

Abstract
Autism spectrum disorders (ASDs) are highly heritable developmental disorders caused by a heterogeneous collection of genetic lesions. Here we use a mouse model to study the effect on cortical connectivity of disrupting the ASD candidate gene PTEN (phosphatase and tensin homolog deleted on chromosome 10). Through Cre-mediated recombination, we conditionally knocked out PTEN expression in a subset of auditory cortical neurons. Analysis of long-range connectivity using channelrhodopsin-2 revealed that the strength of synaptic inputs from both the contralateral auditory cortex and from the thalamus onto PTEN-cko neurons was enhanced compared with nearby neurons with normal PTEN expression. Laser-scanning photostimulation showed that local inputs onto PTEN-cko neurons in the auditory cortex were similarly enhanced. The hyperconnectivity caused by PTEN-cko could be blocked by rapamycin, a specific inhibitor of the PTEN downstream molecule mammalian target of rapamycin complex 1. Together, our results suggest that local and long-range hyperconnectivity may constitute a physiological basis for the effects of mutations in PTEN and possibly other ASD candidate genes.
Introduction
Autism spectrum disorders (ASDs) are a group of widespread developmental disorders with diverse neuropsychiatric symptoms, occurring in 1 of 150 individuals (Fombonne, 2005). ASDs are highly heritable, and dozens of genes have been implicated in their etiology (Sebat et al., 2007; Glessner et al., 2009), but it is unclear how these heterogeneous genetic factors converge to cause the common signatures of ASDs (Veenstra-Vanderweele et al., 2004; Gupta and State, 2007; Schmitz and Rezaie, 2008). It has been hypothesized that disruption of long-range cortical connections could represent the “final pathway” by which diverse ASD genotypes lead to ASD phenotypes (Geschwind and Levitt, 2007). Understanding how neural circuits are altered in animal models of ASDs may therefore provide insight into the mechanisms of ASD.
Here we focus on the ASD candidate gene PTEN (phosphatase and tensin homolog deleted on chromosome 10). Originally identified as a glioma tumor suppressor (Li et al., 1997; Steck et al., 1997), PTEN negatively regulates cell growth, proliferation, polarity, migration, and survival (Leslie et al., 2008; Chalhoub and Baker, 2009) through its inhibition of phosphatidylinositol 3,4,5-trisphosphate-dependent pathways (Maehama and Dixon, 1998). Germline PTEN mutations have been identified in autistic individuals with extreme macrocephaly (Goffin et al., 2001; Butler et al., 2005). Animal studies further support PTEN as an ASD gene (Kwon et al., 2006). Neuronal deletion of PTEN leads to increased soma size, hypertrophic and ectopic dendrites, axonal tracts with increased synapses, and higher excitatory spontaneous activity (Kwon et al., 2006; Luikart et al., 2011). In vivo imaging reveals that deletion of PTEN in mature mice increases the length and tortuosity of apical dendrites of cortical layer 2/3 neurons (Chow et al., 2009). However, little is known about the effect of PTEN disruption on functional connectivity.
ASDs are often associated with difficulties in auditory processing and attention. ASD subjects may show acoustic hyposensitivity or hypersensitivity (Baranek et al., 1997; Dawson et al., 1998) and failure to integrate complex auditory information (Boddaert et al., 2004), and may have problems in spatial attention to sound (Teder-Sälejärvi et al., 2005). The auditory cortex forms functional connections with other sensory cortices and various brainstem areas (Budinger and Scheich, 2009), and plays a critical role in auditory attention and perception (Fritz et al., 2007). Auditory cortical connectivity thus represents a suitable system for understanding circuit-level deficits in animal models of ASDs.
We have assessed the effect of PTEN deletion on functional cortical connectivity. We found that loss of PTEN in cortical neurons enhanced the strength of inputs from both the contralateral auditory cortex and the thalamus, as well as from local inputs. The hyperconnectivity caused by loss of PTEN could be blocked by administration of rapamycin, a specific inhibitor of the PTEN downstream molecule mammalian target of rapamycin complex 1 (mTORC1). Together, our results suggest that local and long-range hyperconnectivity may constitute a physiological basis for the effects of mutations in PTEN and possibly in other ASD candidate genes.
Materials and Methods
Animals and virus.
Animal procedures were approved by the Cold Spring Harbor Laboratory Animal Care and Use Committee. PTENloxP/loxP mice (Trotman et al., 2003) were a gift from Dr. Lloyd Trotman's laboratory (Cold Spring Harbor Laboratory, Cold Spring Harbor, NY) and bred in our own lab. The channelrhodopsin-2 (ChR2)–venus construct was from Dr. Karel Svoboda's laboratory (Janelia Farm Research Campus, Ashburn, VA), and the GFP–IRES–Cre construct was from Dr. Josh Huang's laboratory (Cold Spring Harbor Laboratory, Cold Spring Harbor, NY). The corresponding viruses [adeno-associated virus (AAV) 2/9] were made at the University of North Carolina Gene Therapy Center. Mice were anesthetized by isoflurane (1%, v/v), and virus was injected into mouse auditory cortex or medial geniculate body (MGB) between P18 and P21.
Long-range connection stimulation and recording.
Experiments used PTENloxP/loxP mice under protocols approved by the Cold Spring Harbor Laboratory Animal Committee. We used young adult mice from P30 to P45. Animals were anesthetized and decapitated, and the brains were transferred to a chilled cutting solution composed of the following (in mm): 110 choline chloride, 25 NaHCO3, 25 d-glucose, 11.6 sodium ascorbate, 7 MgCl2, 3.1 sodium pyruvate, 2.5 KCl, 1.25 NaH2PO4, and 0.5 CaCl2. In this study, we made coronal slices. All slices were 350 μm thick and were transferred to artificial CSF (ACSF) containing the following (in mm): 127 NaCl, 25 NaHCO3, 25 d-glucose, 2.5 KCl, 4 MgCl2, 1 CaCl2, and 1.25 NaH2PO4, aerated with 95% O2 5% CO2. The slices were incubated at 34°C for 20–30 min and then kept at room temperature (22°C) during the experiments. Neurons in the slice were visualized using infrared gradient contrast optics and patched with electrodes (4–5 MΩ) containing the following intracellular solution (in mm): 128 potassium methylsulfate, 4 MgCl2, 10 HEPES, 1 EGTA, 4 Na2-ATP, 0.4 Na2-GTP, 10 sodium phosphocreatine, 0.5% Biocytin (Sigma), and 0.015 Alexa Fluor 594 (Invitrogen), pH 7.25, 300 mOsm. The presence of Alexa Fluor 594 in the internal solution rendered cells fluorescent. We confirmed that cells were excitatory by visualizing their dendritic arbor and spines. Whole-cell recordings were made using Axopatch 200B amplifiers (Axons Instruments/Molecular Devices). Excitatory currents were measured at a holding potential of −70 mV, and action potentials were recorded in current-clamp configuration. We delivered light pulses though a lightguide microscope illumination system (Lumen Dynamics) modified to accept a blue-light laser (473 nm, Lasermate Group) in place of the lamp. The laser beam is focused onto the microscope field through the 60× objective during recordings. mEPSCs were recorded in the presence of 1 μm tetrodotoxin and 100 μm picrotoxin, and events were detected and analyzed in Clampfit (Molecular Devices). The t test) was used to assess significance.
