

Abstract
Missense mutations in leucine-rich repeat kinase 2 (LRRK2) cause late-onset Parkinson's disease (PD), and common genetic variation in LRRK2 modifies susceptibility to Crohn's disease and leprosy. High levels of LRRK2 expression in peripheral monocytes and macrophages suggest a role for LRRK2 in these cells, yet little is known about LRRK2 expression and function in immune cells of the brain. Here, we demonstrate a role for LRRK2 in mediating microglial proinflammatory responses and morphology. In a murine model of neuroinflammation, we observe robust induction of LRRK2 in microglia. Experiments with toll-like receptor 4 (TLR4)-stimulated rat primary microglia show that inflammation increases LRRK2 activity and expression, while inhibition of LRRK2 kinase activity or knockdown of protein attenuates TNFα secretion and nitric oxide synthase (iNOS) induction. LRRK2 inhibition blocks TLR4 stimulated microglial process outgrowth and impairs ADP stimulated microglial chemotaxis. However, actin inhibitors that phenocopy inhibition of process outgrowth and chemotaxis fail to modify TLR4 stimulation of TNFα secretion and inducible iNOS induction, suggesting that LRRK2 acts upstream of cytoskeleton control as a stress-responsive kinase. These data demonstrate LRRK2 in regulating responses in immune cells of the brain and further implicate microglial involvement in late-onset PD.
Introduction
The leucine-rich repeat kinase 2 (LRRK2) gene was discovered as part of an evolutionarily conserved family of proteins marked by GTPase domains usually encoded together with kinase domains (Bosgraaf and Van Haastert, 2003). Missense mutations in both the kinase and GTPase domain in LRRK2 cause late-onset Parkinson's disease (PD) with clinical and pathological phenotypes nearly indistinguishable from idiopathic disease, possibly through the upregulation of LRRK2 kinase activity (Paisán-Ruíz et al., 2004; Zimprich et al., 2004; West et al., 2005). Disease penetrance of LRRK2 mutations in PD is incomplete as lifetime risk in clinical populations is estimated at ∼22–32%, suggesting strong modifiers of LRRK2 disease (Goldwurm et al., 2007). A modifying role for the immune system in PD susceptibility is supported by the association of the human leukocyte antigen (HLA) region with late-onset disease (Hamza et al., 2010), and pathological studies of PD brains demonstrate strong microglial and T-cell activation and infiltration in susceptible brain nuclei (McGeer et al., 1988). Genome-wide association studies also highlight LRRK2 in the modification of susceptibility to the chronic autoimmune Crohn's disease and Mycobacterium leprae infection (Zhang et al., 2009; Umeno et al., 2011), raising the possibility that mutations in LRRK2 may modify immunogenic responses in PD.
LRRK2 is expressed in many different cell types in mammals, but the intracellular function of LRRK2 is not clear. In the brain, LRRK2 is expressed in diverse neuronal subtypes and localizes to cytoskeletal structures and a variety of vesicular and membranous organelles (Biskup et al., 2006). In neurons, LRRK2 has been described as a potent regulator of the cytoskeleton where knockdown of protein enhances neurite outgrowth and mutant (overactive) LRRK2 expression inhibits outgrowth (MacLeod et al., 2006). LRRK2 may directly modify microtubule organization and the actin cytoskeleton through phosphorylation of substrates (Gillardon, 2009; Parisiadou et al., 2009). LRRK2 may also play additional kinase-dependent roles in the modification of synaptic vesicle storage and mobilization, in addition to kinase-dependent roles in endocytosis, MAPK signaling, autophagy, and apoptosis (Alegre-Abarrategui et al., 2009; Gloeckner et al., 2009; Piccoli et al., 2011).
