

Abstract
One of the challenges to develop time-resolved fluorescence resonance energy transfer (TR-FRET) assay for serine/threonine (Ser/Thr) protein kinase is to select an optimal peptide substrate and a specific phosphor Ser/Thr antibody. This report describes a multiplexed random screen-based development of TR-FRET assay for ultra-high-throughput screening (uHTS) of small molecule inhibitors for a potent cancer drug target polo-like kinase 1 (Plk1). A screen of a diverse peptide library in a 384-well plate format identified several highly potent substrates that share the consensus motif for phosphorylation by Plk1. Their potencies were comparable to FKD peptide, a designed peptide substrate derived from well-described Plk1 substrate Cdc25C. A specific anti-phosphor Ser/Thr antibody p(S/T)F antibody that detects the phosphorylation of FKD peptide was screened out of 87 antibodies with time-resolved fluorometry technology in a 96-well plate format. Using FKD peptide and p(S/T)F antibody, we successfully developed a robust TR-FRET assay in 384-well plate format, and further miniaturized this assay to 1,536-well plate format to perform uHTS. We screened about 1.2 million compounds for Plk1 inhibitors using a Plk1 deletion mutant that only has the kinase domain and subsequently screened the same compound library using a full-length active-mutant Plk1. These uHTSs identified a number of hit compounds, and some of them had selectivity to either the deletion mutant or the full-length protein. Our results prove that a combination of random screen for substrate peptide and phospho-specific antibodies is very powerful strategy to develop TR-FRET assays for protein kinases.
Introduction
Since protein phosphorylation is one of the major regulation mechanisms for cell growth, differentiation, and survival,1 protein kinases represent one of the most important target classes in therapeutics.2 Protein kinase consists of a large superfamily with the high degree of structural conservation,3 which makes it difficult to develop a kinase inhibitor that is highly specific to the target kinase. One possible way around non-specific kinase inhibitors is to target substrate, or bisubstrate, inhibitors.4 Therefore, it is crucial to take account of specificity and physiological relevance of the assay5 as well as robustness in an assay development for screening of large compound libraries for lead molecule identification for selective inhibitors.
Various detection technologies for lead identification of kinase inhibitor programs have been validated and successfully applied for high-throughput screening (HTS).6,7 Being a homogeneous technology with a non-radioactive, ratiometric, and time-resolved measurement, time-resolved fluorescence resonance energy transfer (TR-FRET) has been most widely used among them.8,9 TR-FRET relies on the resonance energy transfer of photons from a long-lifetime lanthanide donor species to a suitable acceptor fluorophore. This transfer takes place only when the donor and the acceptor are in proximity. In a typical kinase TR-FRET assay, this proximity depends on the interaction mediated by a phospho-specific antibody that binds to the product of the kinase reaction. Therefore, the assay requires an optimal selection of a substrate, typically a synthetic peptide, and an antibody. Many tyrosine kinases accept random copolymers of glutamate and tyrosine as a substrate, and generic antibodies against phosphotyrosine are available whose binding affinities are not influenced by any surrounding residues.8 In contrast, serine/threonine (Ser/Thr) kinases have much higher substrate specificities, and it is challenging to select an optimal peptide substrate containing appropriate recognition motifs and comparable kinetics relative to a native protein. Furthermore, both phosphoserine and phosphothreonine have lower immunogenicity than phosphotyrosine, and each substrate requires different specific antibodies for phosphorylation detection. Therefore, identification of the correct peptide substrate and the corresponding antibody is problematic and often requires lengthy and costly efforts.10,11
A Ser/Thr kinase polo-like kinase 1 (Plk1) plays a crucial role in the precise regulation of cell division in various organisms.12–14 Because human Plk1 is overexpressed in various types of cancer and its expression level correlates to poor patient prognosis, this protein is one of the major drug targets for anti-cancer therapy.15,16 Even though several Plk1 inhibitors have been reported, more selective and efficacious drug without off-target effects needs to be discovered.17
Our goal is to identify novel lead compounds for Plk1 inhibitor by running an ultra-high-throughput screening (uHTS). Several studies have presented in vitro kinase assays for PLK1.18,19 However, these assays are not necessarily suitable for uHTS, being a non-robust radiometric filtration assay or using a substrate without physiological relevance. We employed TR-FRET technology to develop a non-radioisotopic and robust biochemical assay, and identified both a potent substrate peptide with physiological relevance and an antibody specific to the phosphorylated form of the peptide by conducting multiplexed random screenings. First, we screened >800 synthetic peptides with [γ-33P]ATP and a Plk1 deletion mutant that only has the kinase domain, and found a highly potent peptide named FKD. FKD has sequence homology with the region around serine 198 of human Cdc25C, one of Plk1’s physiological substrates in M phase.20 Serine 198 residue is the phosphorylation site of Plk121 and a hydrophobic residue at +1 position and acidic residues at −2 and +3 positions are required for this phosphorylation.22 Subsequently, we tested 87 antibodies in a 96-well format for the detection of the phosphorylated form of FKD using time-resolved fluorometry technology (TRF, also known as DELFIA® technology),23 and found that anti-phospho-(S/T)F antibody efficiently and specifically recognizes phosphorylated FKD peptide. Using FKD peptide and anti-phospho-(S/T)F antibody, we succeeded in developing a robust TR-FRET assay in a 384-well plate format. We miniaturized this assay for a 1,536-well format and performed fully automated uHTSs for about 1.2 million compounds. We screened this library for Plk1 inhibitors with a truncated protein that has only the kinase domain and a constitutively activated full-length protein, and identified a number of hit compounds. Interestingly, some of the hit compounds were active to only one of these 2 forms. We shall propose that a combination of random screening for a substrate peptide and a phospho-specific antibody can be a powerful strategy to develop a specific, physiologically relevant and robust TR-FRET assay for uHTS.
Materials and Methods
Materials
Chemical reagents were from either WAKO (Osaka, Japan) or Sigma-Aldrich (St. Louis, MO), except for the following materials: anti-phospho-(S/T)F antibody-labeled with europium-chelate (Eu-p(S/T)F Ab) and streptavidin–allophycocyanin (SA-APC), PerkinElmer Life Sciences (Boston, MA); K252a, Alomone Labs, Ltd. (Jerusalem, Israel); Kinase Substrate Set (720 peptides), JPT (Berlin, Germany); Kinase Substrate Screening Kit (87 peptides), Cell Signaling Technology (Beverly, MA). Additionally, we designed several biotinylated peptides with the consensus motif of Plk1 phosphorylation site and synthesized them in-house.
Recombinant human Plk1ΔC and Plk1T210D protein that was tagged with glutathione-S-transferase at the N-terminus was prepared with a baculovirus expression system and purified with their affinities to glutathione sepharose. Plk1ΔC is a deletion mutant that has the kinase domain (amino acid residues 1–356), and Plk1T210D is a full-length point mutant whose threonine 210 in the T-loop is mutated to aspartate. We made small aliquots of fractions for the storage at −80°C to avoid repeated freeze-thawing, and freshly thawed the frozen stock before dilution in every single experiment.
We used the following microtiter plates for TR-FRET assay: non-binding surface, black wall, clear-bottom 384-well plate, CORNING (Corning, NY); non-treated, white wall, solid-bottom 1,536-well plate, Greiner America Inc. (Lake Mary, FL).