Laser scanning photostimulation.
Hardware control and data acquisition for laser-scanning photostimulation (LSPS) were performed using ephus (http://www.ephus.org/), as described previously (Shepherd and Svoboda, 2005; Oviedo et al., 2010). Briefly, to the external ACSF solution we added the following (in mm): 0.37 nitroindolinyl-caged glutamate (Tocris Bioscience), 0.005 3-(2-carboxypiperazin-4-yl)-propyl-1-phosphonic acid (Tocris Bioscience), 4 CaCl2, and 4 MgCl2. Caged glutamate was focally photolyzed with a 1 ms light stimulus consisting of 100 pulses from a pulsed ultraviolet laser every 1 s (wavelength, 355 nm with a repetition rate of 100 kHz; DPSS Lasers). The stimulus grid for LSPS mapping in acute brain horizontal slices consisted of a 16 × 16 grid with 75 μm spacing, which resulted in a mapping region of 1.125 × 1.125 mm. To isolate synaptic input responses, the mean current amplitude per stimulus site was calculated in the 7.5–50 ms time window after ultraviolet stimulus and expressed as mean charge transfer [current (pA) × synaptic epoch (ms)]. The values for each stimulus site are represented as pixels in a color map. For every cell, we obtained two to four maps to create an average input map and they were used for all analyses. We performed cell-attached recordings to detect action potentials from L5 cells and constructed excitation profiles. These maps are used to measure how far from the soma an ultraviolet flash can evoke an action potential and to calibrate the laser power across cells. To construct these maps for L5 cells, a smaller stimulus grid was used: an 8 × 16 grid with 50 μm spacing.
Immunostaining and measurement.
To assess PTEN expression in control and PTEN-cko neurons, 60-μm-thick brain slices were prepared and stained with monoclonal anti-PTEN antibody (Millipore) and anti-GFP antibody (Millipore). To visualize the dendritic arbors and spines of biocytin-filled neurons, brain slices were fixed in 4% PFA for >12 h, and we then followed a standard DAB staining protocol with Vectastain Kit (Vector Labs). Reconstructed neuronal morphology was traced and analyzed using ImageJ (NIH).
Rapamycin injection.
Rapamycin powder (LC Laboratory) was dissolved in ethanol and stored at a stock concentration of 25 mg/ml in aliquots at −20°C. A fresh working solution was prepared before use with a final concentration of 1 mg/ml rapamycin in 4% ethanol, 5% Tween 80, and 5% PEG400 (Zhou et al., 2009). Mice were injected intraperitoneally with either rapamycin (10 mg/kg body weight) or saline once per day continuously for 10–14 d.
Results
Conditional knockout of PTEN in mouse left auditory cortex
To examine the role of PTEN in regulating functional cortical connectivity, we used a Cre-lox approach to knock out PTEN expression conditionally in a spatially and temporally controlled manner. We injected an AAV expressing GFP-IRES-Cre into the left auditory cortex of homozygous mutant mice (PTENloxP/loxP) in which PTEN exons 4 and 5 are flanked by loxP sites (Fig. 1A,B) (Trotman et al., 2003). Infected neurons express GFP (as a marker) and Cre recombinase, which renders the PTEN gene defective. PTEN deletion was thus limited to only the subset of infected neurons near the site of injection. Using this strategy, we could assess the direct effect of PTEN deletion by comparing the properties of infected neurons with nearby “wild-type” (uninfected) controls.
Figure 1.
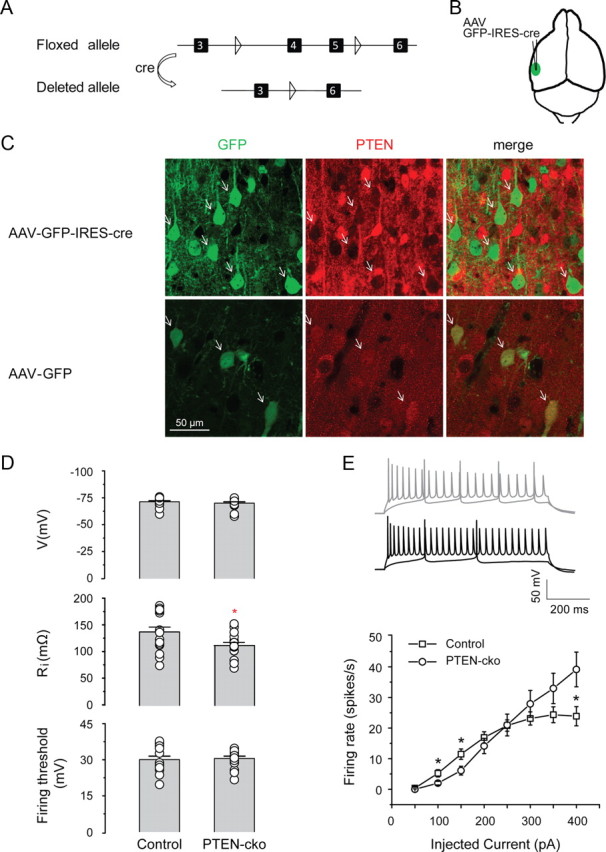
Effect of PTEN deletion on intrinsic membrane properties. A, Map of Cre-mediated deletion of exons 4 and 5 of the PTEN gene. Triangles indicate loxP sites, and black boxes indicate exons. B, Site of injection of AAV-GFP-IRES-Cre into the mouse auditory cortex. C, Immunostaining for PTEN expression indicates PTEN deletion in animals injected with AAV-GFP-IRES-Cre (top row) but not AAV-GFP (bottom row). Arrows indicate example neurons. D, Top, PTEN deletion has no effect on resting membrane potential (−70.3 ± 1.2 mV for control neurons, −71.6 ± 1.0 mV for PTEN-cko neurons, n = 19 for each group, p > 0.05); middle, PTEN deletion decreases input resistance (112.1 ± 5.6 MΩ for control neurons, 134.4 ± 9.0 MΩ for PTEN-cko neurons, n = 16 for each group, *p < 0.05); bottom, PTEN deletion has no effect on excitation threshold (30.0 ± 1.4 mV for control neuron, 30.5 ± 1.0 mV for PTEN-cko neurons, n = 15 for each group, p > 0.05). E, PTEN deletion subtly changed firing patterns of PTEN-cko neurons. Top, Example action potential traces for 100 and 400 pA current injections from PTEN-cko neuron (black) and control neuron (gray). Bottom, Elicited action potential numbers plotted against corresponding injected currents into PTEN-cko neurons and control neurons (n = 16 for each group, *p < 0.05). Data are mean ± SE.
