blocker 2,3-dioxo-6-nitro-1,2,3,4-tetrahydrobenzo[f] quinoxaline-7-sulfonamide disodium salt (NBQX, 10 μm). Miniature EPSCs (mEPSCs) were recorded at −60 mV in the presence of bicuculline and tetrodotoxin (1 μm). Miniature IPSCs (mIPSCs) were recorded at −70 mV in the presence of NBQX, the NMDA receptor blocker d-2-amino-5-phosphonovaleric acid (APV, 100 μm), and tetrodotoxin, using an electrode solution containing the following (in mm): potassium gluconate (78), KCl (70), HEPES (10), GTP (0.2), and MgATP (5). Osmolarity was adjusted to 290 mOsm with sucrose, and pH was adjusted to 7.4 with KOH (Couey et al., 2007). Recordings were rejected when series resistance or holding current changed by 10% or more.
For the STDP experiments, we used an electrode solution containing the following (in mm): potassium gluconate (120), KCl (20), MgCl2 (2), HEPES (10), GTP (0.2) and MgATP (5) (Froemke and Dan, 2002). EPSPs were evoked at 0.1 Hz in layer II/III and layer V pyramidal neurons by extracellular stimulation of layer IV and layer II/III, respectively. After obtaining stable whole-cell recordings, basal EPSPs were recorded for 5 min. The initial 2.5 ms of EPSP slopes were analyzed to study only the monosynaptic components of evoked EPSPs (Froemke and Dan, 2002; Froemke et al., 2005; Couey et al., 2007). For the induction of plasticity, the postsynaptic action potential was evoked ∼5 ms from the onset of the EPSP by applying a current pulse of 1 nA for 2 ms. After 50 pairings (0.1 Hz), postinduction EPSPs were collected for 25 min. Cells were excluded if the input resistance changed >25% during the course of the experiment. Recordings were made using an Axopatch 200B amplifier (Molecular Devices), and digitized by Digidata 1322A (Molecular Devices). Synaptic stimulation was induced using the digital stimulator PG4000A (Cygnus Technology) and the stimulus isolator A365 (World Precision Instruments).
For studying the effect of BDNF on synaptic transmission and STDP, brain slices were treated with BDNF (25 ng/ml) in ACSF bubbled with 95%O2/5%CO2 for 1–1.5 h in a brain slice keeper maintained at room temperature.
Statistics.
Results were expressed as mean ±SEM from all the cells in each group. Two-way ANOVA, repeated measures and t tests were used for statistical analysis (IBM SPSS Statistics 19). The level of significance was p < 0.05.
Results
Fear extinction is altered in BDNFMet/Met mice
Both human and mouse studies suggest increased anxiety-like behavior and fear extinction deficit in BDNFMet carriers (Soliman et al., 2010). However, the earlier fear extinction studies in BDNFMet/Met mice were performed immediately after fear conditioning to mimic human fear extinction studies (Soliman et al., 2010). Since a modification of memory consolidation and fear memory erasure is possible with immediate extinction trials, which might involve a distinct mechanism of extinction compared with a delayed extinction (Myers et al., 2006), we performed fear extinction trials at different times after fear conditioning—24 h (extinction day 1), 48 h (extinction day 2), 72 h (extinction day 3), and 96 h (extinction day 4). Also, we undertook fear extinction experiments in a novel context, given the effect of BDNF Val66Met polymorphism on hippocampus-dependent memory and synaptic plasticity in the hippocampus (Egan et al., 2003; Chen et al., 2006; Ninan et al., 2010). As reported earlier, we did not observe any significant difference in fear conditioning between BDNFMet/Met and wild-type mice (data not shown) (Chen et al., 2006; Soliman et al., 2010). No differences in freezing behavior were observed between genotypes during the early extinction trials (BDNFMet/Met: 75.01 ± 3.98%; wild-type: 75.46 ± 3.18%), but BDNFMet/Met mice exhibited significantly higher freezing behavior during late extinction trials compared with wild-type mice (BDNFMet/Met: 66.23 ± 5.29%; wild-type: 49.4 ± 3.55%), indicative of impaired long-term extinction learning and extinction retention (Fig. 1A,B).
Figure 1.
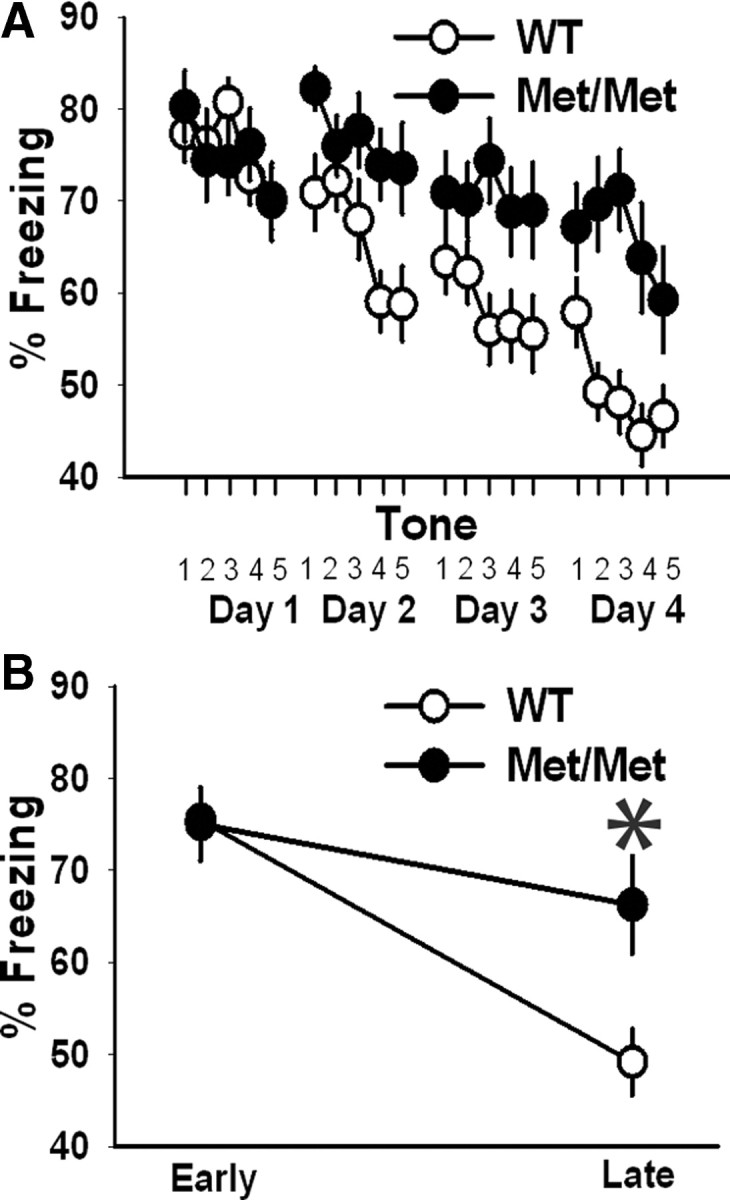
Fear extinction is impaired in BDNFMet/Met mice. A, Average freezing behavior in BDNFMet/Met (n = 16) and wild-type (n = 15) during fear extinction trials 24 h (extinction day 1), 48 h (extinction day 2), 72 h (extinction day 3), and 96 h (extinction day 4) after fear conditioning. BDNFMet/Met mice exhibited significantly higher freezing behavior during late extinction trials compared with wild-type mice. Four days of extinction training resulted in a significant effect of genotype as a function of time (p < 0.05). B, Summary of freezing behavior in BDNFMet/Met and wild-type mice during early and late phase of extinction trials (F(1,29) = 36.4, p < 0.001). Asterisk denotes statistical significance.
Basal glutamatergic transmission is normal in the IL-mPFC pyramidal neurons of BDNFMet/Met mice
A reduction in fear extinction-dependent activation of the ventromedial PFC has been described in the BDNFMet carriers (Soliman et al., 2010), suggesting a potential dysfunction in plasticity. To understand how the Val66Met polymorphism may alter synaptic function in the IL-mPFC, we analyzed basal synaptic transmission in both layer II/III and layer V pyramidal neurons of BDNFMet/Met mice and wild-type littermates. First, we studied glutamatergic transmission in layer II/III pyramidal neurons, which primarily form the callosal and corticocortical projections (DeFelipe and Fariñas, 1992), by comparing the frequency and amplitude of mEPSCs in BDNFMet/Met and wild-type mice. We did not find any significant alteration in either the frequency or amplitude of mEPSCs in BDNFMet/Met (4.7 ± 0.6 Hz and 12.2 ± 0.36 pA) compared with wild-type mice (4.6 ± 0.3 Hz and 13.0 ± 0.25 pA) (Fig. 2A,B). These results suggested that glutamatergic transmission mediated by non-NMDA receptors is normal in BDNFMet/Met mice. These results also suggested normal glutamate release in the BDNFMet/Met mice, as indicated by normal mEPSC frequency. To further investigate basal transmission in BDNFMet/Met mice, we recorded non-NMDA EPSCs at −60 mV in layer II/III pyramidal neurons from BDNFMet/Met and wild-type mice. Non-NMDA EPSCs were evoked by the stimulation of layer IV in the presence of bicuculline. Consistent with the mEPSC experiments, we did not observe any modification of EPSC amplitude in the BDNFMet/Met mice (Fig. 2C,D). These results suggest that the non-NMDA receptor neurotransmission was not affected in the layer II/III pyramidal neurons of BDNFMet/Met mice.