Particularly high LRRK2 expression has been discovered recently in macrophagic and monocytic cells, but not T-cells, leading to speculation of a functional role for LRRK2 in the innate immune system (Thévenet et al., 2011). A number of powerful tools, including highly specific rabbit monoclonal LRRK2 antibodies and potent and selective LRRK2 small-molecule kinase inhibitors, have become available that allow for a careful dissection of LRRK2 function in cells of the immune system. Based on the expression of LRRK2 in monocytes, we hypothesized a role for LRRK2 in the immune cells of the brain. Our results show that LRRK2 is expressed in activated microglia and that LRRK2 modulates proinflammatory responses in these cells. Alterations in LRRK2 function may modify inflammatory responses in neurodegenerative and infectious diseases, potentially leading to disease initiation or modification of progression.
Materials and Methods
Immunohistochemistry and immunofluorescence.
Male 8–12-week-old WT or LRRK2 KO C57BL/6J mice (provided by Heather Melrose, Mayo Clinic, Jacksonville, FL) or Tg(TH-EGFP)DJ76Gsat were perfused with room temperature (RT) PBS, pH 7.4, then 4% paraformaldehyde (PFA) in PBS, and brains removed and post-fixed in 4% PFA in PBS at 4°C for 12 h with agitation, then embedded in 30% sucrose/PBS for 24 h at 4°C, then frozen in isopentane and sectioned at 40 μm width on a freezing microtome. Freshly cut sections were rinsed and immediately treated with 0.3% H2O2 in methanol for 30 min at RT with mild agitation, rinsed, and treated with 10 mm Na-Citrate, pH 6.0, 0.05% Tween, for 30 min at 37°C. Sections were rinsed and blocked first in 3% nonfat milk in PBS with 0.3% Triton X-100 for 1 h RT and then in 10% normal goat serum in PBS with 0.3% Triton X-100 for 1 h. LRRK2 antibody solution [containing 0.2 μg/ml for DAB or 1 μg/ml for immunofluorescence rabbit monoclonal C-41 (Epitomics) 5% goat serum, 0.1% Triton X-100, and 0.01% sodium azide] was applied to sections for 24 h at 4°C with mild agitation. Sections were rinsed and Goat Anti-Rabbit:biotin (Vector Labs), Goat Anti-Rabbit DyLight 649 (Jackson Laboratories), Isolectin-B4:FITC or Isolectin-B4:Biotin (Sigma) was added (as indicated) for 24 h at 4°C. Sections for immunofluorescence were mounted with ProLong Gold (Invitrogen) onto coverslips. Sections with biotinylated markers were developed with the Vectastain Elite ABC kit and Impact DAB (Vector Labs) according to manufacturer's recommendations.
LPS injections.
Five micrograms of LPS (15,000 endotoxin units, Sigma) was stereotactically injected in a 1 μl volume with a flow rate of 0.2 μl/min using a NanoMite pump (Harvard Apparatus) fitted with a 32 gauge fully beveled needle and gas-tight syringe (Hamilton), with a 5 min wait for needle withdrawal, in mice anesthetized with isoflurane. Coordinates were −3.4 anteroposterior (AP), −1.1 ML, and −3.9 DV for substantia nigra pars compacta (SNpc), and +0.4 AP, −1.5 ML, and −2.5 DV for striatum, with respect to bregma. For LRRK2 activity assays, FLAG-LRRK2 BAC mice (Jackson Laboratory strain 012466) were used, and FLAG-M2 resin (Sigma) was used according to manufacturer's recommendations to immunoprecipitate LRRK2 protein. Animal usage was institutionally approved.
Western blotting, ELISA, and chemicals.
Antibodies to LRRK2 (clone C-41, Epitomics), β-actin, GFAP, MBP (Sigma), CD-68 (Serotec), MAP2 (Millipore), GAPDH, interferon regulatory factor-1, voltage-dependent anion channel (VDAC) (Santa Cruz Biotechnology), nitric oxide synthase (iNOS), Phospho-p38, and Ikk-α (Cell Signaling Technology) were used according to manufacturer's suggestions. Antibodies to pT1503 were previously described and were combined with de-phosphopeptide at a concentration of 10 μg/ml during antibody incubations (Webber et al., 2011). Rat TNFα ELISA assays were from eBioscience. Sunitinib (LC Laboratories), L2In1 (provided by Dario Alessi, Medical Research Council, Protein Phosphorylation Unit, Dundee, United Kingdom), and cytochalasin D (Sigma) were dissolved at a concentration of 10 mm in DMSO. DMSO controls represent DMSO concentrations present at the highest amount in the experiment, and did not exceed 0.04% in any experiment.