In brain slices from mice injected with AAV-GFP-IRES-Cre, antibody staining showed no detectable PTEN signal in GFP-positive neurons (Fig. 1C, top), confirming that AAV-mediated expression of Cre recombinase caused functional deletion of the PTEN gene. By contrast, in brain slices from control PTENloxP/loxP mice injected with a virus (AAV-GFP) expressing only GFP but no Cre recombinase, GFP and PTEN were colocalized (Fig. 1C, bottom) in virus-infected neurons, confirming that loss of PTEN depended on the Cre-mediated deletion of exons 4 and 5 and was not simply due to AAV viral infection. In subsequent experiments, we used GFP expression to identify virally injected neurons, which are PTEN-cko (PTEN conditional knockout) in the case of AAV-GFP-IRES-Cre injection.
We first examined the intrinsic physiological properties of PTEN-cko neurons with whole-cell patch-clamp recording, using nearby (<50 μm) GFP-negative neurons as control. Neurons were filled with biocytin to permit subsequent analysis of cell morphology. All cells included in this study were pyramidal neurons across layer 2 to layer 6, as confirmed by morphology. PTEN-cko neurons had resting membrane potentials similar to those of nearby control neurons (−70.3 ± 1.2 and −71.6 ± 1.0 mV, respectively, mean ± SE) (Fig. 1D, top) as well as similar spike thresholds (30.0 ± 1.4 mV for control neurons, 30.5 ± 1.0 mV for PTEN-cko neurons) (Fig. 1D, bottom; but see Fig. 1E). However, the input resistance of PTEN-cko neurons was somewhat lower than that of nearby control neurons (112.1 ± 5.6 and 134.4 ± 9.0 MΩ, respectively) (Fig. 1D, middle).
PTEN-cko neurons receive stronger local inputs in auditory cortex
We next used LSPS to assess how PTEN regulates local connectivity within the auditory cortex (Oviedo et al., 2010). To study local and long-range connectivity in a single cell type, we focused on layer 5 pyramidal neurons, which receive inputs from the thalamus and from the contralateral auditory cortex (Cruikshank et al., 2002; Oviedo et al., 2010). We injected AAV-GFP-IRES-Cre into left auditory cortex of P18–P21 PTENloxP/loxP mice and performed LSPS experiments 10–14 d after virus injection. We minimized the effect of homeostatic compensation by infecting sparsely, so PTEN was deleted in only a small number of neurons. Acute horizontal brain slices were bathed in ACSF containing caged glutamate for local connectivity mapping (Fig. 2A). Using excitation profiles, we found no differences in the number and spatial profile of action potentials evoked per ultraviolet flash between wild-type and PTEN-cko neurons (Fig. 2C) (see Materials and Methods). After achieving a whole-cell recording, a UV laser was flashed briefly (1 ms) to release caged glutamate focally. Short-latency EPSCs following the flash resulted from action potentials elicited in several neurons near the site of uncaging. By flashing the UV beam sequentially over hundreds of spots, the spatial pattern of inputs to each recorded layer 5 pyramidal neurons was obtained (Fig. 2A,B).
Figure 2.

LSPS reveals that PTEN deletion enhances local inputs. A, Uncaging grid overlayed on auditory cortex. B, Example of synaptic inputs onto a layer 5 PTEN-cko neuron, shown as traces (left) and as a color map (right). The white triangle represents the location of the cell body, and blackened pixels represent direct responses excluded from the analysis. C, Excitation profile maps (left panels, n = 3 for each group) and interpolated population synaptic input maps (right panels) from groups of control neurons and PTEN-cko neurons (n = 10 for each group). D, Total input to PTEN-cko neurons is enhanced, with the greatest effect in layer 2.
Across the population, we found that both PTEN-cko neurons and nearby control neurons (<50 μm) received excitatory inputs from other layers in primary auditory cortex, with the largest inputs arising from layer 2 (Fig. 2C). However, the total input onto PTEN-cko neurons was significantly higher than onto nearby controls (Fig. 2D) (2.16 ± 0.28 for control, 3.14 ± 0.34 for PTEN-cko, n = 10 in each group, p < 0.05). This increase was greatest for inputs arising from layer 2 (4.24 ± 0.75 for control, 7.74 ± 1.46 for PTEN-cko, n = 10 in each group, p < 0.05), but was present in all layers. These results indicate that PTEN knockout increased the strength of local inputs, without grossly disrupting the overall pattern of local connections.
ChR2-mediated optical stimulation of callosal inputs to auditory cortex
We then assessed the effect of PTEN knockout on long-range connections onto auditory cortical neurons. The auditory cortex receives inputs from many other regions, including the contralateral auditory, somatosensory, and motor cortices. Using traditional methods, such long-range connections can only be studied in vitro in cases where the anatomy is favorable (i.e., when it is possible to prepare a single planar brain slice preserving the presynaptic elements along with their postsynaptic targets). To circumvent these difficulties, we used ChR2 (Nagel et al., 2003; Zhang et al., 2007). Neurons expressing ChR2 can be excited to fire action potentials by flashing blue light, and presynaptic release from ChR2-positive axon terminals can be induced by flashes even when the axons are severed from their parent somata (Petreanu et al., 2007). Our strategy was therefore to express ChR2 in brain areas known to project to the auditory cortex.
To establish this approach for the auditory cortex, we performed initial control experiments in wild-type mice. We targeted the callosal pathway by injecting AAV-expressing ChR2-venus (AAV-ChR2-venus) into the right auditory cortex of wild-type mice. In acute coronal brain slices prepared 2 weeks after virus injection, ChR2-venus-expressing axon terminals originating from callosally projecting neurons in right auditory cortex could readily be seen in the left auditory cortex (Fig. 3A). We first confirmed that ChR2-positive neurons at the site of the injection could be excited reliably by brief blue light pulses (1 ms, 475 nm) (Fig. 3B). Next, we used whole-cell patch-clamp methods to record light-evoked PSCs in pyramidal neurons in the left auditory cortex, contralateral to the injection. In standard ACSF (2 mm Ca2+, 1 mm Mg2+), light flashes often elicited multipeaked responses (Fig. 3C, left), suggesting a mixture of both direct callosal and recurrent local inputs. Furthermore, under these conditions responses consisted of both excitatory and inhibitory components (Fig. 3C, left); since long-range callosal connections are purely excitatory, the presence of inhibitory currents confirmed that light-evoked responses include a polysynaptic component.
Figure 3.