Figure 2.

Normal basal excitatory synaptic transmission in the IL-mPFC of BDNFMet/Met mice. A, Examples of mEPSCs in the layer II/III pyramidal neurons of BDNFMet/Met and wild-type littermates. B, Average mEPSC frequency and amplitude in the layer II/III pyramidal neurons of BDNFMet/Met (n = 8) and wild-type mice (n = 7). Both the mEPSC frequency and amplitude were not significantly different between BDNFMet/Met and wild-type mice (p > 0.05). C, Examples of non-NMDA EPSCs evoked by 100, 300, and 450 μA stimulation of the layer II/III pyramidal neurons in BDNFMet/Met and wild-type mice. D, Average non-NMDA EPSC amplitude in the layer II/III pyramidal neurons of BDNFMet/Met (n = 8) and wild-type mice (n = 8). The EPSC amplitude was not significantly different between BDNFMet/Met and wild-type mice (p > 0.05). E, Examples of mEPSCs in the layer V pyramidal neurons of BDNFMet/Met and wild-type littermates. F, Average mEPSC frequency and amplitude in the layer V pyramidal neurons of BDNFMet/Met (n = 8) and wild-type mice (n = 8). Both the mEPSC frequency and amplitude were not significantly different between BDNFMet/Met and wild-type mice (p > 0.05). G, Examples of non-NMDA EPSCs evoked by 100, 300, and 450 μA stimulation of the layer V pyramidal neurons in BDNFMet/Met and wild-type mice. H, Average non-NMDA EPSC amplitude in the layer V pyramidal neurons of BDNFMet/Met (n = 7) and wild-type mice (n = 7). The EPSC amplitude was not significantly different between BDNFMet/Met and wild-type mice (p > 0.05).
Next, we examined whether the BDNF Val66Met polymorphism affects glutamatergic transmission in layer V pyramidal neurons, the major projection neurons (DeFelipe and Fariñas, 1992; Gabbott et al., 2005). Similar to the layer II/III pyramidal neurons, the amplitude and frequency of mEPSCs in the layer V pyramidal neurons were not significantly different between BDNFMet/Met mice (6.5 ± 2.2 Hz and 12.4 ± 0.6 pA) and wild-type littermates (7.7 ± 1 Hz and 12.0 ± 0.6 pA), suggesting normal non-NMDA receptor neurotransmission and glutamate release (Fig. 2E,F). To determine the effect of the Val66Met polymorphism on non-NMDA receptor transmission, EPSCs were evoked in the layer V pyramidal neurons by stimulation of layer II/III. The amplitude of non-NMDA EPSCs in layer V pyramidal neurons of BDNFMet/Met mice and wild-type littermates were not significantly different, indicating that non-NMDA receptor transmission was normal in the IL-mPFC of BDNFMet/Met mice (Fig. 2G,H). Thus, the above experiments indicate that the basal synaptic transmission was unaltered in the IL-mPFC pyramidal neurons of BDNFMet/Met mice.
Spike timing-dependent plasticity is impaired in the IL-mPFC pyramidal neurons of BDNFMet/Met mice
Given the critical role of the IL-mPFC in fear extinction, and the reduction in fear extinction-dependent activation of the IL-mPFC in BDNFMet carriers (Soliman et al., 2010), we hypothesized that a deficit in the ability of the IL-mPFC synapses to undergo plasticity might be involved in increased anxiety and fear extinction deficit in BDNFMet/Met mice. To address this possibility, we tested whether STDP is altered in the IL-mPFC pyramidal neurons of BDNFMet/Met mice. We recorded EPSPs from the IL-mPFC layer II/III pyramidal neurons of BDNFMet/Met and wild-type mice before and after the application of a pre-before-post pairing protocol to induce spike timing-dependent long-lasting potentiation (Caporale and Dan, 2008). We observed a significant increase in the EPSP slope after the application of the pairing protocol in wild-type mice (Fig. 3A,B,D). However, this pairing protocol failed to modify EPSP slope in the layer II/III pyramidal neurons of BDNFMet/Met mice (Fig. 3A,C,D). Next, we measured the EPSP slope in the layer V pyramidal neurons before and after the pre-before-post pairing stimulus. Similar to the layer II/III pyramidal neurons, the application of the pairing protocol produced a long-lasting increase in the EPSP slope in the layer V pyramidal neurons of wild-type mice (Fig. 3E). However, as in the case of the layer II/III pyramidal neurons of BDNFMet/Met mice, the layer V pyramidal neurons of BDNFMet/Met mice failed to show any increase in the EPSP slope after the pairing protocol (Fig. 3E). Thus, despite the lack of change in basal excitatory transmission in BDNFMet/Met mice, the Val66Met polymorphism has a profound effect on plasticity in the IL-mPFC pyramidal neurons.
Figure 3.
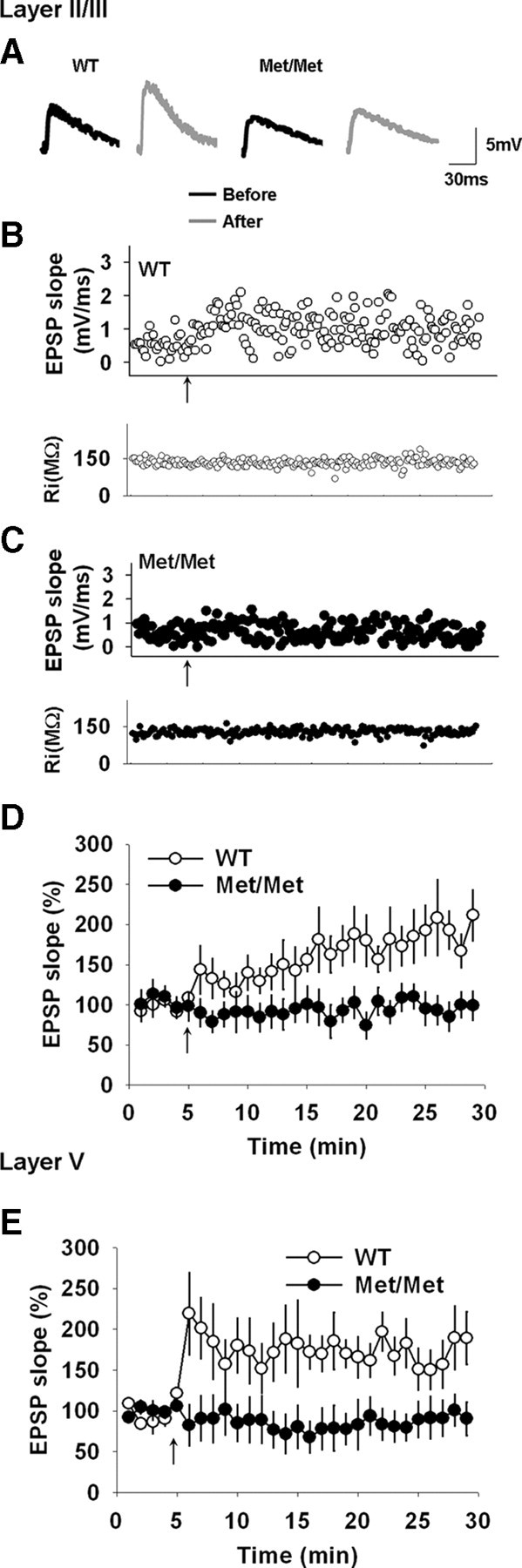
The BDNF Val66Met polymorphism impairs STDP in the IL-mPFC pyramidal neurons. A, Examples of EPSP recordings before and at 20 min after the pairing protocol. B, An example of STDP in the layer II/III pyramidal neuron from wild-type mouse. C, An example of STDP in the layer II/III pyramidal neuron from BDNFMet/Met mouse. D, STDP in the layer II/III pyramidal neurons of BDNFMet/Met (n = 8) and wild-type littermates (n = 8). STDP was absent in BDNFMet/Met mice (F(1,14) = 8.2, p < 0.01). E, STDP in the layer V pyramidal neurons of BDNFMet/Met (n = 9) and wild-type littermates (n = 9). STDP was absent in BDNFMet/Met mice (F(1,16) = 6.6, p < 0.05).
NMDA receptor transmission is reduced in the IL-mPFC pyramidal neurons of BDNFMet/Met mice
Modulation of cortical plasticity can be mediated by both NMDA receptors and GABA receptors (Magee and Johnston, 1997; Markram et al., 1997; Feldman, 2000; Froemke and Dan, 2002; Froemke et al., 2005; Couey et al., 2007; Sakata et al., 2009). Therefore, it is plausible that a modification of NMDA and/or GABA receptor transmission is responsible for the lack of plasticity in the IL-mPFC pyramidal neurons in BDNFMet/Met mice. To test whether IL-mPFC pyramidal neurons in BDNFMet/Met mice exhibited any modification of NMDA receptor neurotransmission, NMDA EPSCs were evoked at +40 mV in the layer II/III pyramidal neurons by stimulation of the layer IV in the presence of NBQX and bicuculline. In contrast to non-NMDA EPSCs, NMDA EPSCs were significantly lower in BDNFMet/Met mice (Fig. 4A,B). Therefore, the Val66Met polymorphism resulted in a decrease in NMDA receptor transmission in the layer II/III pyramidal neurons. Next, we examined whether the NMDA receptor transmission is affected in the layer V pyramidal neurons of BDNFMet/Met mice. NMDA EPSCs were recorded in the layer V pyramidal neurons by the extracellular stimulation of layer II/III. Similar to the layer II/III pyramidal neurons, layer V pyramidal neurons of BDNFMet/Met mice exhibited significant reduction in NMDA EPSC amplitude (Fig. 4C,D). Thus, the Val66Met polymorphism impairs NMDA receptor transmission in both layer II/III and layer V pyramidal neurons of the IL-mPFC.