Screen for a Potent Peptide Substrate for Plk1
In order to identify potent peptide substrate for Plk1, we designed several candidates with the reported consensus motif of Plk1 phosphorylation site22 and synthesized them in-house as biotinylated peptide at the N-terminus. In addition, we obtained the following commercial biotinylated peptide sets: Kinase Substrate Set (720 peptides, JPT) and Kinase Substrate Screening Kit (87 peptides, Cell Signaling Technology, Beverly, MA). Substrate screening with these peptides was performed as follows: 5 µM peptide was incubated with 120 nM Plk1ΔC and 50 µM ATP ([γ-33P]ATP, specific activity = 0.8 Ci/mmol) in 25 µL buffer A [20 mM Tris–HCl (pH 7.4)/10 mM MgCl2/1 mM EDTA/0.5 mM DTT] for 3 h at 25°C. We stopped the reaction with 40 µL Kinase stop buffer (25 mM EDTA in PBS), and transferred 10 µL of the reaction mixture to a 384-well Streptavidin FlashPlate PLUS (PerkinElmer Japan, Tokyo, Japan) that was pre-equilibrated with PBS. After incubating the plate for 30 min at room temperature for the binding of biotin to streptavidin, we washed wells with excess volume of PBS 4 times and measured radioactivity that remained in wells with a TopCount-NXT™ plate reader (PerkinElmer, Japan).
Screen of Anti-Phospho-Specific Antibody
To select a specific antibody for phosphorylated form of biotinylated FKD peptide (biotin-DELMEFpSFKDQEAKV; b-pFKD), we screened 87 phosphor-(Ser/Thr)-specific antibodies from Serine/Threonine Kinase Substrate Screening Kit (Cell Signaling Technology) with time-resolved fluorometry (TRF, also known as DELFIA® technology).23 We incubated b-FKD (biotin-DELMEFSFKDQEAKV) with 50 µM ATP and 120 nM Plk1ΔC in buffer A for 5 h at 25°C, and transferred the reaction mixture to a streptavidin-coated 96-well plate to immobilize b-pFKD. Then we dispensed antibodies, with one per well, and incubated the plate. After adding the secondary antibody labeled with europium (Eu)-chelate and further incubating the plate, we measured relative fluorescence unit (RFU) at 615 nm under the excitation at 340 nm (SIG_pFKD) using a Victor2™ plate reader (PerkinElmer Life Sciences) to quantify the amount of secondary antibody that remained in the wells. To demonstrate the specificity of each antibody for phosphorylation, we incubated b-FKD without Plk1ΔC as background reference (SIG_FKD), and calculated signal-to-background ratio by dividing SIG_pFKD by SIG_FKD. For details, please refer to the instruction manual of the kit. Screening of specific antibodies for the phosphorylated form of a biotinylated ASFA peptide (biotin-DELMEApSFADQEAK; b-pASFA) was conducted with the same procedure, except that b-ASFA (biotin-DELMEASFADQEAK) was used in place of b-FKD.
Development of Plk1 TR-FRET Assay in a 384-Well Format
Plk1 TR-FRET assay with b-FKD and Eu-p(S/T)F Ab was developed in a 384-well plate format (Fig. 1A). The assay reagents were made up as shown below. Each concentration for b-FKD, Eu-p(S/T)F, and SA-APC was determined with a series of optimization studies (data not shown).
Fig. 1.

Plk1 assay protocols used in this study: (A) Original version in a 384-well plate for miniaturization. (B) The modified 384-well version for miniaturization. (C) Time-resolved fluorescence energy transfer (TR-FRET) assay in a 1,536-well plate format. See Materials and Methods for details.
Substrate: 800 nM b-FKD and 20 µM ATP in buffer A.
Enzyme: 0–200 nM (for 0–100 nM in kinase reaction) Plk1ΔC in buffer A.
EDTA: 100 mM EDTA (pH 7.5)
TR-FRET reagent: 2.0 nM Eu-p(S/T)F Ab/200 nM SA-APC/200 mM Tris–HCl (pH 7.8)/20 mg/mL BSA.
Assay was performed at room temperature. We started the reaction by mixing 10 µL each of the substrate and the enzyme. The final concentration of b-FKD, ATP, and Plk1ΔC were 400 nM, 10 µM, and 0–100 nM, respectively. After 60 min, we added 10 µL each of EDTA and TR-FRET reagent to stop the reaction and detect TR-FRET, and further incubated the plate for 60 min. Then, we loaded the plate onto the Victor2™ and measured RFU at 665 nm (RFU665) from APC and 615 nm (RFU615) from europium under the excitation at 337 nm with top read mode. The data were presented as the ratiometric TR-FRET signal as follows:
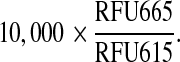 |
Assay Miniaturization of Plk1 TR-FRET to a 1,536-Well Plate Format
To miniaturize the assay to 1,536-well format for uHTS, we modified the original protocol as follows: first, we premixed EDTA and TR-FRET reagent to reduce the number of dispensing steps. Second, in order to accommodate our equipment for uHTS,24 we changed the volumetric ratio among substrate, enzyme, and EDTA/TR-FRET to 3:1:2. The assay reagents in this modified protocol were as follows:
Substrate: 267 nM b-FKD and 13.3 µM ATP in buffer B [20 mM Tris–HCl (pH 7.4)/10 mM MgCl2/1 mM EDTA/0.5 mM DTT/0.0025% (v/v) Tween-20/0.5% (v/v) glycerol/0.1 mg/mL BSA].
Enzyme: 0–400 nM (for 0–100 nM in kinase reaction) Plk1ΔC or Plk1T210D in buffer B.
EDTA/TR-FRET: 1.5 nM Eu-p(S/T)F Ab/150 nM SA-APC/150 mM Tris–HCl (pH 7.8)/60 mM EDTA/ 0.1 mg/mL BSA.
The final concentration of b-FKD, ATP, and Plk1ΔC or T210D in the kinase reaction were 200 nM, 10 µM, and 0–100 nM, respectively.
We validated the modified assay both in a 384- and in a 1,536-well plate format. The volume of substrate, enzyme, and EDTA/TR-FRET were 30, 10, and 20 µL for a 384-well plate and 3, 1, and 2 µL for a 1,536-well plate (Fig. 1B and 1C), respectively. For a 1,536-well plate, we used a Flying Reagent Dispenser I (FRD I, Vertex)25 and a 384-tip pintool (V&P Scientific, San Diego, CA) equipped on a CyBi-DISK (CyBio AG, Jena, Germany) to deliver the assay reagents and compounds, respectively. After adding EDTA/TR-FRET, we further incubated the plate for 45 min, and measured RFU665 and RFU615 under the excitation at 337 nm with the Victor2-V.
Ultra-High-Throughput Screening for Plk1 Inhibitor
In the primary screen, we used a PixSys 3200 Aspirate/Dispense System (Cartesian Technologies, Inc., Irvine, CA) for bulk reagent dispensing and 384-tip pintools equipped on a CyBi Replicator (Cybio AG) for compound transfer.26 Both instruments were under the control of a Robolab fully automated microplate laboratory version 3 (Robocon, Vienna, Austria).
Assay protocol was the same as the one for miniaturization (Fig. 1C). Compounds in the library were stored in 75% (v/v) DMSO solution in 384-well polypropylene plates (REMP, Oberdiessbach, Switzerland). These compound plates had 320 compounds in columns 3–22, with one compound per well. After dispensing the substrate, we transferred 30 nL of each compound from a compound plate to an assay plate with the pintools. Each assay plate received a total of 1,280 compounds from 4 compound plates by quadrants in columns 5–44. To check the carryover of the compounds, we also ran several mock plates which received 75% (v/v) DMSO instead of compounds. The screen concentration was either 15 or 30 µM in 4 µL reaction, depending on the storage concentration in the library. The incubation for reaction and detection was at 25°C in a Cytomat C470 incubator (Kendro, Milford, MA) for 1 and 2 h for Plk1ΔC and T210D, respectively.