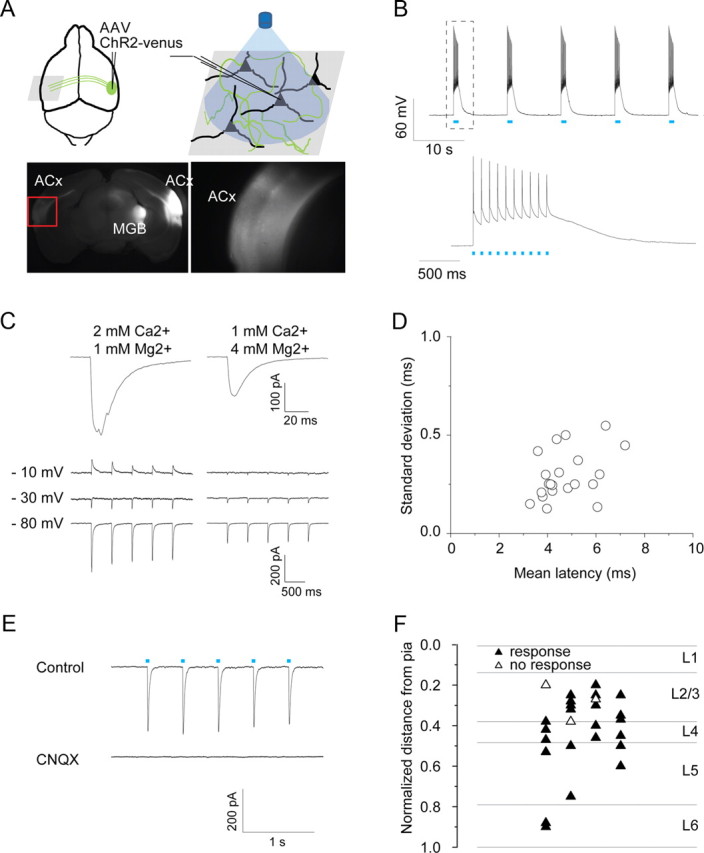
Long-range ChR2 mapping reveals that all layers receive callosal inputs. A, Following AAV-ChR2-venus injection into the right auditory cortex, fluorescent axons and synaptic terminals are seen at the injection site and in projection targets including ipsilateral thalamus and contralateral auditory cortex. Right image is an enlarged picture from red rectangle at left. B, action potentials elicited by blue light pulses (1 ms, 10 Hz, 475 nm) from virus-infected neurons on the right auditory cortex. C, Top, Examples of light-evoked EPSCs recorded at −70 mV for different extracellular divalent ion concentration. Under conditions of high release probability (left), responses are larger and multipeaked, indicating polysynaptic input. Reducing release probability (right) leads to smaller and smoother traces. Bottom, Example traces of single light-evoked EPSCs at different divalent ion concentrations when neurons were held at three voltages. At high release probability, there is a clear polysynaptic inhibitory component that is absent at low release probability. D, Mean latency versus SDs of EPSCs from individual cells. Average latency across cells is 4.7 ± 0.2 ms, average SD is 0.30 ± 0.02 ms (n = 23). E, Light-evoked EPSCs are blocked by application of CNQX (50 μm). Cells were held at −70 mV under voltage-clamp. F, Laminar distribution of neurons tested. The distance from pia to white matter in each brain slice was normalized, and the distance to the pia from each recorded neuron was measured. The horizontal position is randomly assigned. Data are presented as mean ± SE.
Since we were interested in the direct callosal input, we sought conditions under which such polysynaptic activity was absent. To isolate the direct monosynaptic callosal input, we reduced presynaptic release probability by changing the extracellular divalent cation concentration (1 mm Ca2+, 4 mm Mg2+) (Dodge and Rahamimoff, 1967). Under these conditions, light-evoked postsynaptic responses had single peaks (Fig. 3C, right), no inhibitory component (Fig. 3C, right), and short latencies (4.7 ± 0.2 ms; range 3–8 ms) with small jitter (0.30 ± 0.02 ms) (Fig. 3D). These light-evoked EPSCs were completely blocked by CNQX (Fig. 3E), indicating that they were mediated by AMPA-type glutamate receptors. Direct light-evoked EPSCs were observed in neurons (23 of 26) tested in auditory cortex across all layers (Fig. 3F).
PTEN-cko increased neuronal responses to auditory callosal and thalamic inputs
Having established conditions under which we could examine direct callosal inputs to the auditory cortex, we next compared these light-evoked EPSCs in PTEN-cko neurons with EPSCs recorded in nearby wild-type control neurons. In these experiments, AAV-ChR2-venus was injected into the right auditory cortex to deliver ChR2, and AAV-GFP-IRES-Cre was injected into left auditory cortex to knock out PTEN expression. Two weeks after injection, acute coronal brain slices were prepared for electrophysiology recording. GFP-labeled PTEN-cko neurons and nearby (<50 μm) control neurons were recorded (Fig. 4A,B). This paired experimental design normalizes for variability across preparations caused by variable ChR2 injection and other factors. For all 11 pairs of neurons, light-evoked responses in the PTEN-cko neurons were larger than in nearby controls (230.6 ± 44.4 vs 94.2 ± 29.2 pA, p < 0.05) (Fig. 4C). The average enhancement across the population was 3.46 ± 0.61-fold (EPSCPTEN-cko/EPSCControl, n = 11), with no apparent specificity across layers (Fig. 4D).
Figure 4.

PTEN deletion enhances synaptic responses to callosal inputs on PTEN-cko neurons. A, Experimental strategy. AAV-ChR2-venus was injected into right auditory cortex, and AAV-GFP-IRES-Cre was injected into left auditory cortex. B, Example traces of EPSCs elicited by blue light pulses (1 ms, 2 Hz, 475 nm). C, EPSC amplitude scatter plot for each pair of recorded neurons showing enhancement of EPSCs recorded in PTEN-cko neurons (closed circles: control vs control neurons; open circles: control versus PTEN-cko neurons; circles with error bars: mean values and SEs for each group). D, Laminar distribution of control/PTEN-cko pairs (n = 11), indicating relative EPSC enhancement (PTEN-cko/control) at each site.
To determine whether the effect of PTEN was unique to the callosal projection, we applied the same experimental design to the thalamocortical input. AAV-ChR2-venus was injected into left auditory thalamus (MGB), and AAV-GFP-IRES-Cre was injected into the left auditory cortex. Light-evoked EPSCs were recorded from both control and PTEN-cko neurons (Fig. 5A,B). As in the case of the callosal input, light-evoked EPSCs in PTEN-cko neurons were enhanced compared with those in nearby control neurons (158.9 ± 26.5 vs 71.3 ± 20.7 pA, p < 0.05) (Fig. 5C). The average enhancement across the population was 3.34 ± 0.78-fold (EPSCPTEN-cko/EPSCControl, n = 8), and again there was no apparent specificity across layers (Fig. 5D). Thus, knockout of PTEN leads to a robust increase in the efficacy of long-range excitatory synaptic inputs from both the contralateral auditory cortex and the thalamus.
Figure 5.

PTEN deletion enhances synaptic responses to thalamic inputs on PTEN-cko neurons. A, Experimental strategy. AAV-ChR2-venus was injected into left MGB, and AAV-GFP-IRES-Cre was injected into left auditory cortex. B, Example traces of EPSCs elicted by blue light pulses (1 ms, 2 Hz, 475 nm). C, EPSC amplitude scatter plot for each pair of recorded neurons (closed circles: control vs control neurons; open circles: control vs PTEN-cko neurons; circles with error bars: mean values and SEs for each group). D, Laminar distribution of control/PTEN-cko pairs (n = 8), indicating relative EPSC enhancement (PTEN-cko/control) at each site.