Figure 4.

The BDNF Val66Met polymorphism decreases NMDA receptor-transmission in the IL-mPFC pyramidal neurons. A, Examples of NMDA EPSCs evoked by 100, 300, and 450 μA stimulation of the layer II/III pyramidal neurons in BDNFMet/Met and wild-type mice. B, Average NMDA EPSC amplitude in the layer II/III pyramidal neurons of BDNFMet/Met (n = 9) and wild-type mice (n = 8). The NMDA EPSC amplitude was significantly lower in BDNFMet/Met mice (F(1,15) = 38, p < 0.001). C, Examples of NMDA EPSCs evoked by 100, 300, and 450 μA stimulation of the layer V pyramidal neurons in BDNFMet/Met and wild-type mice. D, Average NMDA EPSC amplitude in the layer V pyramidal neurons of BDNFMet/Met (n = 8) and wild-type mice (n = 8). The NMDA EPSC amplitude was significantly lower in BDNFMet/Met mice (F(1,14) = 7, p < 0.05).
GABA receptor transmission is reduced in the IL-mPFC pyramidal neurons of BDNFMet/Met mice
In addition to its role in regulating glutamatergic transmission, BDNF-TrkB signaling is critical for the regulation of GABAergic transmission in the cortex (Huang et al., 1999; Hong et al., 2008; Sakata et al., 2009). Also, alterations in GABA receptor transmission have been implicated in cortical plasticity (Couey et al., 2007; Sakata et al., 2009). However, it is unknown whether the BDNF Val66Met polymorphism affects GABA receptor neurotransmission in the IL-mPFC pyramidal neurons. Inhibitory neurotransmission was therefore assessed by recording mIPSCs at −70 mV in the layer II/III and layer V pyramidal neurons, in the presence of APV, NBQX, and tetrodotoxin. We observed a selective decrease in the frequency (1.5 ± 0.1 Hz and 4.3 ± 1 Hz, respectively, for BDNFMet/Met and wild-type mice) but not the amplitude (24.4 ± 1.8 pA and 25.4 ± 2.8 pA, respectively, for BDNFMet/Met and wild-type mice) of mIPSCs in the layer II/III pyramidal neurons of BDNFMet/Met mice, compared with the wild-type littermates (Fig. 5A–C). Unlike the layer II/III pyramidal neurons, we observed a decrease in both the frequency (2.4 ± 0.3 Hz and 3.1 ± 0.6 Hz, respectively, for BDNFMet/Met and wild-type mice) and amplitude (22.1 ± 1 pA and 27.8 ± 2.3 pA, respectively, for BDNFMet/Met and wild-type mice) of mIPSCs in the layer V pyramidal neurons of BDNFMet/Met mice, compared with the wild-type mice (Fig. 5D,E). These results suggest that the Val66Met polymorphism reduces GABAergic transmission in both the layer II/III and layer V pyramidal neurons of the IL-mPFC. Although the exact mechanism is unclear, layer-specific effects of BDNF, GABA receptor occupancy, or structural plasticity might contribute to the differential effect of Val66Met polymorphism on the frequency and amplitude of mIPSCs in layer II/III and layer V pyramidal neurons (Linnarsson et al., 1997; Perrais and Ropert, 1999; Hájos et al., 2000; Fukumitsu et al., 2006; Yu et al., 2009; Liu et al., 2012).
Figure 5.
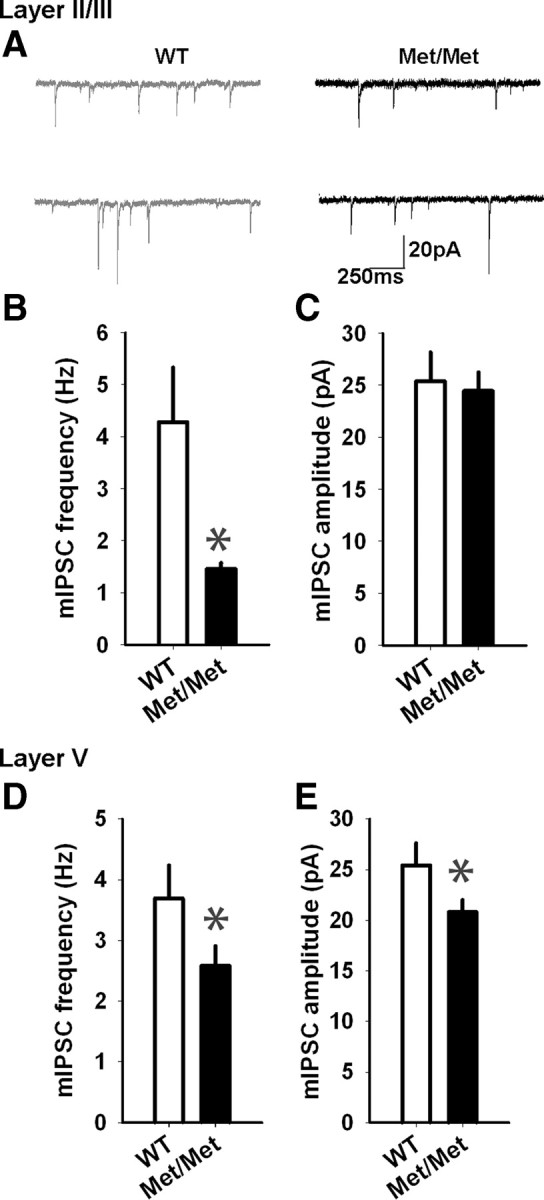
The BDNF Val66Met polymorphism suppresses GABAergic transmission in the IL-mPFC layer II/III and layer V pyramidal neurons. A, Examples of mIPSCs in the layer II/III pyramidal neurons of BDNFMet/Met and wild-type littermates. B, C, Average mIPSC frequency (B) and amplitude (C) in the layer II/III pyramidal neurons of BDNFMet/Met (n = 6) and wild-type mice (n = 6). The mIPSC frequency (p < 0.01) but not the amplitude (p > 0.05) was significantly lower in BDNFMet/Met mice. D, E, Average mIPSC frequency (D) and amplitude (E) in the layer V pyramidal neurons of BDNFMet/Met (n = 10) and wild-type mice (n = 8). Both the mIPSC frequency and amplitude were significantly lower in BDNFMet/Met mice (p < 0.05). Asterisks denote statistical significance.
BDNF treatment reverses plasticity deficit in the IL-mPFC pyramidal neurons of BDNFMet/Met mice
The BDNF Val66Met polymorphism results in a disruption of regulated, but not constitutive, release of BDNF (Chen et al., 2006). Therefore, the decrease in activity-dependent release of BDNF and reduced activation of TrkB might be responsible for plasticity deficit in BDNFMet/Met mice. To test whether the activation of TrkB by BDNF can reverse the impaired STDP in the IL-mPFC of BDNFMet/Met mice, we pretreated BDNFMet/Met brain slices with BDNF (25 ng/ml for 1–1.5 h). BDNF pretreatment reversed the plasticity impairment in BDNFMet/Met mice but did not modify the magnitude of plasticity in either layer II/III or layer V pyramidal neurons of wild-type mice (Fig. 6A–D). These results suggested that acute activation of TrkB was sufficient to reestablish normal plasticity in the IL-mPFC of BDNFMet/Met mice.
Figure 6.

BDNF reverses STDP deficit in the IL-mPFC pyramidal neurons of BDNFMet/Met mice. A, STDP in the layer II/III pyramidal neurons from BDNF-treated (n = 7) and untreated (n = 9) wild-type brain slices. BDNF treatment did not affect STDP in wild-type mice (p > 0.05). B, STDP in the layer II/III pyramidal neurons from BDNF-treated (n = 9) and untreated (n = 10) BDNFMet/Met brain slices. BDNF-treated BDNFMet/Met slices showed significant STDP compared with the untreated BDNFMet/Met slices (F(1,17) = 4.7, p < 0.05). C, STDP in the layer V pyramidal neurons from BDNF-treated (n = 9) and untreated (n = 10) wild-type brain slices. BDNF treatment did not affect STDP in wild-type mice (p > 0.05). D, STDP in the layer V pyramidal neurons from BDNF-treated (n = 9) and untreated (n = 10) BDNFMet/Met brain slices. BDNF-treated BDNFMet/Met slices showed significant STDP compared with the untreated BDNFMet/Met slices (F(1,17) = 8.9, p < 0.01).
BDNF treatment enhances NMDA receptor transmission in the IL-mPFC pyramidal neurons of both wild-type and BDNFMet/Met mice
The decrease in both the plasticity and NMDA receptor transmission in BDNFMet/Met mice is consistent with the earlier findings that NMDA receptor transmission is necessary for STDP (Magee and Johnston, 1997; Markram et al., 1997; Bi and Poo, 1998; Feldman, 2000; Froemke and Dan, 2002; Froemke et al., 2005; Caporale and Dan, 2008). Therefore, we tested whether the reversal of STDP deficit in BDNFMet/Met mice after BDNF treatment also involves rescue of normal NMDA receptor transmission in BDNFMet/Met mice. BDNF treatment enhanced NMDA receptor transmission in both the layer II/III and layer V pyramidal neurons from wild-type mice (Fig. 7A,C). Our results are consistent with the earlier studies showing enhancement of NMDA receptor transmission by BDNF in the rat visual cortex (Carmignoto et al., 1997). Similar to the effect of BDNF on STDP, BDNF-treated slices from BDNFMet/Met mice showed a robust enhancement of NMDA receptor transmission in both the layer II/III and layer V pyramidal neurons (Fig. 7B,D). Therefore, it is possible that the BDNF-induced reversal of plasticity deficit in BDNFMet/Met mice is due to a reestablishment of normal NMDA receptor transmission in the IL-mPFC pyramidal neurons.