To confirm the positive results in primary uHTS, we picked the primary hits from the library and re-plated them to 384-well polypropylene plates. Using these plates, we re-tested the hit compounds in 3 cycles to create triplicate results.
Assay Quality Assessment and Data Analysis
we analyzed data in Excel and the nonlinear regression software package GraphPad Prism (GraphPad Software Inc., San Diego, CA).
To assess the quality of the primary HTS with TR-FRET, we calculated the following 3 values for each plate with the equations shown below. The window is the ratio of the median of RFU ratio between the compound population and the inhibition control population.
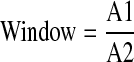 |
A1: the median of RFU ratio of the compound population
A2: the median of RFU ratio of the 100% inhibition population
The percentage coefficient of variance (% CV) is the percentage ratio of the standard deviation (std) over the mean of RFU ratio of the compound population.
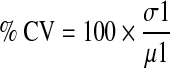 |
σ1: the standard deviation of RFU ratio of the compound population
µ1: the mean of RFU ratio of the compound population
The Z factor has been described previously.27
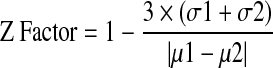 |
σ2: the standard deviation of RFU ratio of the 100% inhibition population
µ2: the mean of RFU ratio of the 100% inhibition population
To determine the activity of each compound, we calculated percentage inhibition (% INH) with the equation below. We normalized data with the median of RFU ratio from the compound field and the inhibition population in the same plate for 0 and 100, respectively.
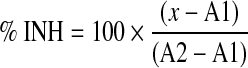 |
To select the primary hits, we targeted data variation at the 99.73% confidence limit and used the mean plus 3 times the standard deviation of the % INH values (mean + 3 std)27,28 of the compound population for each plate.
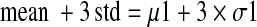 |
For the confirmatory assay of the primary hits, we calculated the same values, except that the 0% inhibition control was used in place of the median of the compound sample field for normalization. The equations are as follows.27
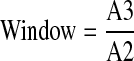 |
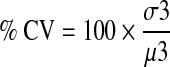 |
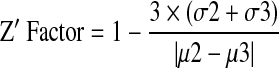 |
A3: the median of RFU ratio of the 0% inhibition population
σ3: the standard deviation of RFU ratio of the 0% inhibition population
µ3: the mean of RFU ratio of the 0% inhibition population
% INH activity of each compound was normalized with the median of RFU ratio from the 0% inhibition population.
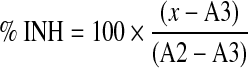 |
Results
Screen of Random Peptide Library for a Highly Potent Substrate of Plk1 Phosphorylation
To develop a robust TR-FRET assay to measure Plk1 activity, we searched a biotinylated peptide substrate that shows high potency. Plk1 phosphorylates various cellular proteins including cyclin B1 and Cdc25C.21,29,30 Nakajima et al. identified the consensus motif (D/E)x(S/T)φx(D/E) (S/T, serine/threonine to be phosphorylated; φ, a hydrophobic amino acid; x, any amino acid) for phosphorylation by Plk1 and found that a hydrophobic residue at +1 position and acidic residues at −2 and +3 positions are essential22 (Fig. 2A). Referring the sequence of these known substrates, we designed several candidates with the consensus sequence and synthesized them in-house. Additionally, we obtained 2 peptide libraries from commercial resources to avoid bias, and made up a library of >800 peptides with diverse sequences.
Fig. 2.

Search for potent peptide substrates for Plk1. (A) Consensus motif around Plk1 phosphorylation site. The following conserved residues are shown in one-letter code: “S/T,” serine or threonine at position 0 to be phosphorylated; “D/E,” an acidic amino acid residue at positions −2 and +3; “Φ,” a hydrophobic residue at +1. Phosphorylation site and the surrounding conserved residues are indicated with arrow and asterisks, respectively. (B) Results of 5 highly potent peptides. See Materials and Methods for the detailed assay procedure. Relative incorporation of radioactivity (expressed in CPM, or counts per minute) into these peptides is presented, with normalizing to Cdc25C-FKD data. Error bars demonstrate standard deviation of the averages for n = 4 and 2 for Cdc25C-FKD and others, respectively. “Whole” indicates the average of the results of the whole group. (C) Sequences of the peptides shown in panel (B) and alignment of the sequences with the consensus motif. Cdc25C-FKD was designed with the modification of Cdc25C phosphorylation site (Nakajima et al., 2003)22 and synthesized in-house. Other 4 peptides were from the peptide library.
Plk1 has an N-terminal kinase domain and a C-terminal regulatory domain composed of 2 polo-boxes. The C-terminal domain binds to the N-terminal domain and inhibits its kinase activity, and a truncated protein with C-terminal deletion has higher activity in several folds than a full-length protein.31 To screen a potent peptide substrate, we prepared Plk1ΔC, a C-terminal deletion mutant with accelerated kinase activity. With this mutant and [γ-33P]ATP, we tested the peptide library in a radiometric FlashPlate assay in a 384-well plate format, and identified several peptides that showed efficient incorporation of [33P]Pi (Fig. 2B). The top 5 peptides that showed the highest efficiencies consisted of an in-house designed peptide, Cdc25C-FKD, and 4 peptides from the commercial peptide libraries (Fig. 2C). Sequence alignment of these peptides with the deduced phosphorylation sites indicate that all share the sequence similarity to the consensus motif, which was identified by Nakajima et al. (Fig. 2C), and there was a correlation between the level of incorporation of [33P]Pi and the similarity of the peptide sequence to the consensus motif. Among them, the sequences of the following 3 peptides that scored the highest incorporation show the complete match: Cdc25C-FKD, biotin-DELMEFSFKDQEAKV; N-MYC (1), biotin-SGEDTLSDSDDED; N-MYC (2), biotin-TSGEDTLSDSDDE (double and single underlines indicate the deduced phosphorylation site and other conserved residues, respectively). These results prove the importance of this motif for Plk1 phosphorylation and the reliability of our screen results. As Cdc25C has been extensively characterized as a biological substrate for Plk1 in M phase,20 we selected Cdc25C-FKD for further assay development.
Anti-Phospho-Specific Antibody Screen to Detect Phosphorylated Substrate
To find a specific antibody for the phosphorylated FKD peptide, we screened a collection of anti-phospho-specific antibodies with DELFIA® technology23 (see Materials and Methods for details). Among the 87 antibodies tested, anti-phospho-(S/T)F antibody (p(S/T)F Ab) showed the highest specificity to b-pFKD over b-FKD (Fig. 3B, lower panel).
Fig. 3.