PTEN deletion promotes outgrowth of dendrites and spines, and increased synaptic activity
Knockout of PTEN has previously been shown to induce proliferation of axons and dendrites (Kwon et al., 2006; Chow et al., 2009; Luikart et al., 2011). Morphological analysis of biocytin-filled recorded neurons confirmed increases in dendritic total length and branch number (Fig. 6A,B), and in the density of spines (Fig. 6C), all of which might contribute to the observed enhancement of synaptic input. Analysis of miniature spontaneous EPSCs revealed increased mEPSC frequency (Fig. 6D), consistent with the increased dendritic branch number and spine density. We also observed an increase in mEPSC amplitude, which might further contribute to the observed enhancement of evoked synaptic efficacy.
Figure 6.

PTEN deletion promoted spine outgrowth and increased mEPSCs. A, Morphology of a biocytin-filled neuron (top), neurite traces in ImageJ (middle), and spine images (bottom). B, Quantification of total neurite length (top graph) and branches (bottom graph) of reconstructed neurons. Total length (μm), control neurons 980.26 ± 148.29, PTEN-cko neurons 1611.17 ± 218.30 (*p < 0.05, n = 6 neurons for each group). Branches number, control neurons 11.0 ± 0.8, PTEN-cko neurons 15.8 ± 1.3 (*p < 0.05, n = 6 neurons for each group). C, Spine densities (counts/10 μm) for control (4.1 ± 0.2) and PTEN-cko (9.1 ± 1.1) neurons (**p < 0.001, n = 15 for control neurons and n = 7 for PTEN-cko neurons). D, Top, mEPSC sample traces recorded from control and PTEN-cko neurons. Bottom left, Quantification of mEPSC frequency, control neuron 5.0 ± 0.6, PTEN-cko 14.2 ± 1.5 (**p < 0.001, n = 17 for control neurons and n = 15 for PTEN-cko neurons), and quantification of mEPSC mean amplitude, control neuron 5.6 ± 0.2, PTEN-cko 13.1 ± 0.3 (**p < 0.001, n = 17 for control neurons and n = 15 for PTEN-cko neurons). Data are mean ± SE. Bottom right, Cumulative histograms of mEPSCs from control and PTEN-cko neurons.
PTEN deletion effect can be blocked by rapamycin
PTEN is a negative regulator of the mTOR/PI3K (phosphatidylinositol 3-kinase) pathway. Downregulation of PTEN increases mTORC1 kinase activity, which promotes protein translation and cell growth through downstream effectors (Zoncu et al., 2011). To test whether the PTEN-cko effects we observed were mediated by mTORC1, we administered the specific mTORC1 inhibitor rapamycin. Ten days of intraperitoneal rapamycin injection blocked the effect of PTEN knockout on spine number (Fig. 7A,B), and on the PTEN-cko-mediated increase in the strength of the callosal projection (Fig. 7C,D). Together, the complete suppression of PTEN-cko-mediated effects on spine number and synaptic strength suggests that these effects are mediated largely or wholly by the mTORC1 pathway.
Figure 7.

Rapamycin antagonized PTEN-cko effects on auditory cortical neurons. A, Images of biocytin staining for neuron morphology reconstruction. B, Quantified spine densities (counts/10 μm) for control and PTEN-cko neurons from saline- or rapamycin-injected mice (right). In the saline group, control neurons 4.6 ± 0.5, PTEN-cko neurons 9.8 ± 0.6 (**p < 0.001, n = 4 for control neurons and n = 6 for PTEN-cko neurons). In the rapamycin group, control neurons 4.7 ± 0.8, PTEN-cko neurons 6.1 ± 0.3 (p > 0.05, n = 4 for control neurons and n = 5 for PTEN-cko neurons). C, EPSC amplitude scatter plot for each pair of recorded neurons (closed circle: rapamycin-injected mice; open circle: saline-injected mice). D, For each pair of neurons, EPSC amplitudes were normalized to the value from control neuron. In saline group, 3.39 ± 0.75 (*p < 0.01, n = 6). In rapamycin group, 1.11 ± 0.17 (p > 0.05, n = 8). Open circles show increase for individual pairs (EPSCPTEN-cko/EPSCControl), black bars are the mean increase for each injection group. Data are mean ± SE.
Discussion
We have studied the effects of spatiotemporally restricted PTEN deletion on cortical connectivity. Using viral delivery of Cre recombinase, we deleted PTEN after early development in a subset of neurons in the mouse auditory cortex, with nearby uninfected neurons as an internal control. Our main conclusions are as follows: (1) deletion of PTEN causes a rapid and robust increase in the strength of both long-range and local excitatory inputs; (2) deletion of PTEN causes an increase in dendritic length and spine density; and (3) these effects are blocked by rapamycin, suggesting that they occur through the mTORC1 pathway. Our study is the first functional comparison of how perturbing an ASD affects both local and long-range synaptic connectivity. Our findings suggest that hyperconnectivity may constitute a physiological basis for the effects of mutations in PTEN and possibly in other ASD candidate genes.
PTEN hyperconnectivity and ASDs
Autism is defined by the core triad of symptoms: impaired language, impaired social interaction, and restricted and repetitive behaviors. Because these symptoms affect behaviors that are unique to or highly specialized in humans, a central challenge in developing animal models of ASD is to find circuit-level endophenotypes. ASDs may arise from disruption of any of dozens of candidate genes (Sebat et al., 2007; Glessner et al., 2009), but it remains unclear what these diverse genetic pathways have in common. Convergent lines of evidence have led to the hypothesis that local hyperconnectivity and long-range hypoconnectivity represent a unifying mechanism for the core cognitive and behavioral deficits in ASD (Courchesne et al., 2005; Geschwind and Levitt, 2007).
We found using LSPS that PTEN deletion increased the strength of local inputs onto layer 5 neurons in auditory cortex, supporting the local hyperconnectivity hypothesis. Comparable increases in efficacy were obtained using similar methods for another ASD candidate gene, the receptor tyrosine kinase MET (Qiu et al., 2011), although in that case the changes were specific to a subset of layer 5 neurons. Another ASD candidate gene, CNTNAP2, was found using functional magnetic resonance imaging to correlate with increased local frontal connections in autistic patients (Scott-Van Zeeland et al., 2010). However, other ASD candidate genes, including FMR1 (Bureau et al., 2008) and MeCP2 (Wood et al., 2009), are associated with decreased local connectivity. Thus, it appears that disruption of ASD candidate genes can cause both hyperconnectivity and hypoconnectivity.
Using ChR2 to target long-range projections (Petreanu et al., 2007) to auditory cortex, we found that PTEN disruption caused a consistent increase in the strength of inputs from both the contralateral auditory cortex and the auditory thalamus. This increase was comparable for both projections across cortical recipient layers, suggesting that these effects are not pathway specific, but are likely to be similar for other inputs as well.