Figure 7.

BDNF enhances NMDA receptor transmission in the IL-mPFC pyramidal neurons of both wild-type and BDNFMet/Met mice. A, NMDA receptor transmission in the layer II/III pyramidal neurons from BDNF-treated (n = 7) and untreated (n = 9) wild-type brain slices. BDNF-treated wild-type slices showed significantly higher NMDA receptor transmission compared with the untreated wild-type slices at stimulation strength 100, 150, and 200 μA (p < 0.05). B, NMDA receptor transmission in the layer II/III pyramidal neurons from BDNF-treated (n = 9) and untreated (n = 10) BDNFMet/Met brain slices. BDNF-treated BDNFMet/Met slices showed significantly higher NMDA receptor transmission compared with the untreated BDNFMet/Met slices (F(1,17) = 21.3, p < 0.001). C, NMDA receptor transmission in the layer V pyramidal neurons from BDNF-treated (n = 7) and untreated (n = 9) wild-type brain slices. BDNF-treated wild-type slices showed significantly higher NMDA receptor transmission compared with the untreated wild-type slices above the stimulation strength of 250 μA (p < 0.01). D, NMDA receptor transmission in the layer V pyramidal neurons from BDNF-treated (n = 8) and untreated (n = 9) BDNFMet/Met brain slices. BDNF-treated BDNFMet/Met slices showed significantly higher NMDA receptor transmission compared with the untreated BDNFMet/Met slices (F(1,15) = 21, p < 0.001). Asterisks denote statistical significance.
BDNF treatment suppresses GABA receptor transmission in the IL-mPFC pyramidal neurons of wild-type mice but not BDNFMet/Met mice
Earlier studies indicated that BDNF suppresses GABAergic transmission in hippocampal neurons (Frerking et al., 1998; Brünig et al., 2001; Wardle and Poo, 2003; Jovanovic et al., 2004). Although we observed a decrease in GABAergic transmission in the IL-mPFC of BDNFMet/Met mice, it is possible that exogenous BDNF might further suppress GABAergic transmission in BDNFMet/Met mice, which could play a role in the reversal of plasticity deficit (Couey et al., 2007; Sakata et al., 2009). Therefore, we measured mIPSCs in the layer II/III and layer V pyramidal neurons in both wild-type and BDNFMet/Met brain slices after BDNF treatment. BDNF suppressed the mIPSC frequency but not the amplitude in wild-type layer II/III pyramidal neurons (Fig. 8A,B). However, BDNF suppressed both the frequency and amplitude of mIPSCs in the layer V pyramidal neurons from wild-type slices (Fig. 8C,D). Unlike the rescue of STDP and NMDA receptor transmission in BDNFMet/Met mice after BDNF treatment, GABA receptor transmission was unaffected in BDNF-treated BDNFMet/Met slices, as indicated by the absence of modification of frequency or amplitude of mIPSCs in layer II/III and layer V pyramidal neurons (Fig. 8A–D). The selective rescue of plasticity and NMDA receptor transmission but not GABA receptor transmission in BDNFMet/Met mice after BDNF treatment suggest that there may be a potential developmental deficit in GABAergic transmission due to the Val66Met polymorphism.
Figure 8.

BDNF suppressed GABA receptor transmission in the IL-mPFC pyramidal neurons of wild-type mice but not BDNFMet/Met mice. A, Average mIPSC frequency in the layer II/III pyramidal neurons of untreated wild-type (n = 14), untreated BDNFMet/Met (n = 15), BDNF-treated wild-type (n = 15), and BDNF-treated BDNFMet/Met brain slices (n = 14). BDNF suppressed mIPSC frequency in wild-type mice (p < 0.001) but not in BDNFMet/Met mice (p > 0.05). B, Average mIPSC amplitude in the layer II/III pyramidal neurons of untreated wild-type (n = 14), untreated BDNFMet/Met (n = 15), BDNF-treated wild-type (n = 15), and BDNF-treated BDNFMet/Met brain slices (n = 14). BDNF failed to modify mIPSC amplitude in wild-type and BDNFMet/Met mice (p > 0.05). C, Average mIPSC frequency in the layer V pyramidal neurons of untreated wild-type (n = 13), untreated BDNFMet/Met (n = 17), BDNF-treated wild-type (n = 15), and BDNF-treated BDNFMet/Met brain slices (n = 18). BDNF suppressed mIPSC frequency in wild-type mice (p < 0.05) but not in BDNFMet/Met mice (p > 0.05). D, Average mIPSC amplitude in the layer V pyramidal neurons of untreated wild-type (n = 13), untreated BDNFMet/Met (n = 17), BDNF-treated wild-type (n = 15) and BDNF-treated BDNFMet/Met brain slices (n = 18). BDNF suppressed mIPSC amplitude in wild-type neurons (p < 0.05) but not in BDNFMet/Met neurons (p > 0.05). Asterisks denote statistical significance.
BDNF treatment enhances non-NMDA receptor transmission in the IL-mPFC pyramidal neurons of wild-type mice but not BDNFMet/Met mice
Although both presynaptic and postsynaptic mechanisms have been implicated in BDNF-induced enhancement of excitatory synaptic transmission, a predominant effect of BDNF is upon presynaptic glutamate release (Carmignoto et al., 1997; Li et al., 1998; Wardle and Poo, 2003; Wu et al., 2004; Yano et al., 2006; Caldeira et al., 2007b; Madara and Levine, 2008). Given that both NMDA and non-NMDA glutamate receptors are believed to be colocalized at synapses, a presynaptic modulation by BDNF would affect synaptic responses mediated by both NMDA and non-NMDA receptors similarly (Perkel and Nicoll, 1993). Therefore, it is possible that an increase in glutamate release after BDNF treatment could enhance both the NMDA and non-NMDA receptor transmission in both wild-type and BDNFMet/Met mice. Given that the BDNF treatment enhanced NMDA receptor transmission in both wild-type and BDNFMet/Met neurons, we tested whether an enhancement of the non-NMDA receptor transmission occurs in the IL-mPFC pyramidal neurons from wild-type and BDNFMet/Met mice after BDNF treatment. Layer II/III and layer V pyramidal neurons from wild-type mice showed significant enhancement of non-NMDA EPSC amplitude in BDNF-treated slices compared with untreated slices (Fig. 9A,C). However, surprisingly, we did not observe any enhancement of non-NMDA EPSC amplitude in either layer II/III or layer V pyramidal neurons from BDNFMet/Met mice after BDNF treatment (Fig. 9B,D). These results suggest that BDNFMet/Met mice are resistant to BDNF-induced long-lasting enhancement of non-NMDA EPSC amplitude (Carmignoto et al., 1997). Furthermore, these results also suggest that the BDNF-induced reversal of NMDA EPSC deficit in BDNFMet/Met mice is not due to an increase in glutamate release.
Figure 9.

BDNF enhanced non-NMDA receptor transmission in the IL-mPFC pyramidal neurons of wild-type mice but not BDNFMet/Met mice. A, Average non-NMDA EPSC amplitudes in the layer II/III pyramidal neurons from BDNF-treated (n = 6) and untreated wild-type brain slices (n = 10). BDNF treatment significantly enhanced non-NMDA receptor transmission in the wild-type neurons (F(1,14) = 14.7, p < 0.01). B, Average non-NMDA EPSC amplitudes in the layer II/III pyramidal neurons from BDNF-treated (n = 9) and untreated BDNFMet/Met brain slices (n = 7). BDNF treatment failed to enhance non-NMDA receptor transmission in the BDNFMet/Met neurons (p > 0.05). C, Average non-NMDA EPSC amplitudes in the layer V pyramidal neurons from BDNF-treated (n = 7) and untreated wild-type brain slices (n = 5). BDNF treatment significantly enhanced non-NMDA receptor transmission in the wild-type neurons at stimulation strength >250 μA (F(1,10) = 5.9, p < 0.05). D, Average non-NMDA EPSC amplitudes in the layer V pyramidal neurons from BDNF-treated (n = 6) and untreated BDNFMet/Met brain slices (n = 6). BDNF treatment failed to enhance non-NMDA receptor transmission in the BDNFMet/Met neurons (p > 0.05).
Discussion
Our results demonstrate a selective effect of the BDNF Val66Met polymorphism on NMDA, but not non-NMDA, receptor transmission in both layer II/III and layer V pyramidal neurons of the IL-mPFC. Recent experiments also revealed a similar decrease in NMDA receptor transmission in the CA1 pyramidal neurons of BDNFMet/Met mice (Ninan et al., 2010). Although the mechanism is unclear, BDNF is known to alter phosphorylation and activity of NMDA receptors (Suen et al., 1997; Levine et al., 1998; Lin et al., 1998; Crozier et al., 2008). Also, the role of BDNF in the regulation of expression and trafficking of NMDA receptors has been suggested recently (Caldeira et al., 2007a). Therefore, it is possible that activity-dependent release of BDNF is necessary for normal NMDA receptor transmission. Although a recent study demonstrated altered dendritic spine morphology in the mPFC pyramidal neurons of BDNFMet/Met mice (Liu et al., 2012), our results showed normal non-NMDA receptor transmission in both the layer II/III and layer V pyramidal neurons from BDNFMet/Met mice.