Antibody screening for phosphor-FKD-specific antibodies. (A) Sequence of the peptide substrates b-FKD and b-ASFA. Double underlines indicate the phosphorylation site. Single underlines show the difference of amino acid residues at positions −1 and +2. b-ASFA lacks the C-terminal valine. (B) Affinity and specificity of various phospho-(S/T) antibodies to phosphorylated b-FKD (b-pFKD) were evaluated by quantifying the amount of bounded antibody with DELFIA® technology. See Materials and Methods for details. White and black dots demonstrate relative intensity unit (RFU) at 615 nm of wells with phosphorylated (◯ SIG_pFKD) and unphosphorylated (● SIG_FKD) b-FKD, respectively, and gray bar represents antibody’s specificity to pFKD by showing SIG_pFKD/SIG_FKD ratio. Those with highest specificities were marked with Greek letters: α, anti-phospho-(S/T)F; β, anti-phospho-PKCΔ/θ(Ser643/676); γ, anti-phospho-mTOR(Ser2481); Δ, anti-phospho-(S/T)PDK1 Docking Motif. In addition to α, there were 3 wells for anti-phospho-(S/T)F antibody (denoted with asterisk) and each of them also showed significant specificity to pFKD. Bottom presents the results of the top 4 antibodies marked with Greek letters in the graph. RFU with pFKD (SIG_pFKD) and FKD (SIG_FKD) and their ratio (specificity) are presented. Bottom row shows the median value of total 87 antibodies tested. (C) Results with phosphorylated b-ASFA (b-pASFA). The identical screening to panel (B) was performed except that b-ASFA replaced b-FKD. White and black dots demonstrate relative fluorescence unit (RFU) at 615 nm of wells with phosphorylated (◯ SIG_pASFA) and unphosphorylated (● SIG_ASFA) b-ASFA, respectively, and gray bar represents antibody’s specificity to pASFA by showing SIG_pASFA/SIG_ASFA ratio. Those with highest specificities were marked with Greek letters: α, anti-phospho-PKCΔ/θ(Ser643/676); β, anti-phospho-(Ser) R-X-Y/F-X-pS Motif; γ, anti-phospho-(S/T)F; Δ, anti-phospho-mTOR(Ser2481). Three asterisks show the same 3 wells for anti-phospho-(S/T)F antibody shown in panel (B). Bottom presents the results of the top 4 antibodies marked with Greek letters in the graph. RFU with pASFA (SIG_pASFA) and ASFA (SIG_ASFA) and their ratio (specificity) are presented. Bottom row shows the median value of total 87 antibodies tested.
After the antibody screening with b-pFKD was conducted, we identified another potent peptide substrate for Plk1.32 This peptide, namely b-ASFA, is derived from b-FKD. b-ASFA has alanine at −1 and +2 positions instead of phenylalanine and lysine and lacks valine at the C-termini (biotin-DELMEA ADQEAK (double and single underlines indicate the deduced phosphorylation site and other conserved residues, respectively) (Fig. 3A). b-ASFA showed higher potency than b-FKD by several folds as Plk1 substrate in radiometric assay.32 Therefore, we tested the identical library of 87 antibodies with b-pASFA and found that 3 antibodies, including p(S/T)F Ab, recognized both b-pFKD and b-pASFA peptides, whereas a few antibodies recognized only one of them (Fig. 3C). As the combination of b-FKD and p(S/T)F Ab showed the highest specificity in these 2 screenings, we decided to use them for development of Plk1 TR-FRET assay.
ADQEAK (double and single underlines indicate the deduced phosphorylation site and other conserved residues, respectively) (Fig. 3A). b-ASFA showed higher potency than b-FKD by several folds as Plk1 substrate in radiometric assay.32 Therefore, we tested the identical library of 87 antibodies with b-pASFA and found that 3 antibodies, including p(S/T)F Ab, recognized both b-pFKD and b-pASFA peptides, whereas a few antibodies recognized only one of them (Fig. 3C). As the combination of b-FKD and p(S/T)F Ab showed the highest specificity in these 2 screenings, we decided to use them for development of Plk1 TR-FRET assay.
Assay Development of Plk1 TR-FRET in a 384-Well Plate Format
Using b-FKD peptide and p(S/T)F antibody, we developed a Plk1 TR-FRET assay in a 384-well plate format. First, we evaluated the potency of p(S/T)F to detect b-pFKD in this plate density with chemically synthesized b-pFKD peptide (Fig. 4). To check the linearity of detection, we prepared a 400 nM mixture of b-FKD and b-pFKD with various ratios in the reaction buffer, and mixed it with an equal volume of the detection reagent which contained p(S/T)F Ab labeled with Eu-chelate (Eu-p(S/T)F Ab) and streptavidin–allophycocyanin (SA-APC) as donor and acceptor for FRET, respectively (final concentration of peptide mixture was 200 nM). After a 60-min incubation at room temperature, we measured RFU at 665 (RFU665) and 615 nm (RFU615) from APC and europium, respectively, with the excitation at 337 nm, and calculated the TR-FRET signal. The signal rose as the concentration of b-pFKD in the peptide mixture increased (Fig. 4A). Although the titration curve lost linearity when the concentration of b-pFKD was higher than 40 nM (Fig. 4A), S/B ratio27 was already higher than 4.7 with 10 nM b-pFKD and Z′ value27 was constantly higher than 0.8 (Fig. 4B). Therefore, we judged that this detection system was robust in this conversion range, and moved on to the validation of the Plk1 assay.
Fig. 4.

Development of Plk1 time-resolved fluorescence energy transfer (TR-FRET) assay in a 384-well format. (A) shows the signal dynamic range of the assay. The 400 nM of total mixture of b-FKD and b-pFKD with the indicated b-pFKD concentration was dispensed into 384-well plate in 10 µL of buffer A. After receiving 10 µL of TR-FRET reagent including Eu-p(S/T)F and SA-APC, the plate was further incubated for 45 min. Subsequently, relative fluorescence unit in 665 (RFU665) and 615 nm (RFU615) from APC and europium, respectively, with the excitation at 337 nm, were measured with Victor2™, and the TR-FRET signal was calculated (A). Error bars demonstrate standard deviation of the averages for n = 4. (B) S/B ratio (◯) and Z′ factor (●) at various b-pFKD concentrations. (C) Plk1 TR-FRET assay time-course experiment. See Materials and Methods for details. The 400 nM of b-FKD was incubated with 50 (◯) or 100 nM (●) Plk1ΔC in 20 µL for the indicated time. Error bars demonstrate standard deviation of the averages for n = 4.
Figure 4C shows a time-course experiment with 50 and 100 nM Plk1ΔC. The increase of the TR-FRET signal was observed in a reaction time-dependent manner, although it started to lose linearity after 15 min with both concentrations. As the TR-FRET signal with 50 nM Plk1ΔC at 15 min was already higher than that with 10 nM b-pFKD (Fig. 4A), we concluded that our TR-FRET assay using b-FKD and p(S/T)F Ab could detect Plk1 enzymatic activity in a 384-well format.
Miniaturization of Plk1 TR-FRET Assay in a 1,536-Well Plate Format
To run an uHTS of a compound library for Plk1 inhibitors, we miniaturized the assay to a 1,536-well format. First, we checked a dynamic range of the TR-FRET signal with a 500-nM mixture of b-FKD and b-pFKD (Fig. 5A). The signal in a 1,536-well plate started to plateau with higher than 50 nM b-pFKD (●), which is consistent with the result of the original 384-well assay (Fig. 4A). We observed the same trend with the modified 384-well assay format (◯) and the signal range was wider in a 1,536-well plate than in a 384-well plate with the same volumetric ratio (Fig. 5A).
Fig. 5.