ASD has been hypothesized to arise from a partial disruption during development of long-range connections (Geschwind and Levitt, 2007). Our finding that PTEN-cko neurons in the auditory cortex received stronger inputs from both the contralateral cortex and the thalamus are consistent with this model. Furthermore, our results suggest that disruption can occur not only by weakening of connections but also by enhancement, suggesting that it is the precise balance of local and long-range connections that is essential to normal function.
PTEN in neuronal development and circuitry maintenance
PTEN has previously been implicated in the development and maintenance of neuronal circuits. The effect of knocking out PTEN depends on the developmental stage at which the knockout occurs. PTEN expresses very early in embryonic development, and PTEN knockout leads to embryonic lethality at E7.5–E9.5 (Di Cristofano et al., 1998). In our study, we deleted PTEN at P18–P21, when auditory cortical neurons already have established most local synaptic connections (Oswald and Reyes, 2008; Barkat et al., 2011), and assessed the resulting phenotype at P30–P45. Our findings are consistent with previous results showing neuronal hypertrophy and an increase of the density of dendritic spines after PTEN knockout at either P7 or at P42–P56 (Luikart et al., 2011), and with the increase in dendritic growth observed after PTEN knockout at P28 (Chow et al., 2009). Thus, our results may reflect mainly the role of PTEN in maintaining and refining, rather than establishing, neural connections.
In our experiments, we knocked out PTEN in relatively sparse subset of cortical neurons. This approach has the advantage that it allows for a direct comparison between PTEN-cko neurons and nearby control neurons in the same slice, but requires that we limit our conclusions largely to cell-autonomous postsynaptic effects of PTEN misregulation. To the extent that the sparse and dense models can be compared directly—the effect on dendritic morphology and spine density—the two approaches agree; but the results of the present study may not extrapolate directly to the case in which a germline mutation leads to a genetically uniform neuronal population. Moreover, the effect of PTEN may be different in different subsets of neurons (e.g., inhibitory interneurons). Nevertheless, an understanding of the cell autonomous effects provides a foundation for understanding the role of PTEN in regulating neuronal circuits.
Mechanisms of PTEN hyperconnectivity
Consistent with previous studies in other brain regions (Kwon et al., 2006; Fraser et al., 2008; Chow et al., 2009; Luikart et al., 2011), we found (Fig. 6) that PTEN deletion caused morphological changes in neurons, including increased neurite branching and higher spine density. These morphological changes could contribute to the observed increase in the frequency of mEPSCs (Fig. 6D) (Luikart et al., 2011), but cannot readily explain the observed increase in mEPSC amplitude (Fig. 6D), suggesting that PTEN directly regulates synaptic release machinery. Both morphological and other changes could contribute to the observed increases in local and long-range excitatory connectivity.
However, in a previous report Fraser et al. (2008) showed that conditional deletion of PTEN weakened synaptic transmission at CA3–CA1 excitatory synapses in hippocampus. This contradiction could be due to the differences in examined brain areas, the deletion onset time, and the deletion populations. Further studies may be done to clarify PTEN functions in various brain areas and in different developmental stages.
Signaling pathways underlying PTEN hyperconnectivity
Many genes have been implicated in the etiology of ASDs, including molecules in the PI3K pathway. In addition to PTEN, the tuberous sclerosis complex (TSC) is the major upstream mTORC1-negative regulator (Manning and Cantley, 2007). Mutations of TSC genes, in addition to giving rise to an inheritable disease syndrome, have also been implicated in ASDs (de Vries, 2010; Ehninger and Silva, 2011; Numis et al., 2011; Waltereit et al., 2011; Ehninger et al., 2012).
Neurofibromatosis type 1 is a Ras-antagonizing tumor suppressor, whose loss can result in activation of the PI3K pathway to produce gliomas (Hambardzumyan et al., 2011), and its mutations have been associated with ASD (Ey et al., 2011). Different downstream targets in PI3K/Akt pathways lead to distinct anatomical and cellular effects. One group of downstream targets, the small GTPases of the Rac, Cdc42, and Arf families, are involved in PTEN-mediated effects on cell polarity and migration (Liliental et al., 2000; Raftopoulou et al., 2004; Leslie et al., 2007; Dey et al., 2008). Yet, the major node in the PTEN pathway is the oncogenic Akt kinase, which has a wide variety targets, including mTORC1, that promote protein synthesis, cell growth, and proliferation (Manning and Cantley, 2007). Our observation that inhibition of mTORC1 by rapamycin abolished the effects of PTEN-cko (Fig. 7) indicates that mTORC1-dependent signaling axis is the primary mechanism for these effects under our conditions. It has also been shown that rapamycin injection can prevent and reverse macrocephaly, neuronal hypertrophy, and abnormal behaviors in PTEN mutant mice (Zhou et al., 2009), presumably through inhibition of mTORC1 activity. Together, these data suggest that the mTORC1 may represent a therapeutic target for certain cases of PTEN-mediated brain disorders.
Our study not only provides new evidence for a role of PTEN in regulating cortical connectivity, but also demonstrates a general paradigm for assessing the effect of ASD candidate genes. Such information will provide insights into the mechanisms of ASD, and guide the development of novel therapeutic strategies and approaches.
Footnotes
This work was supported by grants from the National Institutes of Health (A.M.Z.) and Autism Speaks (A.M.Z., Q.X.). We thank B. J. Burbach, T. Hromadka, S. Jaramillo, A. P. Reid, and M. Zeeman for invaluable technical help; and K. Borges, S. Chattopadhyay for discussion.