Consistent with the decrease in NMDA receptor transmission, we observed significant deficit in STDP in the IL-mPFC of BDNFMet/Met mice. Earlier studies have shown that STDP required the activation of NMDA receptors (Magee and Johnston, 1997; Markram et al., 1997; Bi and Poo, 1998; Feldman, 2000; Froemke and Dan, 2002; Froemke et al., 2005; Caporale and Dan, 2008). Although BDNF-TrkB signaling has been implicated in activity-dependent synaptic plasticity (Kang and Schuman, 1995; Figurov et al., 1996; Yano et al., 2006), little is known about its role in STDP (Mu and Poo, 2006). A recent study on a mouse that lacks promoter IV driven Bdnf transcription (BDNF-KIV) exhibited an increase in STDP (Sakata et al., 2009). In contrast, we observed a complete lack of STDP in both the layer II/III and layer V pyramidal neurons in BDNFMet/Met mice, suggesting that the Val66Met polymorphism can interfere with the IL-mPFC function by decreasing plasticity. Unlike BDNF-KIV mice that showed decrease in GABAergic transmission without affecting NMDA receptor transmission (Sakata et al., 2009), BDNFMet/Met mice showed not only a decrease in GABAergic transmission but also a decrease in NMDA receptor transmission. Therefore, the effect of the Val66Met polymorphism on STDP is likely to be mediated by the decrease in NMDA receptor neurotransmission in both layer II/III and layer V pyramidal neurons.
Apart from the decrease in NMDA receptor-mediated transmission, we observed a significant deficit in GABAergic neurotransmission in both the layer II/III and layer V pyramidal neurons of BDNFMet/Met mice. While layer II/III pyramidal neurons showed a selective decrease in mIPSC frequency, layer V pyramidal neurons exhibited a decrease in both the frequency and amplitude of mIPSCs. Our results are consistent with the critical role of BDNF in the regulation of GABAergic neurotransmission in the cortex (Rutherford et al., 1997; Huang et al., 1999; Woo and Lu, 2006; Hong et al., 2008; Sakata et al., 2009; Zheng et al., 2011). BDNF has been known to affect presynaptic GABA release, postsynaptic GABA receptor function, and the expression of chloride transporters (Rivera et al., 2002; Aguado et al., 2003; Rivera et al., 2004; Gottmann et al., 2009; Peng et al., 2010; Zheng et al., 2011). Disruption of Bdnf promoter IV function selectively impairs GABAergic synapses (Hong et al., 2008; Sakata et al., 2009; Jiao et al., 2011). Although protein kinase C and phosphatase-2A have been proposed in the action of BDNF on GABAergic synapses (Jovanovic et al., 2004), little is known about how BDNF regulates GABAergic transmission. Nonetheless, our results imply that decreased activity-dependent release of BDNF could alter GABAergic transmission in the IL-mPFC.
Synaptic impairments in BDNFMet/Met mice could be due to either the lack of BDNF availability or a developmental modification of synapses. To address this issue, we tested whether acute activation of BDNF-TrkB could reverse any of these synaptic deficits. Interestingly, BDNF treatment reestablished normal plasticity and NMDA receptor transmission in the IL-mPFC pyramidal neurons of BDNFMet/Met mice. Given the role of NMDA receptors in STDP (Bi and Poo, 1998; Caporale and Dan, 2008), the reestablishment of NMDA receptor transmission might have led to the reversal of plasticity deficit in BDNFMet/Met mice. Other studies have shown that BDNF modulates phosphorylation, activity, expression, and trafficking of NMDA receptors (Carmignoto et al., 1997; Suen et al., 1997; Levine et al., 1998; Lin et al., 1998; Caldeira et al., 2007a; Crozier et al., 2008). Also, a recent study showed that BDNF infusion into the IL-mPFC enhances fear extinction in an NMDA receptor-dependent fashion (Peters et al., 2010). Therefore, decreased NMDA receptor transmission and plasticity in BDNFMet/Met mice may be due to the lack of availability of BDNF.
Congruent with the earlier studies (Frerking et al., 1998; Brünig et al., 2001; Wardle and Poo, 2003; Jovanovic et al., 2004), BDNF treatment decreased GABA receptor transmission in the IL-mPFC pyramidal neurons of wild-type mice. Both presynaptic and postsynaptic mechanisms might be involved in BDNF-induced suppression of inhibitory transmission (Jovanovic et al., 2004; Lemtiri-Chlieh and Levine, 2010). Surprisingly, BDNFMet/Met neurons were resistant to modification of GABA receptor transmission after BDNF treatment, suggesting that the BDNF Val66Met polymorphism might affect synapses by other mechanisms. Given the effect of the BDNF Val66Met polymorphism on dendritic complexity in the mPFC (Yu et al., 2009; Liu et al., 2012), it is plausible that morphological changes may contribute to the deficits in inhibitory transmission in BDNFMet/Met mice. Further studies will be needed to define the mechanism of BDNF-induced suppression of GABA receptor transmission in the cortex and how the Val66Met polymorphism interferes with this process.
BDNF-induced enhancement of excitatory synaptic transmission has been well described in several brain regions (Carmignoto et al., 1997; Li et al., 1998; Wardle and Poo, 2003; Wu et al., 2004; Yano et al., 2006; Caldeira et al., 2007b; Madara and Levine, 2008). We consistently observed an enhancement of non-NMDA receptor transmission in the IL-mPFC pyramidal neurons from wild-type mice after BDNF treatment (Carmignoto et al., 1997). However, this effect was absent in the IL-mPFC pyramidal neurons of BDNFMet/Met mice. Given that BDNF selectively enhanced NMDA but not non-NMDA receptor transmission in BDNFMet/Met mice, a presynaptic mechanism is unlikely to play a role in the differential effect of BDNF on NMDA receptor transmission in BDNFMet/Met neurons (Perkel and Nicoll, 1993). It has been shown that BDNF could modulate non-NMDA receptor transmission by regulating the expression and synaptic insertion of AMPA receptors (Narisawa-Saito et al., 1999; Caldeira et al., 2007b). The Val66Met polymorphism could interfere with BDNF-mediated expression and/or synaptic insertion of AMPA receptors in the IL-mPFC pyramidal neurons.
Abnormal regulation of fear extinction is implicated in major depression and anxiety-like disorders (Kendler et al., 2004; Hettema, 2008; Peters et al., 2009; Sandi and Richter-Levin, 2009). Recent studies have suggested that the top-down regulation of the amygdala by the IL-mPFC regulates fear extinction (Morgan and LeDoux, 1995; Bremner et al., 1999; Quirk et al., 2000, 2006; Milad and Quirk, 2002; Phelps et al., 2004; Santini et al., 2004, 2008; Amano et al., 2010). The IL-mPFC projections terminate in the basolateral amygdala and the GABAergic intercalated cell masses of the amygdala (Russchen, 1982; Gabbott and Bacon, 1996; McDonald et al., 1996; McDonald, 1998; Likhtik et al., 2005). Extinction-dependent synaptic and intrinsic plasticity in the IL-mPFC have been recently reported (Herry and Garcia, 2002; Burgos-Robles et al., 2007; Santini et al., 2008). Therefore, an impairment of IL-mPFC plasticity could result in dysregulation of affective behaviors. One potential mechanism for impaired synaptic plasticity and altered affective behaviors is reduced levels of BDNF (Chen et al., 2006; Soliman et al., 2010). The reported decrease in IL-mPFC activation in BDNFMet carriers is likely due to decreased NMDA receptor transmission and the resulting impairment of plasticity (Soliman et al., 2010). The present study indicates that enhancement of BDNF-TrkB signaling can reverse deficits in both the NMDA receptor transmission and plasticity in the IL-mPFC of BDNFMet/Met mice. Infusion of BDNF in the IL-mPFC enhanced fear extinction in an NMDA receptor-manner (Peters et al., 2010). Therefore, BDNF signaling might regulate NMDA receptor-mediated transmission and plasticity in the IL-mPFC, which is critical for normal affective behaviors.
In conclusion, our results show that the BDNF Val66Met polymorphism impairs NMDA and GABA receptor transmission. Also, the Val66Met polymorphism significantly alters STDP in the IL-mPFC. These synaptic deficits may play an important role in affective disorders. Furthermore, acute enhancement of BDNF signaling reversed the impairment of plasticity and NMDA receptor transmission in BDNFMet/Met mice without affecting either GABA or non-NMDA receptor transmission, suggesting that the IL-mPFC synapses are profoundly affected by BDNF.
Footnotes
This work was supported by the DeWitt-Wallace Fund of the New York Community Trust (F.S.L.), Burroughs Wellcome Foundation (F.S.L.), NARSAD (I.N., K.G.B.), and NIH Grants HD-055177 (S.S.P.), NS-21072 (M.V.C.), MH079513 (F.S.L.), and NS052819 (F.S.L.). We thank Dr. Robert Froemke for his comments on an earlier version of this manuscript.