Quantitative RT-PCR.
Total RNA was extracted with Trizol reagent (Invitrogen), and first strand cDNA was generated with Superscript III (Invitrogen). qPCR was performed using Taqman assays Rn01455646_m1 TATA-binding protein (TBP) and Rn00562055_m1 TNFα primer sets (Invitrogen), and iQ Powermix (Bio-Rad). Thermocycling was performed on a Bio-Rad CFX96 machine.
LRRK2 kinase assays.
Kinase assays were performed as previously described (Sen et al., 2009). Recombinant purified human LRRK2 (Invitrogen) was combined into kinase buffer with LRRKtide substrate (Enzo Bioscience), and activity was measured by scintillation counting of P-81 Whatman phosphocellulose paper.
Lentiviral purification.
Lentivirus preparation was performed as previously described (Tomlinson, 2008). HEK293-FT cells were transfected with pLP1, pLP2, pVSV-G, and lentiviral expression vectors pLKO.1_LRRK2 [plasmids TRCN0000022655 (shRNA-A) and TRCN0000022658 (shRNA-B), Open Biosystems] or pLKO.1_Noncoding (NC) shRNA (Addgene plasmid 1864), or cFUGW (no RNAi) control. For determination of titer, RNA was extracted and cDNA was synthesized using the SuperScript VILO cDNA synthesis kit (Invitrogen). Real-time PCR was performed using primers that target the Rev response element (F-GCA GCA GGA AGC ACT ATG; R-CGC CTC AAT AGC CCT CAG C). Cycle threshold values obtained from the virus were compared with the plasmid standard curve to determine the number of copies of virus per microliter. EGFP epifluorescence was used to verify that >90% of cells were transduced for the entirety of the experiment.
Primary microglia cell cultures.
Primary mixed glial cultures were isolated from the forebrains of 2-d-old Swiss Webster rats of either sex using a differential detachment method. Forebrains were digested with HBSS (Invitrogen) containing 0.01% trypsin and 10 μg/ml DNase, and triturated with DMEM (Invitrogen) containing 20% heat-inactivated fetal bovine serum (FBS; Hyclone) and 1% penicillin-streptomycin. The dissociated cell suspension was plated onto poly-d-lysine-coated flasks. Media changes with DMEM containing 20% fetal bovine serum and 1% penicillin-streptomycin were performed every other day for 7 d. Microglia were separated by shaking the flasks for 1 h at 200 rpm. The resulting microglial cell suspension was removed and plated at a density of ∼1 × 105 cells/cm2 in DMEM supplemented with 10% FBS and 10 ng/ml granulocyte macrophage-CSF (Peprotech). The purity of microglia was verified by anti-rat CD-68 (Serotec) immunolabeling and Western blot.
Morphological assessment.
Randomized captured phase contrast images were derived from live cultures in a 37°C humidified chamber at 5% CO2 on a Carl Zeiss Cell Observer using Axiovision 4.7 Mark and Find controller. Before image collection, cells were incubated with 2.5 μm propidium iodide and 10 μm Hoechst 33342 stain for 10 min. The resultant images were analyzed by an observer blinded to experimental identity using ImageJ software to calculate process length in microglia cells.
Chemotaxis assay.
A total of 90,000 primary microglia cells were added to transwell plates (8 μm pore, 24-well inserts, Corning) immediately after microglial removal from astrocyte beds and were allowed to adhere to the upper chamber for 6 h. The lower chamber was then supplemented with 100 μm ADP (Sigma) to encourage migration through the membrane to the lower chamber, and experimental drug or DMSO was added to both the upper and lower chambers. After 30 additional hours, media was removed and the total number of cells counted, after incubation with 10 μm Hoechst 33342 for 10 min on a Carl Zeiss Cell Observer using Mark and Find software.