References
- Baranek GT, Foster LG, Berkson G. Tactile defensiveness and stereotyped behaviors. Am J Occup Ther. 1997;51:91–95. doi: 10.5014/ajot.51.2.91. [DOI] [PubMed] [Google Scholar]
- Barkat TR, Polley DB, Hensch TK. A critical period for auditory thalamocortical connectivity. Nat Neurosci. 2011;14:1189–1194. doi: 10.1038/nn.2882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boddaert N, Chabane N, Belin P, Bourgeois M, Royer V, Barthelemy C, Mouren-Simeoni MC, Philippe A, Brunelle F, Samson Y, Zilbovicius M. Perception of complex sounds in autism: abnormal auditory cortical processing in children. Am J Psychiatry. 2004;161:2117–2120. doi: 10.1176/appi.ajp.161.11.2117. [DOI] [PubMed] [Google Scholar]
- Budinger E, Scheich H. Anatomical connections suitable for the direct processing of neuronal information of different modalities via the rodent primary auditory cortex. Hear Res. 2009;258:16–27. doi: 10.1016/j.heares.2009.04.021. [DOI] [PubMed] [Google Scholar]
- Bureau I, Shepherd GM, Svoboda K. Circuit and plasticity defects in the developing somatosensory cortex of FMR1 knock-out mice. J Neurosci. 2008;28:5178–5188. doi: 10.1523/JNEUROSCI.1076-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler MG, Dasouki MJ, Zhou XP, Talebizadeh Z, Brown M, Takahashi TN, Miles JH, Wang CH, Stratton R, Pilarski R, Eng C. Subset of individuals with autism spectrum disorders and extreme macrocephaly associated with germline PTEN tumour suppressor gene mutations. J Med Genet. 2005;42:318–321. doi: 10.1136/jmg.2004.024646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalhoub N, Baker SJ. PTEN and the PI3-kinase pathway in cancer. Annu Rev Pathol. 2009;4:127–150. doi: 10.1146/annurev.pathol.4.110807.092311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow DK, Groszer M, Pribadi M, Machniki M, Carmichael ST, Liu X, Trachtenberg JT. Laminar and compartmental regulation of dendritic growth in mature cortex. Nat Neurosci. 2009;12:116–118. doi: 10.1038/nn.2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courchesne E, Redcay E, Morgan JT, Kennedy DP. Autism at the beginning: microstructural and growth abnormalities underlying the cognitive and behavioral phenotype of autism. Dev Psychopathol. 2005;17:577–597. doi: 10.1017/S0954579405050285. [DOI] [PubMed] [Google Scholar]
- Cruikshank SJ, Rose HJ, Metherate R. Auditory thalamocortical synaptic transmission in vitro. J Neurophysiol. 2002;87:361–384. doi: 10.1152/jn.00549.2001. [DOI] [PubMed] [Google Scholar]
- Dawson G, Meltzoff AN, Osterling J, Rinaldi J. Neuropsychological correlates of early symptoms of autism. Child Dev. 1998;69:1276–1285. [PMC free article] [PubMed] [Google Scholar]
- de Vries PJ. Targeted treatments for cognitive and neurodevelopmental disorders in tuberous sclerosis complex. Neurotherapeutics. 2010;7:275–282. doi: 10.1016/j.nurt.2010.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dey N, Crosswell HE, De P, Parsons R, Peng Q, Su JD, Durden DL. The protein phosphatase activity of PTEN regulates SRC family kinases and controls glioma migration. Cancer Res. 2008;68:1862–1871. doi: 10.1158/0008-5472.CAN-07-1182. [DOI] [PubMed] [Google Scholar]
- Di Cristofano A, Pesce B, Cordon-Cardo C, Pandolfi PP. Pten is essential for embryonic development and tumour suppression. Nat Genet. 1998;19:348–355. doi: 10.1038/1235. [DOI] [PubMed] [Google Scholar]
- Dodge FA, Jr, Rahamimoff R. Co-operative action a calcium ions in transmitter release at the neuromuscular junction. J Physiol. 1967;193:419–432. doi: 10.1113/jphysiol.1967.sp008367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehninger D, Silva AJ. Rapamycin for treating tuberous sclerosis and autism spectrum disorders. Trends Mol Med. 2011;17:78–87. doi: 10.1016/j.molmed.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehninger D, Sano Y, de Vries PJ, Dies K, Franz D, Geschwind DH, Kaur M, Lee YS, Li W, Lowe JK, Nakagawa JA, Sahin M, Smith K, Whittemore V, Silva AJ. Gestational immune activation and Tsc2 haploinsufficiency cooperate to disrupt fetal survival and may perturb social behavior in adult mice. Mol Psychiatry. 2012;17:62–70. doi: 10.1038/mp.2010.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ey E, Leblond CS, Bourgeron T. Behavioral profiles of mouse models for autism spectrum disorders. Autism Res. 2011;4:5–16. doi: 10.1002/aur.175. [DOI] [PubMed] [Google Scholar]
- Fombonne E. Epidemiology of autistic disorder and other pervasive developmental disorders. J Clin Psychiatry. 2005;66(Suppl 10):3–8. [PubMed] [Google Scholar]
- Fraser MM, Bayazitov IT, Zakharenko SS, Baker SJ. Phosphatase and tensin homolog, deleted on chromosome 10 deficiency in brain causes defects in synaptic structure, transmission and plasticity, and myelination abnormalities. Neuroscience. 2008;151:476–488. doi: 10.1016/j.neuroscience.2007.10.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritz JB, Elhilali M, David SV, Shamma SA. Auditory attention—focusing the searchlight on sound. Curr Opin Neurobiol. 2007;17:437–455. doi: 10.1016/j.conb.2007.07.011. [DOI] [PubMed] [Google Scholar]
- Geschwind DH, Levitt P. Autism spectrum disorders: developmental disconnection syndromes. Curr Opin Neurobiol. 2007;17:103–111. doi: 10.1016/j.conb.2007.01.009. [DOI] [PubMed] [Google Scholar]
- Glessner JT, Wang K, Cai G, Korvatska O, Kim CE, Wood S, Zhang H, Estes A, Brune CW, Bradfield JP, Imielinski M, Frackelton EC, Reichert J, Crawford EL, Munson J, Sleiman PM, Chiavacci R, Annaiah K, Thomas K, Hou C, et al. Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature. 2009;459:569–573. doi: 10.1038/nature07953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goffin A, Hoefsloot LH, Bosgoed E, Swillen A, Fryns JP. PTEN mutation in a family with Cowden syndrome and autism. Am J Med Genet. 2001;105:521–524. doi: 10.1002/ajmg.1477. [DOI] [PubMed] [Google Scholar]
- Gupta AR, State MW. Recent advances in the genetics of autism. Biol Psychiatry. 2007;61:429–437. doi: 10.1016/j.biopsych.2006.06.020. [DOI] [PubMed] [Google Scholar]
- Hambardzumyan D, Parada LF, Holland EC, Charest A. Genetic modeling of gliomas in mice: new tools to tackle old problems. Glia. 2011;59:1155–1168. doi: 10.1002/glia.21142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon CH, Luikart BW, Powell CM, Zhou J, Matheny SA, Zhang W, Li Y, Baker SJ, Parada LF. Pten regulates neuronal arborization and social interaction in mice. Neuron. 2006;50:377–388. doi: 10.1016/j.neuron.2006.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leslie NR, Yang X, Downes CP, Weijer CJ. PtdIns(3,4,5)P(3)-dependent and -independent roles for PTEN in the control of cell migration. Curr Biol. 2007;17:115–125. doi: 10.1016/j.cub.2006.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leslie NR, Batty IH, Maccario H, Davidson L, Downes CP. Understanding PTEN regulation: PIP2, polarity and protein stability. Oncogene. 2008;27:5464–5476. doi: 10.1038/onc.2008.243. [DOI] [PubMed] [Google Scholar]
- Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang SI, Puc J, Miliaresis C, Rodgers L, McCombie R, Bigner SH, Giovanella BC, Ittmann M, Tycko B, Hibshoosh H, Wigler MH, Parsons R. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science. 1997;275:1943–1947. doi: 10.1126/science.275.5308.1943. [DOI] [PubMed] [Google Scholar]
- Liliental J, Moon SY, Lesche R, Mamillapalli R, Li D, Zheng Y, Sun H, Wu H. Genetic deletion of the Pten tumor suppressor gene promotes cell motility by activation of Rac1 and Cdc42 GTPases. Curr Biol. 2000;10:401–404. doi: 10.1016/s0960-9822(00)00417-6. [DOI] [PubMed] [Google Scholar]
- Luikart BW, Schnell E, Washburn EK, Bensen AL, Tovar KR, Westbrook GL. Pten knockdown in vivo increases excitatory drive onto dentate granule cells. J Neurosci. 2011;31:4345–4354. doi: 10.1523/JNEUROSCI.0061-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maehama T, Dixon JE. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem. 1998;273:13375–13378. doi: 10.1074/jbc.273.22.13375. [DOI] [PubMed] [Google Scholar]
- Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagel G, Szellas T, Huhn W, Kateriya S, Adeishvili N, Berthold P, Ollig D, Hegemann P, Bamberg E. Channelrhodopsin-2, a directly light-gated cation-selective membrane channel. Proc Natl Acad Sci U S A. 2003;100:13940–13945. doi: 10.1073/pnas.1936192100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Numis AL, Major P, Montenegro MA, Muzykewicz DA, Pulsifer MB, Thiele EA. Identification of risk factors for autism spectrum disorders in tuberous sclerosis complex. Neurology. 2011;76:981–987. doi: 10.1212/WNL.0b013e3182104347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oswald AM, Reyes AD. Maturation of intrinsic and synaptic properties of layer 2/3 pyramidal neurons in mouse auditory cortex. J Neurophysiol. 2008;99:2998–3008. doi: 10.1152/jn.01160.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oviedo HV, Bureau I, Svoboda K, Zador AM. The functional asymmetry of auditory cortex is reflected in the organization of local cortical circuits. Nat Neurosci. 2010;13:1413–1420. doi: 10.1038/nn.2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petreanu L, Huber D, Sobczyk A, Svoboda K. Channelrhodopsin-2-assisted circuit mapping of long-range callosal projections. Nat Neurosci. 2007;10:663–668. doi: 10.1038/nn1891. [DOI] [PubMed] [Google Scholar]
- Qiu S, Anderson CT, Levitt P, Shepherd GM. Circuit-specific intracortical hyperconnectivity in mice with deletion of the autism-associated met receptor tyrosine kinase. J Neurosci. 2011;31:5855–5864. doi: 10.1523/JNEUROSCI.6569-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raftopoulou M, Etienne-Manneville S, Self A, Nicholls S, Hall A. Regulation of cell migration by the C2 domain of the tumor suppressor PTEN. Science. 2004;303:1179–1181. doi: 10.1126/science.1092089. [DOI] [PubMed] [Google Scholar]
- Schmitz C, Rezaie P. The neuropathology of autism: where do we stand? Neuropathol Appl Neurobiol. 2008;34:4–11. doi: 10.1111/j.1365-2990.2007.00872.x. [DOI] [PubMed] [Google Scholar]
- Scott-Van Zeeland AA, Abrahams BS, Alvarez-Retuerto AI, Sonnenblick LI, Rudie JD, Ghahremani D, Mumford JA, Poldrack RA, Dapretto M, Geschwind DH, Bookheimer SY. Altered functional connectivity in frontal lobe circuits is associated with variation in the autism risk gene CNTNAP2. Sci Transl Med. 2010;2:56ra80. doi: 10.1126/scitranslmed.3001344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebat J, Lakshmi B, Malhotra D, Troge J, Lese-Martin C, Walsh T, Yamrom B, Yoon S, Krasnitz A, Kendall J, Leotta A, Pai D, Zhang R, Lee YH, Hicks J, Spence SJ, Lee AT, Puura K, Lehtimäki T, Ledbetter D, et al. Strong association of de novo copy number mutations with autism. Science. 2007;316:445–449. doi: 10.1126/science.1138659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd GM, Svoboda K. Laminar and columnar organization of ascending excitatory projections to layer 2/3 pyramidal neurons in rat barrel cortex. J Neurosci. 2005;25:5670–5679. doi: 10.1523/JNEUROSCI.1173-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steck PA, Pershouse MA, Jasser SA, Yung WK, Lin H, Ligon AH, Langford LA, Baumgard ML, Hattier T, Davis T, Frye C, Hu R, Swedlund B, Teng DH, Tavtigian SV. Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat Genet. 1997;15:356–362. doi: 10.1038/ng0497-356. [DOI] [PubMed] [Google Scholar]
- Teder-Sälejärvi WA, Pierce KL, Courchesne E, Hillyard SA. Auditory spatial localization and attention deficits in autistic adults. Brain Res Cogn Brain Res. 2005;23:221–234. doi: 10.1016/j.cogbrainres.2004.10.021. [DOI] [PubMed] [Google Scholar]
- Trotman LC, Niki M, Dotan ZA, Koutcher JA, Di Cristofano A, Xiao A, Khoo AS, Roy-Burman P, Greenberg NM, Van Dyke T, Cordon-Cardo C, Pandolfi PP. Pten dose dictates cancer progression in the prostate. PLoS Biol. 2003;1:E59. doi: 10.1371/journal.pbio.0000059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veenstra-Vanderweele J, Christian SL, Cook EH., Jr Autism as a paradigmatic complex genetic disorder. Annu Rev Genomics Hum Genet. 2004;5:379–405. doi: 10.1146/annurev.genom.5.061903.180050. [DOI] [PubMed] [Google Scholar]
- Waltereit R, Japs B, Schneider M, de Vries PJ, Bartsch D. Epilepsy and tsc2 haploinsufficiency lead to autistic-like social deficit behaviors in rats. Behav Genet. 2011;41:364–372. doi: 10.1007/s10519-010-9399-0. [DOI] [PubMed] [Google Scholar]
- Wood L, Gray NW, Zhou Z, Greenberg ME, Shepherd GM. Synaptic circuit abnormalities of motor-frontal layer 2/3 pyramidal neurons in an RNA interference model of methyl-CpG-binding protein 2 deficiency. J Neurosci. 2009;29:12440–12448. doi: 10.1523/JNEUROSCI.3321-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Aravanis AM, Adamantidis A, de Lecea L, Deisseroth K. Circuit-breakers: optical technologies for probing neural signals and systems. Nat Rev Neurosci. 2007;8:577–581. doi: 10.1038/nrn2192. [DOI] [PubMed] [Google Scholar]
- Zhou J, Blundell J, Ogawa S, Kwon CH, Zhang W, Sinton C, Powell CM, Parada LF. Pharmacological inhibition of mTORC1 suppresses anatomical, cellular, and behavioral abnormalities in neural-specific Pten knock-out mice. J Neurosci. 2009;29:1773–1783. doi: 10.1523/JNEUROSCI.5685-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011;12:21–35. doi: 10.1038/nrm3025. [DOI] [PMC free article] [PubMed] [Google Scholar]