References
- Aguado F, Carmona MA, Pozas E, Aguil ó A, Martínez-Guijarro FJ, Alcantara S, Borrell V, Yuste R, Ibañez CF, Soriano E. BDNF regulates spontaneous correlated activity at early developmental stages by increasing synaptogenesis and expression of the K+/Cl- co-transporter KCC2. Development. 2003;130:1267–1280. doi: 10.1242/dev.00351. [DOI] [PubMed] [Google Scholar]
- Amano T, Unal CT, Par é D. Synaptic correlates of fear extinction in the amygdala. Nat neuroscience. 2010;13:489–494. doi: 10.1038/nn.2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi GQ, Poo MM. Synaptic modifications in cultured hippocampal neurons: dependence on spike timing, synaptic strength, and postsynaptic cell type. J Neurosci. 1998;18:10464–10472. doi: 10.1523/JNEUROSCI.18-24-10464.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bremner JD, Staib LH, Kaloupek D, Southwick SM, Soufer R, Charney DS. Neural correlates of exposure to traumatic pictures and sound in Vietnam combat veterans with and without posttraumatic stress disorder: a positron emission tomography study. Biol Psychiatry. 1999;45:806–816. doi: 10.1016/s0006-3223(98)00297-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brünig I, Penschuck S, Berninger B, Benson J, Fritschy JM. BDNF reduces miniature inhibitory postsynaptic currents by rapid downregulation of GABA(A) receptor surface expression. The EurJ neuroscience. 2001;13:1320–1328. doi: 10.1046/j.0953-816x.2001.01506.x. [DOI] [PubMed] [Google Scholar]
- Burgos-Robles A, Vidal-Gonzalez I, Santini E, Quirk GJ. Consolidation of fear extinction requires NMDA receptor-dependent bursting in the ventromedial prefrontal cortex. Neuron. 2007;53:871–880. doi: 10.1016/j.neuron.2007.02.021. [DOI] [PubMed] [Google Scholar]
- Caldeira MV, Melo CV, Pereira DB, Carvalho RF, Carvalho AL, Duarte CB. BDNF regulates the expression and traffic of NMDA receptors in cultured hippocampal neurons. Mol Cell Neurosci. 2007a;35:208–219. doi: 10.1016/j.mcn.2007.02.019. [DOI] [PubMed] [Google Scholar]
- Caldeira MV, Melo CV, Pereira DB, Carvalho R, Correia SS, Backos DS, Carvalho AL, Esteban JA, Duarte CB. Brain-derived neurotrophic factor regulates the expression and synaptic delivery of alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptor subunits in hippocampal neurons. J Biol Chem. 2007b;282:12619–12628. doi: 10.1074/jbc.M700607200. [DOI] [PubMed] [Google Scholar]
- Caporale N, Dan Y. Spike timing-dependent plasticity: a Hebbian learning rule. Ann Rev Neurosci. 2008;31:25–46. doi: 10.1146/annurev.neuro.31.060407.125639. [DOI] [PubMed] [Google Scholar]
- Carmignoto G, Pizzorusso T, Tia S, Vicini S. Brain-derived neurotrophic factor and nerve growth factor potentiate excitatory synaptic transmission in the rat visual cortex. J Physiol. 1997;498:153–164. doi: 10.1113/jphysiol.1997.sp021848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ZY, Jing D, Bath KG, Ieraci A, Khan T, Siao CJ, Herrera DG, Toth M, Yang C, McEwen BS, Hempstead BL, Lee FS. Genetic variant BDNF (Val66Met) polymorphism alters anxiety-related behavior. Science. 2006;314:140–143. doi: 10.1126/science.1129663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couey JJ, Meredith RM, Spijker S, Poorthuis RB, Smit AB, Brussaard AB, Mansvelder HD. Distributed network actions by nicotine increase the threshold for spike-timing-dependent plasticity in prefrontal cortex. Neuron. 2007;54:73–87. doi: 10.1016/j.neuron.2007.03.006. [DOI] [PubMed] [Google Scholar]
- Crozier RA, Bi C, Han YR, Plummer MR. BDNF modulation of NMDA receptors is activity dependent. J Neurophysiol. 2008;100:3264–3274. doi: 10.1152/jn.90418.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeFelipe J, Fariñas I. The pyramidal neuron of the cerebral cortex: morphological and chemical characteristics of the synaptic inputs. Prog Neurobiol. 1992;39:563–607. doi: 10.1016/0301-0082(92)90015-7. [DOI] [PubMed] [Google Scholar]
- Drevets WC, Price JL, Furey ML. Brain structural and functional abnormalities in mood disorders: implications for neurocircuitry models of depression. Brain structure and function. 2008;213:93–118. doi: 10.1007/s00429-008-0189-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dwivedi Y. Brain-derived neurotrophic factor: role in depression and suicide. Neuropsychiatr Dis Treat. 2009;5:433–449. doi: 10.2147/ndt.s5700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dwivedi Y, Rizavi HS, Conley RR, Roberts RC, Tamminga CA, Pandey GN. Altered gene expression of brain-derived neurotrophic factor and receptor tyrosine kinase B in postmortem brain of suicide subjects. Arch Gen Psychiatry. 2003;60:804–815. doi: 10.1001/archpsyc.60.8.804. [DOI] [PubMed] [Google Scholar]
- Egan MF, Kojima M, Callicott JH, Goldberg TE, Kolachana BS, Bertolino A, Zaitsev E, Gold B, Goldman D, Dean M, Lu B, Weinberger DR. The BDNF val66met polymorphism affects activity-dependent secretion of BDNF and human memory and hippocampal function. Cell. 2003;112:257–269. doi: 10.1016/s0092-8674(03)00035-7. [DOI] [PubMed] [Google Scholar]
- Fanselow MS. Conditioned and unconditional components of post-shock freezing. Pavlov J Biol Sci. 1980;15:177–182. doi: 10.1007/BF03001163. [DOI] [PubMed] [Google Scholar]
- Feldman DE. Timing-based LTP and LTD at vertical inputs to layer II/III pyramidal cells in rat barrel cortex. Neuron. 2000;27:45–56. doi: 10.1016/s0896-6273(00)00008-8. [DOI] [PubMed] [Google Scholar]
- Figurov A, Pozzo-Miller LD, Olafsson P, Wang T, Lu B. Regulation of synaptic responses to high-frequency stimulation and LTP by neurotrophins in the hippocampus. Nature. 1996;381:706–709. doi: 10.1038/381706a0. [DOI] [PubMed] [Google Scholar]
- Frerking M, Malenka RC, Nicoll RA. Brain-derived neurotrophic factor (BDNF) modulates inhibitory, but not excitatory, transmission in the CA1 region of the hippocampus. J Neurophysiol. 1998;80:3383–3386. doi: 10.1152/jn.1998.80.6.3383. [DOI] [PubMed] [Google Scholar]
- Froemke RC, Dan Y. Spike-timing-dependent synaptic modification induced by natural spike trains. Nature. 2002;416:433–438. doi: 10.1038/416433a. [DOI] [PubMed] [Google Scholar]
- Froemke RC, Poo MM, Dan Y. Spike-timing-dependent synaptic plasticity depends on dendritic location. Nature. 2005;434:221–225. doi: 10.1038/nature03366. [DOI] [PubMed] [Google Scholar]
- Fukumitsu H, Ohtsuka M, Murai R, Nakamura H, Itoh K, Furukawa S. Brain-derived neurotrophic factor participates in determination of neuronal laminar fate in the developing mouse cerebral cortex. J Neurosci. 2006;26:13218–13230. doi: 10.1523/JNEUROSCI.4251-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabbott PL, Bacon SJ. Local circuit neurons in the medial prefrontal cortex (areas 24a,b,c, 25 and 32) in the monkey: II. Quantitative areal and laminar distributions. J Comp Neurol. 1996;364:609–636. doi: 10.1002/(SICI)1096-9861(19960122)364:4<609::AID-CNE2>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- Gabbott PL, Warner TA, Jays PR, Salway P, Busby SJ. Prefrontal cortex in the rat: projections to subcortical autonomic, motor, and limbic centers. J Comp Neurol. 2005;492:145–177. doi: 10.1002/cne.20738. [DOI] [PubMed] [Google Scholar]
- Gonul AS, Akdeniz F, Taneli F, Donat O, Eker C, Vahip S. Effect of treatment on serum brain-derived neurotrophic factor levels in depressed patients. Eur Arch Psychiatry Clin Neurosci. 2005;255:381–386. doi: 10.1007/s00406-005-0578-6. [DOI] [PubMed] [Google Scholar]
- Gottmann K, Mittmann T, Lessmann V. BDNF signaling in the formation, maturation and plasticity of glutamatergic and GABAergic synapses. Exp Brain Res. 2009;199:203–234. doi: 10.1007/s00221-009-1994-z. [DOI] [PubMed] [Google Scholar]
- Hájos N, Nusser Z, Rancz EA, Freund TF, Mody I. Cell type- and synapse-specific variability in synaptic GABAA receptor occupancy. Eur J Neurosci. 2000;12:810–818. doi: 10.1046/j.1460-9568.2000.00964.x. [DOI] [PubMed] [Google Scholar]
- Herry C, Garcia R. Prefrontal cortex long-term potentiation, but not long-term depression, is associated with the maintenance of extinction of learned fear in mice. J Neurosci. 2002;22:577–583. doi: 10.1523/JNEUROSCI.22-02-00577.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hettema JM. What is the genetic relationship between anxiety and depression? Am J Med Genet C Semin Med Genet. 2008;148C:140–146. doi: 10.1002/ajmg.c.30171. [DOI] [PubMed] [Google Scholar]