Miniaturization of Plk1 time-resolved fluorescence energy transfer (TR-FRET) assay in 1,536-well plate format. See Materials and Methods for details. The same batch of reagents was delivered to a 384-well plate with a Multidrop dispenser and a 1,536-well plate with a Flying Reagent Dispenser I. Y-axis shows the ratio of relative fluorescence unit (RFU) at 665 and 615 nm under the excitation of 337 nm measured with the Victor V. (A) Signal dynamic range. A 384- (◯) and 1,536 (●)-well plate received 250 nM mixture of b-FKD and b-pFKD with the indicated b-pFKD concentration in 10 µM ATP/buffer A, and EDTA/TR-FRET containing SA-APC and Eu-p(S/T)F. The volume of the peptide mixture and TR-FRET was 40 and 20 µL for 384-well plate and 4 and 2 µL for 1,536-well plate. The plate was incubated for 45 min before measuring RFUs with the Victor V. Error bars indicate standard deviation of the medians for n = 48 for a 384-well plate, and n = 192 for a 1,536-well plate. (B) Enzyme titration. The assay protocol is shown in Figure 1B and 1C. Indicated concentration of Plk1ΔC was incubated for 60 min at 25°ºC with 250 nM b-FKD peptide in the absence (□, ◯) or presence (■, ●) of 10 µM ATP for a 384- (■, □) and 1,536 (●, ◯)-well plate, respectively. Each plate received EDTA/TR-FRET, and was loaded onto the Victor2™ after 45 min. Error bars demonstrate standard deviation of the medians for n = 176 (●) and 16 (◯) for a 1,536-well plate, and n = 44 (■) and 4 (□) for a 384-well plate. (C) Time course. 9 nM Plk1ΔC was incubated for the indicated time with b-FKD peptide in the absence (□, ◯) or presence (■, ●) of ATP for a 384- (■, □) and 1,536 (●, ◯)-well plate, respectively. Error bars demonstrate standard deviation of the medians for n = 176 (●) and 16 (◯) for a 1,536-well plate, and n = 44 (■) and 4 (□) for a 384-well plate. Data at 0 min was from background wells that did not have Plk1ΔC.
Titration of Plk1ΔC concentration showed that the TR-FRET signal increased up to 28 nM and started to plateau with 7 nM (Fig. 5B). Reaction with 9 nM enzyme was linear up to 60 min (Fig. 5C).
To screen out the compounds that target outside of the kinase domain as well, we also validated our TR-FRET assay with a full-length protein. Phosphorylation of threonine 210 in the T-loop in the kinase domain is the key regulation for Plk1 activity, and the mutation of this residue to aspartate mimics this phosphothreonine and elevates kinase activity.31,33 We prepared Plk1T210D, a full-length protein with this mutation, and compared the activity to ΔC in our system. T210D was less active than ΔC in a 60-min incubation (Fig. 6A), and the phosphorylation depends on the concentration up to 88 nM in both a 60- and a 120-min incubation (Fig. 6B). We tested the potency of staurosporine and K252a with 4 nM ΔC in a 60-min incubation and with 66 nM T210D in a 120-min incubation (Fig. 6C). Even though the concentration and incubation time was different, dose–response curve overlapped between these 2 proteins. The IC50 value of staurosporine was about 60 nM on ΔC and 100 nM on T210D, and the same value for K252a was about 100 nM on ΔC and 60 nM on T210D. To complete the off-line validation and determine the screen condition, we carried out a whole plate experiment (Fig. 7). Panel A shows the assay plate map. Columns 1–4 and 45–48 were used for quality control: the upper half of rows in columns 1 and 2 and the entire rows in columns 3 and 4, “0% inhibition” with DMSO; the lower half of rows in columns 1 and 2, “50% inhibition” with the reference inhibitor (not published); 45 and 46, “100% inhibition” with the reference inhibitor; 47 and 48, “Background” with no ATP. The median of the signal from “100% inhibition” wells was used as an inhibition control for normalization. The total 1,280 wells in columns 5–44 would receive one test compound per well in a virtual screening. In the validation, we added DMSO to the final concentration of 0.5% (v/v) in these columns. To normalize percentage inhibition (% inhibition) for each well, we used the median value of the RFU ratio from the compound field in columns 5–44 and the inhibition population in columns 45 and 46 for 0 and 100, respectively (see Materials and Methods for details). Panel B shows scatter plot for the %INH of the 60-min reaction with 4 nM ΔC (left) and the 120-min reaction with 66 nM T210D (right), respectively. With the exception of 3 false-positives, columns 5–44 did not have any significant inhibition in both cases. These false-positives may have been brought in by a dispensing error of the liquid handler. Window, % CV, and the Z factor of these plates were 7.4, 5.0, and 0.70 for ΔC and 7.2, 5.5, and 0.70 for T210D, respectively. Because these 3 values proved the high assay quality with both proteins, we concluded that assay development with b-FKD and Eu-p(S/T)F Ab was successful and judged that our assay was robust enough for uHTS using both ΔC and T210D. On the basis of these values, we locked the screening condition with 4 nM ΔC and 66 nM T210D, and moved forward to uHTS.
Fig. 6.

Plk1 time-resolved fluorescence energy transfer (TR-FRET) assay with T210D mutant. (A) Enzyme titration in a 384-well plate format. Indicated concentration of Plk1ΔC (◯) and Plk1T210D (●) was incubated with ATP and b-FKD for 60 min at 25°C. The assay plate received EDTA/TR-FRET, and was loaded onto the Victor2™ after 45 min. Error bars demonstrate standard error of the medians for n = 15. (B) Enzyme titration in the assay in 1,536-well plate. Indicated concentration of Plk1T210D was incubated in 60 (◯) and 120 (●) min. Error bars demonstrate standard error of the medians for n = 44. (C) Dose–response to the known inhibitors. Indicated concentration of staurosporine (left) and K252a (right) was added to the 60-min reaction with 4 nM Plk1ΔC (◯) and 66 nM Plk1T210D (●). Error bars demonstrate standard error of the medians for n = 8.
Fig. 7.

Off-line validation of Plk1 time-resolved fluorescence energy transfer (TR-FRET) assay in a 1,536-well plate format. (A) Assay plate map. Columns 1–4 and 45–48 were used for quality control: Upper half of rows in columns 1 and 2 and the entire rows in columns 3 and 4, “0% inhibition” with dimethyl sulfoxide (DMSO); lower half of rows in columns 1 and 2, “50% inhibition” with the reference inhibitor (not published); 45 and 46, “100% inhibition” with the reference inhibitor; 47 and 48, “background” with no ATP. (B) A typical scatter plot of the percent inhibition of a DMSO plate with (left) 4 nM Plk1ΔC and (right) 66 nM Plk1T210D. In these plates, 0.5% (v/v) DMSO replaced compounds in columns 5–44. Inserted numbers in the left panel correspond to those shown in the column 1 of the table in panel (A), except for 3 that were wells with DMSO, not compounds.
Ultra-High-Throughput Screening
We screened a library of about 1.2 million compounds with ΔC and T210D independently. Prior to each screening, we performed an overnight pilot screening with a portion of our compound library. The screening concentration of compounds was either 15 or 30 µM, depending on the concentration of the stock in the library. For quality control, we ran DMSO plates in-between the assay plates that received DMSO instead of compounds. We also included several spike plates in which the reference inhibitor was spiked in a couple of wells in columns 5–44 of DMSO plates. Figure 8 presents a scatter plot of one of the spike plates (left) and subsequent DMSO plate (right) for ΔC (A) and T210D (B) assay. In both cases, all the inhibition with spiked wells were detected (encircled with a dotted line), but there were no false-positive wells in the DMSO plate. This result demonstrated that there was no carryover of compound between assay plates. In this screening, assay window, % CV and the Z factor with the median of the data from columns 5 to 44 were consistently ∼5.5–6, ∼7.5, and ∼0.70 for ΔC and ∼5–5.5, ∼5.0, and ∼0.65 for T210D, respectively. These values were comparable to those we obtained in the off-line experiment (Fig. 7), and did not change with the working reagents that had been stored on ice overnight. We concluded that our assay was robust in the automated system, and moved on to the primary screenings of the entire library.
Fig. 8.

Robotics validation of Plk1 assay. A scatter plot of the percent inhibition from an overnight test run with Plk1ΔC (A) and Plk1T210D (B) is presented. For each panel, left plot is the result of a spike plate in which the control inhibitor was spiked in several wells in columns 5–44 (encircled), and right plot is the result of a subsequently run dimethyl sulfoxide (DMSO) plate that did not have inhibitors in the same field. See Figure 7 for the plate map for columns 1, 2, 47, and 48.