Statistics.
Data from all experiments were analyzed with GraphPad Prism and InStat software.
Results
LRRK2 expression in toll-like receptor 4-activated microglia
High LRRK2 expression in peripheral mouse monocytes and macrophages led us to examine whether LRRK2 may be expressed in brain-resident macrophage cells (i.e., microglia). We first applied recently characterized and highly specific rabbit monoclonal antibodies directed against LRRK2 to normal mouse brain tissue and failed to detect any cells positive for LRRK2 with morphology consistent with microglia, despite strong LRRK2 immunoreactivity in several neuronal populations. Since LRRK2 has been hypothesized as a stress-responsive kinase, we analyzed brain tissue from mice subjected to an intracranial injection of the potent toll-like receptor 4 (TLR4) agonist lipopolysaccharide (LPS) for a period of 24 h. Although we observed no loss of tyrosine hydroxylase (TH)-positive cells at this time point, the intensity of TH expression was slightly diminished (Fig. 1A–C). Whereas only blood vessels were labeled by isolectin B4 in the contralateral side to LPS injection (Fig. 1D), numerous small strongly stained cells consistent with activated microglia were detected in the LPS-treated SNpc (Fig. 1E,F). LRRK2 staining in the LPS-injected SNpc revealed a strong induction of LRRK2 immunoreactivity in small cells with a morphology and size consistent with the activated microglia identified by isolectin B4 (Fig. 1H). This staining was abolished in LRRK2 knock-out mice (Fig. 1I). LRRK2 protein was undetectable in white matter tracts in normal brain tissue, although many LRRK2-positive small cells were found in the corpus callosum of mice after an intrastriatal LPS injection (Fig. 1K). To rule out nonspecific or cross-reactive labeling, nonimmune rabbit IgG was also applied at concentrations comparable to LRRK2 antibody-treated sections; negligible immunoreactivity was observed in these negative control sections, and none were reminiscent of microglia cells. Finally, staining in LRRK2 KO mice revealed the LRRK2 monoclonal antibody to be specific for LRRK2 (Fig. 1I,L).
Figure 1.
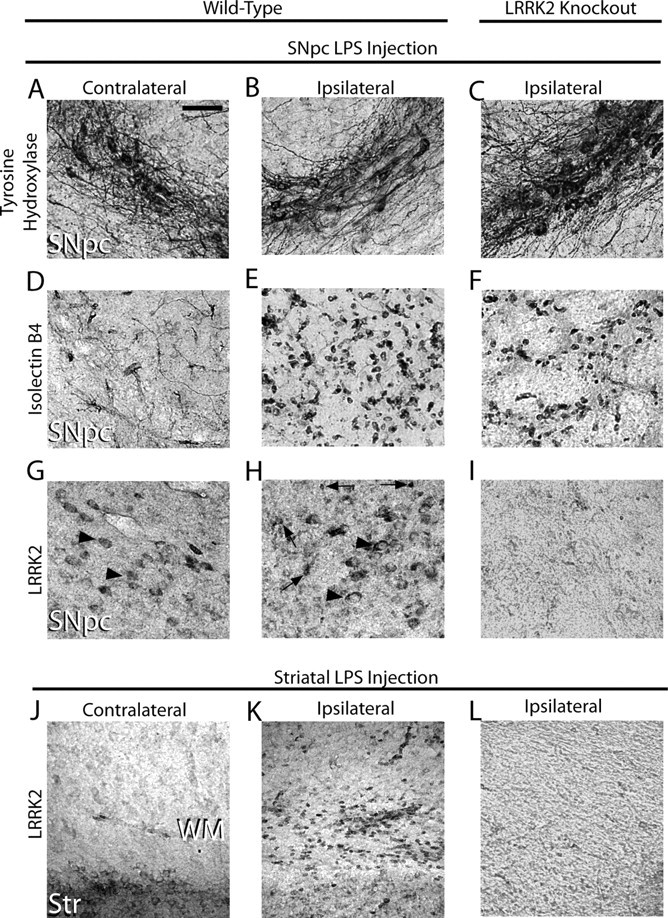
TLR4 stimulationPer triggers LRRK2 expression in microglia cells. Five micrograms of LPS (Escherichia coli 0111:B4) was unilaterally injected into the SNpc or striatum of 12-week-old male WT and LRRK2 KO C57BL/6J 12 mice. A–L, Immunohistochemistry for TH (A–C), isolectin B4 (marker for microglia and endothelial cells) (D–F), or LRRK2 was performed on serial coronal sections spanning the SNpc and striatum (G–L). Arrowheads indicate LRRK2 immunoreactivity on cells in the SNpc with the size and location of TH-positive neurons on both the contralateral and ipsilateral injection sides. Arrows indicate intense LRRK2 staining in numerous small cells observed exclusively on the ipsilateral side. WM, White matter; Str, striatum. No specific cellular staining in these areas was observed when primary antibodies were replaced with species-matched whole IgG (data not shown). Scale bar: A–L (in A), 50 μm.