- Hong EJ, McCord AE, Greenberg ME. A biological function for the neuronal activity-dependent component of Bdnf transcription in the development of cortical inhibition. Neuron. 2008;60:610–624. doi: 10.1016/j.neuron.2008.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YY, Simpson E, Kellendonk C, Kandel ER. Genetic evidence for the bidirectional modulation of synaptic plasticity in the prefrontal cortex by D1 receptors. Proc Natl Acad Sci U S A. 2004;101:3236–3241. doi: 10.1073/pnas.0308280101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang ZJ, Kirkwood A, Pizzorusso T, Porciatti V, Morales B, Bear MF, Maffei L, Tonegawa S. BDNF regulates the maturation of inhibition and the critical period of plasticity in mouse visual cortex. Cell. 1999;98:739–755. doi: 10.1016/s0092-8674(00)81509-3. [DOI] [PubMed] [Google Scholar]
- Hwang JP, Tsai SJ, Hong CJ, Yang CH, Lirng JF, Yang YM. The Val66Met polymorphism of the brain-derived neurotrophic-factor gene is associated with geriatric depression. Neurobiol Aging. 2006;27:1834–1837. doi: 10.1016/j.neurobiolaging.2005.10.013. [DOI] [PubMed] [Google Scholar]
- Jiang X, Xu K, Hoberman J, Tian F, Marko AJ, Waheed JF, Harris CR, Marini AM, Enoch MA, Lipsky RH. BDNF variation and mood disorders: a novel functional promoter polymorphism and Val66Met are associated with anxiety but have opposing effects. Neuropsychopharmacology. 2005;30:1353–1361. doi: 10.1038/sj.npp.1300703. [DOI] [PubMed] [Google Scholar]
- Jiao Y, Zhang Z, Zhang C, Wang X, Sakata K, Lu B, Sun QQ. A key mechanism underlying sensory experience-dependent maturation of neocortical GABAergic circuits in vivo. Proc Natl Acad Sci U S A. 2011;108:12131–12136. doi: 10.1073/pnas.1105296108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jovanovic JN, Thomas P, Kittler JT, Smart TG, Moss SJ. Brain-derived neurotrophic factor modulates fast synaptic inhibition by regulating GABA(A) receptor phosphorylation, activity, and cell-surface stability. J Neurosci. 2004;24:522–530. doi: 10.1523/JNEUROSCI.3606-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang H, Schuman EM. Long-lasting neurotrophin-induced enhancement of synaptic transmission in the adult hippocampus. Science. 1995;267:1658–1662. doi: 10.1126/science.7886457. [DOI] [PubMed] [Google Scholar]
- Karege F, Bondolfi G, Gervasoni N, Schwald M, Aubry JM, Bertschy G. Low brain-derived neurotrophic factor (BDNF) levels in serum of depressed patients probably results from lowered platelet BDNF release unrelated to platelet reactivity. Biol Psychiatry. 2005;57:1068–1072. doi: 10.1016/j.biopsych.2005.01.008. [DOI] [PubMed] [Google Scholar]
- Kendler KS, Kuhn J, Prescott CA. The interrelationship of neuroticism, sex, and stressful life events in the prediction of episodes of major depression. Am J Psychiatry. 2004;161:631–636. doi: 10.1176/appi.ajp.161.4.631. [DOI] [PubMed] [Google Scholar]
- Koponen E, Rantamäki T, Voikar V, Saarelainen T, MacDonald E, Castrén E. Enhanced BDNF signaling is associated with an antidepressant-like behavioral response and changes in brain monoamines. Cell Mol Neurobiol. 2005;25:973–980. doi: 10.1007/s10571-005-8468-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemtiri-Chlieh F, Levine ES. BDNF evokes release of endogenous cannabinoids at layer 2/3 inhibitory synapses in the neocortex. J Neurophysiol. 2010;104:1923–1932. doi: 10.1152/jn.00472.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine ES, Crozier RA, Black IB, Plummer MR. Brain-derived neurotrophic factor modulates hippocampal synaptic transmission by increasing N-methyl-d-aspartic acid receptor activity. Proc Natl Acad Sci U S A. 1998;95:10235–10239. doi: 10.1073/pnas.95.17.10235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YX, Zhang Y, Lester HA, Schuman EM, Davidson N. Enhancement of neurotransmitter release induced by brain-derived neurotrophic factor in cultured hippocampal neurons. J Neurosci. 1998;18:10231–10240. doi: 10.1523/JNEUROSCI.18-24-10231.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Likhtik E, Pelletier JG, Paz R, Par é D. Prefrontal control of the amygdala. J Neurosci. 2005;25:7429–7437. doi: 10.1523/JNEUROSCI.2314-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin SY, Wu K, Levine ES, Mount HT, Suen PC, Black IB. BDNF acutely increases tyrosine phosphorylation of the NMDA receptor subunit 2B in cortical and hippocampal postsynaptic densities. Brain Res Mol Brain Res. 1998;55:20–27. doi: 10.1016/s0169-328x(97)00349-5. [DOI] [PubMed] [Google Scholar]
- Linnarsson S, Björklund A, Ernfors P. Learning deficit in BDNF mutant mice. Eur J Neurosci. 1997;9:2581–2587. doi: 10.1111/j.1460-9568.1997.tb01687.x. [DOI] [PubMed] [Google Scholar]
- Liu RJ, Lee FS, Li XY, Bambico F, Duman RS, Aghajanian GK. Brain-derived neurotrophic factor Val66Met allele impairs basal and ketamine-stimulated synaptogenesis in prefrontal cortex. Biol Psychiatry. 2012 doi: 10.1016/j.biopsych.2011.09.030. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madara JC, Levine ES. Presynaptic and postsynaptic NMDA receptors mediate distinct effects of brain-derived neurotrophic factor on synaptic transmission. J Neurophysiol. 2008;100:3175–3184. doi: 10.1152/jn.90880.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magee JC, Johnston D. A synaptically controlled, associative signal for Hebbian plasticity in hippocampal neurons. Science. 1997;275:209–213. doi: 10.1126/science.275.5297.209. [DOI] [PubMed] [Google Scholar]
- Markram H, Lübke J, Frotscher M, Sakmann B. Regulation of synaptic efficacy by coincidence of postsynaptic APs and EPSPs. Science. 1997;275:213–215. doi: 10.1126/science.275.5297.213. [DOI] [PubMed] [Google Scholar]
- McDonald AJ. Cortical pathways to the mammalian amygdala. Prog Neurobiol. 1998;55:257–332. doi: 10.1016/s0301-0082(98)00003-3. [DOI] [PubMed] [Google Scholar]
- McDonald AJ, Mascagni F, Guo L. Projections of the medial and lateral prefrontal cortices to the amygdala: a Phaseolus vulgaris leucoagglutinin study in the rat. Neuroscience. 1996;71:55–75. doi: 10.1016/0306-4522(95)00417-3. [DOI] [PubMed] [Google Scholar]
- Milad MR, Quirk GJ. Neurons in medial prefrontal cortex signal memory for fear extinction. Nature. 2002;420:70–74. doi: 10.1038/nature01138. [DOI] [PubMed] [Google Scholar]
- Morgan MA, LeDoux JE. Differential contribution of dorsal and ventral medial prefrontal cortex to the acquisition and extinction of conditioned fear in rats. Behav Neurosci. 1995;109:681–688. doi: 10.1037//0735-7044.109.4.681. [DOI] [PubMed] [Google Scholar]
- Mu Y, Poo MM. Spike timing-dependent LTP/LTD mediates visual experience-dependent plasticity in a developing retinotectal system. Neuron. 2006;50:115–125. doi: 10.1016/j.neuron.2006.03.009. [DOI] [PubMed] [Google Scholar]
- Myers KM, Ressler KJ, Davis M. Different mechanisms of fear extinction dependent on length of time since fear acquisition. Learn Mem. 2006;13:216–223. doi: 10.1101/lm.119806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narisawa-Saito M, Carnahan J, Araki K, Yamaguchi T, Nawa H. Brain-derived neurotrophic factor regulates the expression of AMPA receptor proteins in neocortical neurons. Neuroscience. 1999;88:1009–1014. doi: 10.1016/s0306-4522(98)00496-5. [DOI] [PubMed] [Google Scholar]
- Nibuya M, Morinobu S, Duman RS. Regulation of BDNF and trkB mRNA in rat brain by chronic electroconvulsive seizure and antidepressant drug treatments. J Neurosci. 1995;15:7539–7547. doi: 10.1523/JNEUROSCI.15-11-07539.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ninan I, Bath KG, Dagar K, Perez-Castro R, Plummer MR, Lee FS, Chao MV. The BDNF Val66Met polymorphism impairs NMDA receptor-dependent synaptic plasticity in the hippocampus. J Neurosci. 2010;30:8866–8870. doi: 10.1523/JNEUROSCI.1405-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Par é D, Quirk GJ, Ledoux JE. New vistas on amygdala networks in conditioned fear. J Neurophysiol. 2004;92:1–9. doi: 10.1152/jn.00153.2004. [DOI] [PubMed] [Google Scholar]