We ran the primary screenings for 24 h/day, topping off the fresh reagent in every 12 h. Figure 9 shows the plate chart of assay window, % CV, and the Z factor of the entire screen with ΔC (A) and T210D (B). The assay window was about 6 and 5 for ΔC and T210D, respectively (left). In both screenings, % CV was about 10 (middle) and the Z factor was around 0.55 (right). We re-ran the assay plates with the Z factor of lower than 0.5.27 Each screening completed in 2 weeks, and the screen throughput was approximately 9 × 104 compounds per day.
Fig. 9.

Primary ultra-high-throughput screening (uHTS) for Plk1 inhibitors. Charts of 3 values of all of the plates in the complete screen with Plk1ΔC (A) and Plk1T210D (B) are presented. For each panel, left, center, and right demonstrates Window, % CV, and Z factor, respectively. See text for details.
The mean plus 3 times the standard deviation (mean + 3 std) of the global % inhibition from all of the plates was approximately 0.0 + 3 × 10.0 = 30.0 for ΔC and 0.0 + 3 × 11.8 = 35.4 for T210D. With 35 as a global cutoff for hit selection for both proteins, 7,610 and 13,096 compounds resulted as active for ΔC and T210D, respectively. This criterion lead a hit rate of about 0.7% and 1.3%, respectively.
Confirmatory uHTS of the Primary Hits for Lead Identification
We selected a total of 12,585 compounds out of the hit list on the basis of availability, and re-tested them with both proteins to eliminate false-positive inhibitors that possible malfunction of equipment or measurement variation brought in. We ran these confirmatory screenings in 3 cycles, and used the median of the 3 replicate results as a representation of the activity. Unlike the whole compound population in the primary screening that follows the normal distribution, this hit population was biased and a large number of compounds were active. Therefore, we normalized the data with the median of “0% inhibition” wells instead of “compounds,” and employed the same hit criterion for the primary screening. A Venn diagram in Figure 10 summarizes the results. A total of 4,679 compounds were confirmed, with the confirmation rate of about 36%, and the number of confirmed hits was 271 for ΔC, 1,543 for T210D, and 2,863 for both proteins. The inserted graph presents the correlation of plots of confirmatory uHTSs of 2,863 compounds that were confirmed in both the ΔC and T210D screenings. The coefficient of multiple determination (R2 value) between these screenings was 0.55.
Fig. 10.

Summary for 2 ultra-high-throughput screening (uHTS) campaigns for Plk1 inhibitors. Each step to reduce the primary hits to select lead compounds is presented. The inserted graph shows the correlation of plots of confirmatory uHTSs about 2,863 compounds that were confirmed in both. Venn diagram in the bottom shows the overlap of the hits between these 2 campaigns.
Discussion
The rate-limiting step for development of Ser/Thr kinase TR-FRET assay is to search an optimal pair of a peptide substrate for the target and an antibody for the phosphorylated substrate. In this study, we identified FKD peptide and p(S/T)F antibody in a week by screening >800 peptides and 87 antibodies with a multiplexed assay (Figs. 2 and 3) and developed a highly specific system for uHTS for Plk1 inhibitors. We propose that a series of multiplexed random screening for substrate peptides and specific antibodies is a quite powerful strategy to develop a TR-FRET assay.
The sequence of FKD is derived from phosphorylation site of Cdc25C, a major physiological substrate proteins of Plk120 (Fig. 2). Recent study suggested that a hydrophobic residue at +1 and acidic residues at −2 and +3 positions in the consensus motif are essential22 (Fig. 2A). The dependency on these acidic residues is a very unique feature to Plk1, as major Ser/Thr kinases, including PKA and PKC family members, require basic residues at the equivalent positions.34 Assay specificity is important to develop a highly specific inhibitor that does not have any off-target effect on other kinases. As FKD peptide conserves these residues (Fig. 2B), we believe that our assay has much higher specificity and corroborates its biological relevance than other Plk assay systems with generic substrates.
Besides FKD, 4 peptides from the commercial libraries presented high potencies (Fig. 2B). Since these libraries are not biased and most peptides were almost completely inactive (Fig. 2B), it is striking that all of these 4 peptides conserve the essential residues of consensus motif of Plk1 phosphorylation (Fig. 2C). Therefore, we are confident about the high efficacy of our peptide screening, and conclude that a random screen of peptide libraries, as well as a rational-based peptide design, is a powerful strategy to identify potent and physiologically relevant substrates. Among the origins of these 4 peptides, N-MYC and HPV16 E7 proteins have relevance to human cancer35,36 and doublecortin functions in microtubule assembly.37 As Plk1 is relevant to cancer and mitosis, these proteins may be novel physiological substrates of Plk1.
We also screened the same set of antibodies using b-ASFA peptide, and found that p(S/T)F antibody, anti-phospho-PKCΔ/θ antibody, and anti-phospho-mTOR(Ser2481) antibody showed high specificity to phosphorylated form of both peptide (Fig. 3B and 3C). Even though the amino acid sequence of ASFA is also derived from Cdc25C and is quite similar to FKD, we observed interesting difference of the results. For instance, p(S/T)F antibody scored best specificity in the screening with b-pFKD, whereas anti-phospho-PKCΔ/Θ antibody scored best in the screening with b-pASFA. Furthermore, anti-phospho-(Ser/Thr) PDK1 Docking Motif antibody scored only to b-pFKD, whereas anti-phospho-(Ser) Arg-X-Tyr/Phe-X-pSer motif antibody showed the second highest specificity to b-pASFA but were negative to b-pFKD (Fig. 3B and 3C). The selectivity of anti-phospho-(Ser/Thr) PDK1 Docking Motif antibody and anti-phospho-(Ser) Arg-X-Tyr/Phe-X-pSer motif antibody to only one of the peptides is particularly interesting because it may be difficult to find a specific antibody that distinguishes these peptides by rational-based antibody selection. These results support our conclusion that a random screening of antibody is a powerful approach to identify a substrate–antibody combination of higher specificity.
Several studies described HTS for PLK1 inhibitors. Santamaria et al. reported scintillation proximity assay using casein and [γ-33P]ATP.19 Sharlow et al. developed a HTS assay using immobilized metal affinity for phosphochemicals (IMAP) technology and screened 97,101 compounds in a 384-well plate format.38 We succeeded in miniaturizing our TR-FRET assay to a 1,536-well format for uHTS for 1.2 million compounds, and performed 2 fully automated uHTSs with the kinase domain and an activated full-length protein. Each screening was completed in 10 days, and the statistics proved the robustness of our assay (Fig. 9). We confirmed 4,679 compounds, and observed that 271 and 2,863 compounds were active only for the kinase domain and the full-length protein, respectively (Fig. 10). We concluded that to use a physiologically relevant substrate was the major key for the development of robust assay, subsequent miniaturization, and identification of many hit compounds.
In addition to an assay specificity to the target kinase, expectable advantage to use a substrate with physiological relevance is to get more chance to screen out non-ATP-competitive inhibitors. Plk1 has 603 residues with N-terminal kinase domain (aa residues 53–303) and C-terminal polo-box domain (PBD) (aa 345–603) containing 2 polo-boxes (PB1 and 2) and a phosphopeptide-binding motif. Current hypothesis is that PBD may guide Plk1 to specific subcellular locations and interacts with the target with phosphopeptide-binding motif. Without this interaction, PBD is speculated to fold back to the kinase domain to inhibit substrate binding.16,39 ATP-competitive inhibitors that hit kinase domain have more chance to cause off-target effect due to the high degree of structural conservation of the ATP-binding site among all kinase superfamily members. The unique PBD is expected to be a target for non-ATP-competitive inhibitors that block the protein–protein interaction. It is tempting to speculate that some of 1,500 lead compounds that hit only T210D might target PBD including the phosphopeptide-binding motif.