To colocalize LRRK2 with microglial markers in TLR4-activated microglia, a triple-staining protocol that uses a single fluorescently labeled antibody was developed and applied to mice LPS-treated in either the SNpc or striatum (Fig. 2). Twenty-four hours postinjection, microglial cells rapidly accumulated and surrounded TH-positive neurons (Fig. 2A). Microglia could not be detected in the SNpc in animals that did not receive an LPS injection (Fig. 2B). Similar to the SNpc, LRRK2 colocalized to microglial cells present in the white matter tract post-striatal LPS injection (Fig. 2C). Resident microglia with resting morphologies in noninjected animals failed to demonstrate immunoreactivity for LRRK2 (Fig. 2D). As in Figure 1, whole-rabbit IgG control stained sections confirmed specificity of staining, and the LRRK2 antibody produces a single band of the correct size on Western blot analysis (Fig. 2E).
Figure 2.
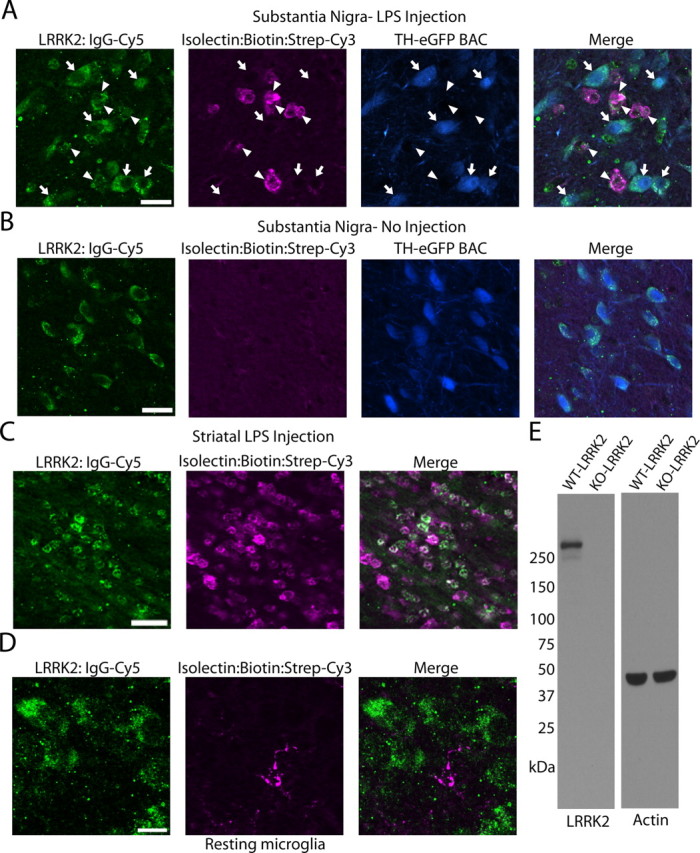
LRRK2 colocalizes with TLR4-stimulated microglia. TH-EGFP BAC mice were unilaterally injected with 5 μg of LPS (E. coli 0111:B4) into the SNpc or striatum. A, Using a triple-staining protocol (anti-rabbit IgG-Cy5 to detect LRRK2 antibody, ExtraAvidin-Cy3 to detect Isolectin-B4:biotin-positive cells, EGFP epifluorescence in TH-positive cells), LRRK2-labeled cells were observed in the SNpc as either large (arrows) or small (arrowheads) cells that colocalized with EGFP (arrows) or isolectin (arrowheads). Scale bar, 20 μm. B, LRRK2 staining in the SNpc in control no-injection mice. Scale bar, 20 μm. C, In striatal injected mice, LRRK2 was observed colocalized with most microglial cells in the white matter projection tract. Scale bar, 30 μm. D, LRRK2 staining could not be detected in microglia with resting morphology. Scale bar, 10 μm. Overlap of green (LRRK2) and magenta (microglia) is white, and overlap of blue (TH-positive cells) and green (LRRK2) is cyan. E, Western blot for LRRK2 with 20 μg of total protein lysate loaded per well that was derived from LRRK2 KO or WT whole-brain tissue.
LRRK2 induction in TLR4-stimulated cells