- Peng YR, Zeng SY, Song HL, Li MY, Yamada MK, Yu X. Postsynaptic spiking homeostatically induces cell-autonomous regulation of inhibitory inputs via retrograde signaling. J Neurosci. 2010;30:16220–16231. doi: 10.1523/JNEUROSCI.3085-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkel DJ, Nicoll RA. Evidence for all-or-none regulation of neurotransmitter release: implications for long-term potentiation. J Physiol. 1993;471:481–500. doi: 10.1113/jphysiol.1993.sp019911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrais D, Ropert N. Effect of zolpidem on miniature IPSCs and occupancy of postsynaptic GABAA receptors in central synapses. J Neurosci. 1999;19:578–588. doi: 10.1523/JNEUROSCI.19-02-00578.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters J, Kalivas PW, Quirk GJ. Extinction circuits for fear and addiction overlap in prefrontal cortex. Learn Mem. 2009;16:279–288. doi: 10.1101/lm.1041309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters J, Dieppa-Perea LM, Melendez LM, Quirk GJ. Induction of fear extinction with hippocampal-infralimbic BDNF. Science. 2010;328:1288–1290. doi: 10.1126/science.1186909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phelps EA, Delgado MR, Nearing KI, LeDoux JE. Extinction learning in humans: role of the amygdala and vmPFC. Neuron. 2004;43:897–905. doi: 10.1016/j.neuron.2004.08.042. [DOI] [PubMed] [Google Scholar]
- Piccinni A, Marazziti D, Catena M, Domenici L, Del Debbio A, Bianchi C, Mannari C, Martini C, Da Pozzo E, Schiavi E, Mariotti A, Roncaglia I, Palla A, Consoli G, Giovannini L, Massimetti G, Dell'Osso L. Plasma and serum brain-derived neurotrophic factor (BDNF) in depressed patients during 1 year of antidepressant treatments. J Affect Disord. 2008;105:279–283. doi: 10.1016/j.jad.2007.05.005. [DOI] [PubMed] [Google Scholar]
- Quirk GJ, Russo GK, Barron JL, Lebron K. The role of ventromedial prefrontal cortex in the recovery of extinguished fear. J Neurosci. 2000;20:6225–6231. doi: 10.1523/JNEUROSCI.20-16-06225.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quirk GJ, Garcia R, González-Lima F. Prefrontal mechanisms in extinction of conditioned fear. Biol Psychiatry. 2006;60:337–343. doi: 10.1016/j.biopsych.2006.03.010. [DOI] [PubMed] [Google Scholar]
- Rantamäki T, Hendolin P, Kankaanp ää A, Mijatovic J, Piepponen P, Domenici E, Chao MV, Männist ö PT, Castrén E. Pharmacologically diverse antidepressants rapidly activate brain-derived neurotrophic factor receptor TrkB and induce phospholipase-Cgamma signaling pathways in mouse brain. Neuropsychopharmacology. 2007;32:2152–2162. doi: 10.1038/sj.npp.1301345. [DOI] [PubMed] [Google Scholar]
- Rauch SL, Shin LM, Phelps EA. Neurocircuitry models of posttraumatic stress disorder and extinction: human neuroimaging research–past, present, and future. Biol Psychiatry. 2006;60:376–382. doi: 10.1016/j.biopsych.2006.06.004. [DOI] [PubMed] [Google Scholar]
- Rivera C, Li H, Thomas-Crusells J, Lahtinen H, Viitanen T, Nanobashvili A, Kokaia Z, Airaksinen MS, Voipio J, Kaila K, Saarma M. BDNF-induced TrkB activation down-regulates the K+-Cl- cotransporter KCC2 and impairs neuronal Cl- extrusion. J Cell Biol. 2002;159:747–752. doi: 10.1083/jcb.200209011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera C, Voipio J, Thomas-Crusells J, Li H, Emri Z, Sipil ä S, Payne JA, Minichiello L, Saarma M, Kaila K. Mechanism of activity-dependent downregulation of the neuron-specific K-Cl cotransporter KCC2. J Neurosci. 2004;24:4683–4691. doi: 10.1523/JNEUROSCI.5265-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russchen FT. Amygdalopetal projections in the cat. I. Cortical afferent connections. A study with retrograde and anterograde tracing techniques. J Comp Neurol. 1982;206:159–179. doi: 10.1002/cne.902060206. [DOI] [PubMed] [Google Scholar]
- Russo-Neustadt A, Beard RC, Cotman CW. Exercise, antidepressant medications, and enhanced brain derived neurotrophic factor expression. Neuropsychopharmacology. 1999;21:679–682. doi: 10.1016/S0893-133X(99)00059-7. [DOI] [PubMed] [Google Scholar]
- Rutherford LC, DeWan A, Lauer HM, Turrigiano GG. Brain-derived neurotrophic factor mediates the activity-dependent regulation of inhibition in neocortical cultures. J Neurosci. 1997;17:4527–4535. doi: 10.1523/JNEUROSCI.17-12-04527.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakata K, Woo NH, Martinowich K, Greene JS, Schloesser RJ, Shen L, Lu B. Critical role of promoter IV-driven BDNF transcription in GABAergic transmission and synaptic plasticity in the prefrontal cortex. Proc Natl Acad Sci U S A. 2009;106:5942–5947. doi: 10.1073/pnas.0811431106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandi C, Richter-Levin G. From high anxiety trait to depression: a neurocognitive hypothesis. Trends Neurosci. 2009;32:312–320. doi: 10.1016/j.tins.2009.02.004. [DOI] [PubMed] [Google Scholar]
- Santini E, Ge H, Ren K, Peña de Ortiz S, Quirk GJ. Consolidation of fear extinction requires protein synthesis in the medial prefrontal cortex. J Neurosci. 2004;24:5704–5710. doi: 10.1523/JNEUROSCI.0786-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santini E, Quirk GJ, Porter JT. Fear conditioning and extinction differentially modify the intrinsic excitability of infralimbic neurons. J Neurosci. 2008;28:4028–4036. doi: 10.1523/JNEUROSCI.2623-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacher J, Jamra RA, Becker T, Ohlraun S, Klopp N, Binder EB, Schulze TG, Deschner M, Schmäl C, Höfels S, Zobel A, Illig T, Propping P, Holsboer F, Rietschel M, Nöthen MM, Cichon S. Evidence for a relationship between genetic variants at the brain-derived neurotrophic factor (BDNF) locus and major depression. Biol Psychiatry. 2005;58:307–314. doi: 10.1016/j.biopsych.2005.04.006. [DOI] [PubMed] [Google Scholar]
- Soliman F, Glatt CE, Bath KG, Levita L, Jones RM, Pattwell SS, Jing D, Tottenham N, Amso D, Somerville LH, Voss HU, Glover G, Ballon DJ, Liston C, Teslovich T, Van Kempen T, Lee FS, Casey BJ. A genetic variant BDNF polymorphism alters extinction learning in both mouse and human. Science. 2010;327:863–866. doi: 10.1126/science.1181886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suen PC, Wu K, Levine ES, Mount HT, Xu JL, Lin SY, Black IB. Brain-derived neurotrophic factor rapidly enhances phosphorylation of the postsynaptic N-methyl-d-aspartate receptor subunit 1. Proc Natl Acad Sci U S A. 1997;94:8191–8195. doi: 10.1073/pnas.94.15.8191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terracciano A, Tanaka T, Sutin AR, Deiana B, Balaci L, Sanna S, Olla N, Maschio A, Uda M, Ferrucci L, Schlessinger D, Costa PJ., Jr BDNF Val66Met is associated with introversion and interacts with 5-HTTLPR to influence neuroticism. Neuropsychopharmacology. 2010;35:1083–1089. doi: 10.1038/npp.2009.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson Ray M, Weickert CS, Wyatt E, Webster MJ. Decreased BDNF, trkB-TK+ and GAD67 mRNA expression in the hippocampus of individuals with schizophrenia and mood disorders. J Psychiatry Neurosci. 2011;36:195–203. doi: 10.1503/jpn.100048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wardle RA, Poo MM. Brain-derived neurotrophic factor modulation of GABAergic synapses by postsynaptic regulation of chloride transport. J Neurosci. 2003;23:8722–8732. doi: 10.1523/JNEUROSCI.23-25-08722.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo NH, Lu B. Regulation of cortical interneurons by neurotrophins: from development to cognitive disorders. Neuroscientist. 2006;12:43–56. doi: 10.1177/1073858405284360. [DOI] [PubMed] [Google Scholar]
- Wu K, Len GW, McAuliffe G, Ma C, Tai JP, Xu F, Black IB. Brain-derived neurotrophic factor acutely enhances tyrosine phosphorylation of the AMPA receptor subunit GluR1 via NMDA receptor-dependent mechanisms. Brain Res Mol Brain Res. 2004;130:178–186. doi: 10.1016/j.molbrainres.2004.07.019. [DOI] [PubMed] [Google Scholar]
- Yano H, Ninan I, Zhang H, Milner TA, Arancio O, Chao MV. BDNF-mediated neurotransmission relies upon a myosin VI motor complex. Nat Neurosci. 2006;9:1009–1018. doi: 10.1038/nn1730. [DOI] [PubMed] [Google Scholar]
- Yu H, Wang Y, Pattwell S, Jing D, Liu T, Zhang Y, Bath KG, Lee FS, Chen ZY. Variant BDNF Val66Met polymorphism affects extinction of conditioned aversive memory. J Neurosci. 2009;29:4056–4064. doi: 10.1523/JNEUROSCI.5539-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng K, An JJ, Yang F, Xu W, Xu ZQ, Wu J, Hökfelt TG, Fisahn A, Xu B, Lu B. TrkB signaling in parvalbumin-positive interneurons is critical for gamma-band network synchronization in hippocampus. Proc Natl Acad Sci U S A. 2011;108:17201–17206. doi: 10.1073/pnas.1114241108. [DOI] [PMC free article] [PubMed] [Google Scholar]