The caveat is that there must be false-positive artifacts in our data that TR-FRET specifically produces. Such compounds include those that cause assay interference, like inner filter effect and quenching.40 We found that a significant portion of our hits was negative in other screens that had been conducted with the same library using the TR-FRET technology, and prioritized these unique compounds for further investigation. Several studies argued the data concordance between different detection technologies.41–43 Therefore, we are currently testing these leads with several other technologies (Scintillation Proximity Assay, radiometric filtration assay, etc.) for further prioritization for optimization, and will describe those results in future reports.
Conclusion
To develop a robust TR-FRET assay for uHTS for small molecule inhibitors of a major cancer target Plk1, we performed multiplexed screens of a peptide library and phospho-specific antibodies, and obtained a highly potent substrate Cdc25C-FKD and anti-phospho-(S/T)F antibody. These peptide and antibody brought us a robust Plk1 TR-FRET assay in a 1,536-well plate format and a successful uHTS campaign. We conclude that to use highly specific substrate instead of generic peptide was the major key for our success to identify many hit compounds for Plk1 inhibitors, and propose that random screen of peptide and antibody libraries is a powerful strategy to develop a TR-FRET assay with a physiological relevance and good robustness.
Abbreviations
- % CV
percentage coefficient of variation
- APC
allophycocyanin
- b-FKD
biotinylated FKD peptide
- DELFIA®
dissociation-enhanced lanthanide fluorescence immunoassay
- b-ASFA
biotinylated ASFA peptide
- DMSO
dimethyl sulfoxide
- Eu-p(S/T)F Ab
anti-phosphor-(S/T)F antibody labeled with europium-chelate
- FI
fluorescence intensity
- HTS
high-throughput screening
- HPV16
human papillomavirus type 16
- IC50
a half-maximal inhibitory concentration
- mean + 3 std
the assay mean plus 3 times the standard deviation of percentage inhibition
- Plk1ΔC
polo-like kinase 1 deletion mutant with the N-terminal kinase domain
- Plk1T210D
polo-like kinase 1 point mutant with threonine 210 mutated to aspartate
- RFU
relative fluorescence unit
- std
standard deviation
- SA
streptavidin
- TRF
time-resolved fluorometry
- TR-FRET
time-resolved fluorescence energy transfer
- uHTS
ultra-high-throughput screening.
Acknowledgments
We thank Edward Hudakk and Kevin Nguen (Merck Research Laboratories) for picking the primary hits from the compound library for the confirmatory screening, and Ikuko Takahashi (Banyu Pharmaceutical) for invaluable comments on the manuscript. Banyu Pharmaceutical Co. and Rosetta Inpharmatics are wholly owned subsidiaries of Merck & Co.
Author Disclosure Statement
K.T., K.F., and H.K. are employees of TAIHO Pharmaceutical Co., Ltd. M.K. is an employee of Astellas Research Technologies Co., Ltd. M.I. is an employee of Banyu Pharmaceutical Co., Ltd. E.S., J.C., and K.R. are employees of Merck & Co., Inc. P.C. and P.H. are employees of Scripps Florida. T.T. is an employee of Memorial Sloan-Kettering Cancer Center.
References
- 1.Hunter T. Signaling—2000 and beyond. Cell. 2000;100:113–127. doi: 10.1016/s0092-8674(00)81688-8. [DOI] [PubMed] [Google Scholar]
- 2.Cohen P. Protein kinases—the major drug targets of the twenty-first century? Nat Rev Drug Discov. 2002;1:309–315. doi: 10.1038/nrd773. [DOI] [PubMed] [Google Scholar]
- 3.Manning G. Whyte DB. Martinez R. Hunter T. Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298:1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- 4.Scapin G. Protein kinase inhibition: Different approaches to selective inhibitor design. Curr Drug Targets. 2006;7:1443–1454. doi: 10.2174/1389450110607011443. [DOI] [PubMed] [Google Scholar]
- 5.Johnson EF. Stewart KD. Woods KW. Giranda VL. Luo Y. Pharmacological and functional comparison of the polo-like kinase family: Insight into inhibitor and substrate specificity. Biochemistry. 2007;46:9551–9563. doi: 10.1021/bi7008745. [DOI] [PubMed] [Google Scholar]
- 6.von Ahsen O. Bomer U. High-throughput screening for kinase inhibitors. Chembiochem. 2005;6:481–490. doi: 10.1002/cbic.200400211. [DOI] [PubMed] [Google Scholar]
- 7.Wesche H. Xiao SH. Young SW. High throughput screening for protein kinase inhibitors. Comb Chem High Throughput Screen. 2005;8:181–195. doi: 10.2174/1386207053258514. [DOI] [PubMed] [Google Scholar]
- 8.Kolb AJ. Kaplita PV. Hayes DJ. Park Y-W. Pernell C. Major JS, et al. Tyrosine kinase assays adapted to homogeneous time-resolved fluorescence. Drug Disc Today. 1998;3:333–342. [Google Scholar]
- 9.Klumpp M. Boettcher A. Becker D. Meder G. Blank J. Leder L, et al. Readout technologies for highly miniaturized kinase assays applicable to high-throughput screening in a 1536-well format. J Biomol Screen. 2006;11:617. doi: 10.1177/1087057106288444. [DOI] [PubMed] [Google Scholar]
- 10.Kemp BE. Pearson RB. Design and use of peptide substrates for protein kinases. Methods Enzymol. 1991;200:121–134. doi: 10.1016/0076-6879(91)00134-i. [DOI] [PubMed] [Google Scholar]
- 11.Panse S. Dong L. Burian A. Carus R. Schutkowski M. Reimer U, et al. Profiling of generic anti-phosphopeptide antibodies and kinases with peptide microarrays using radioactive and fluorescence-based assays. Mol Divers. 2004;8:291–299. doi: 10.1023/b:modi.0000036240.39384.eb. [DOI] [PubMed] [Google Scholar]
- 12.Barr FA. Silljé HHW. Nigg EA. Polo-like kinases and the orchestration of cell division. Nat Rev Mol Cell Biol. 2004;5:429–440. doi: 10.1038/nrm1401. [DOI] [PubMed] [Google Scholar]
- 13.Xie S. Xie B. Lee MY. Dai W. Regulation of cell cycle checkpoints by polo-like kinases. Oncogene. 2005;24:277–286. doi: 10.1038/sj.onc.1208218. [DOI] [PubMed] [Google Scholar]
- 14.van Vugt MA. Medema RH. Getting in and out of mitosis with Polo-like kinase-1. Oncogene. 2005;24:2844–2859. doi: 10.1038/sj.onc.1208617. [DOI] [PubMed] [Google Scholar]
- 15.Takai N. Hamanaka R. Yoshimatsu J. Miyakawa I. Polo-like kinases (Plks) and cancer. Oncogene. 2005;24:287–291. doi: 10.1038/sj.onc.1208272. [DOI] [PubMed] [Google Scholar]
- 16.Strebhardt K. Ullrich A. Targeting polo-like kinase 1 for cancer therapy. Nat Rev Cancer. 2006;6:321–328. doi: 10.1038/nrc1841. [DOI] [PubMed] [Google Scholar]
- 17.McInnes C. Mezna M. Fischer PM. Progress in the discovery of polo-like kinase inhibitors. Curr Topics Med Chem. 2005;5:181–197. doi: 10.2174/1568026053507660. [DOI] [PubMed] [Google Scholar]
- 18.Zhang W. Fletcher L. Muschel RJ. The role of polo-like kinase1 in the inhibition of centrosome separation after ionizing radiation. J Biol Chem. 2005;280:42994–42999. doi: 10.1074/jbc.M505450200. [DOI] [PubMed] [Google Scholar]
- 19.Santamaria A. Neef R. Eberspächer U. Eis K. Husemann M. Mumberg D, et al. Use of the novel Plk1 inhibitor ZK-thiazolidinone to elucidate the functions of Plk1 in early and late stages of mitosis. Mol Biol Cell. 2007;18:4024–4036. doi: 10.1091/mbc.E07-05-0517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Karlsson-Rosenthal C. Millar JBA. Cdc25: mechanisms of checkpoint inhibition and recovery. Trends Cell Biol. 2006;16:285–292. doi: 10.1016/j.tcb.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 21.Toyoshima-Morimoto F. Taniguchi E. Nishida E. Plk1 promotes nuclear translocation of human Cdc25C during prophase. EMBO Rep. 2002;3:341–348. doi: 10.1093/embo-reports/kvf069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nakajima H. Toyoshima-Morimoto F. Taniguchi E. Nishida E. Identification of a consensus motif for Plk (Polo-like kinase) phosphorylation reveals Myt1 as a Plk1 substrate. J Biol Chem. 2003;278:25277–25280. doi: 10.1074/jbc.C300126200. [DOI] [PubMed] [Google Scholar]
- 23.Sadler TM. Achilleos M. Ragunathan S. Pitkin A. LaRocque J. Morin J, et al. Development and comparison of two nonradioactive kinase assays for I kappa B kinase. Anal Biochem. 2004;326:106–113. doi: 10.1016/j.ab.2003.11.021. [DOI] [PubMed] [Google Scholar]
- 24.Chung CC. Ohwaki K. Schneeweis JE. Stec E. Varnerin JP. Goudreau PN, et al. A fluorescence-based thiol quantification assay for ultra-high-throughput screening for inhibitors of coenzyme A production. Assay Drug Dev Technol. 2008;6:361–374. doi: 10.1089/adt.2007.105. [DOI] [PubMed] [Google Scholar]
- 25.Rodems SM. Hamman BD. Lin C. Zhao J. Shah S. Heidary D, et al. A FRET-based assay platform for ultra-high density drug screening of protein kinases and phosphatases. Assay Drug Dev Technol. 2002;1:9–19. doi: 10.1089/154065802761001266. [DOI] [PubMed] [Google Scholar]
- 26.Hodder P. Mull R. Cassaday J. Berry K. Strulovici B. Miniaturization of intracellular calcium functional assays to 1536-well plate format using a fluorometric imaging plate reader. J Biomol Screen. 2004;9:417–426. doi: 10.1177/1087057104264038. [DOI] [PubMed] [Google Scholar]
- 27.Zhang JH. Chung TDY. Oldenburg KR. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J Biomol Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 28.Hodder P. Cassaday J. Peltier R. Berry K. Inglese J. Feuston B, et al. Identification of metabotropic glutamate receptor antagonists using an automated high-throughput screening system. Anal Biochem. 2003;313:246–254. doi: 10.1016/s0003-2697(02)00608-5. [DOI] [PubMed] [Google Scholar]
- 29.Toyoshima-Morimoto E. Taniguchi E. Shinya N. Iwamatsu A. Nishida E. Polo-like kinase 1 phosphorylates cyclin B1 and targets it to the nucleus during prophase. Nature. 2001;410:215–220. doi: 10.1038/35065617. [DOI] [PubMed] [Google Scholar]
- 30.Yuan J. Eckerdt F. Bereiter-Hahn J. Kurunci-Csacsko E. Kaufmann M. Strebhardt K. Cooperative phosphorylation including the activity of polo-like kinase 1 regulates the subcellular localization of cyclin B1. Oncogene. 2002;21:8282–8292. doi: 10.1038/sj.onc.1206011. [DOI] [PubMed] [Google Scholar]
- 31.Jang YJ. Lin CY. Ma S. Erikson RL. Functional studies on the role of the C-terminal domain of mammalian polo-like kinase. Proc Natl Acd Sci USA. 2002;99:1984–1989. doi: 10.1073/pnas.042689299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Arai T. Haze K. Iimura-Morita Y. Machida T. Iida M. Tanaka K, et al. Identification of beta-catenin as a novel substrate of polo-like kinase 1. Cell Cycle. 2008;7:3556–3563. doi: 10.4161/cc.7.22.7072. [DOI] [PubMed] [Google Scholar]
- 33.Jang YJ. Ma S. Terada Y. Erikson RL. Phosphorylation of threonine 210 and the role of serine 137 in the regulation of mammalian polo-like kinase 1. J Biol Chem. 2002;277:44115–44120. doi: 10.1074/jbc.M202172200. [DOI] [PubMed] [Google Scholar]
- 34.Kemp BE. Pearson RB. Protein kinase recognition sequence motif. Trends Biochem Sci. 1990;15:342–346. doi: 10.1016/0968-0004(90)90073-k. [DOI] [PubMed] [Google Scholar]
- 35.Nesbit CE. Tersak JM. Prochownik EV. MYC oncogenes and human neoplastic disease. Oncogene. 1999;18:3004–3016. doi: 10.1038/sj.onc.1202746. [DOI] [PubMed] [Google Scholar]
- 36.Fehrmann F. Laimins LA. Human papillomaviruses: targeting differentiating epithelial cells for malignant transformation. Oncogene. 2003;22:5201–5207. doi: 10.1038/sj.onc.1206554. [DOI] [PubMed] [Google Scholar]
- 37.Horesh D. Sapir T. Francis F. Wolf SG. Caspi M. Elbaum M, et al. Doublecortin, a stabilizer of microtubules. Hum Mol Genet. 1999;8:1599–1610. doi: 10.1093/hmg/8.9.1599. [DOI] [PubMed] [Google Scholar]
- 38.Sharlow ER. Leimgruber S. Shun TY. Lazo JS. Development and implementation of a miniaturized high-throughput time-resolved fluorescence energy transfer assay to identify small molecule inhibitors of polo-like kinase 1. Assay Drug Dev Technol. 2007;5:723–735. doi: 10.1089/adt.2007.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pawson T. Nash P. Assembly of cell regulatory systems through protein interaction domains. Science. 2003;300:445–452. doi: 10.1126/science.1083653. [DOI] [PubMed] [Google Scholar]
- 40.Comley J. Assay interference: a limiting factor in HTS? Drug Disc World. 2003:91–98. Summer. [Google Scholar]
- 41.Zhang JH. Wu X. Sills MA. Probing the primary screening by multiple replicate testing: A quantitative analysis of hit confirmation and false screening results of a biochemical assay. J Biomol Screen. 2005;10:695–704. doi: 10.1177/1087057105279149. [DOI] [PubMed] [Google Scholar]
- 42.Wu X. Sills MA. Zhang JH. Further comparison of primary hit identification by different assay technologies and effects of assay measurement viability. J Biomol Screen. 2005;10:581–589. doi: 10.1177/1087057105275628. [DOI] [PubMed] [Google Scholar]
- 43.von Ahsen O. Schmidt A. Klotz M. Parczyk K. Assay concordance between SPA and TR-FRET in high-throughput-screening. J Biomol Screen. 2006;11:606–616. doi: 10.1177/1087057106288183. [DOI] [PubMed] [Google Scholar]