TLR4 activation stimulates signal transduction pathways responsible for the upregulation of proinflammatory factors. To further explore LRRK2 activity in response to TLR4 stimulation, mice transgenic for a FLAG-LRRK2 bacterial artificial chromosome insert were injected (bilateral) with LPS in the SNpc, and the SNpc and immediately surrounding regions were removed after 24 h. After LRRK2 immunoprecipitation from this tissue, LRRK2 was allowed to autophosphorylate for 15 min in the presence of 100 μm ATP, and protein eluted and transferred to membranes and probed with the recently described autophosphorylation-specific antibody pT1503 (Webber et al., 2011). LPS treatment significantly increased the proportion of LRRK2 in an activated state as revealed by an enhanced proportion of autophosphorylated LRRK2 (Fig. 2B). To ensure the pT1503 antibody could not cross-react with non-autophosphorylated LRRK2 protein, recombinant protein harboring mutations in the 1503 autophosphoraytion site or in the kinase domain (kinase dead, D1994A) was derived from transiently transfected cells and evaluated by Western blot (Fig. 2C). The pT1503 antibody could not detect signal in the kinase dead or the T1503A mutant LRRK2 protein, suggesting a high degree of specificity for this antibody.
To address whether LRRK2 expression also becomes upregulated during TLR4 activation, primary microglia in culture were treated with increasing concentrations of LPS for 12 h and LRRK2 protein levels determined by Western blot (Fig. 3E). A concentration of 100 ng/ml LPS was sufficient to increase levels of LRRK2 protein obtained in SDS-solubilized cell lysates, while higher concentrations of LPS failed to further increase LRRK2 induction. These primary microglia cultures were free from other cell types, and >99% of cells in culture were CD-68 positive (data not shown). Quantification of LRRK2 protein levels across primary microglia, astrocytes, hippocampal neurons, and oligodendrocytes, all derived from postnatal day 2 rats, unexpectedly revealed LRRK2 expression in primary astrocytes despite the lack of expression we could observe in these cells in vivo. LRRK2 expression in primary microglia cells is comparable to that of primary neurons in culture (Fig. 3D).
Figure 3.

LRRK2 induction by TLR4 stimulation. A, Five micrograms of LPS or no LPS control was bilaterally injected into the SNpc of FLAG-LRRK2 mice (Jackson Laboratory strain 012466), the SNpc was dissected after 24 h, and FLAG-LRRK2 was immunoprecipitated and treated with ATP. Eluted protein was analyzed by Western blot with either a total LRRK2 antibody- or autophosphorylation-specific pT1503 antibody. B, Quantification of pT1503-autophosphorylated LRRK2 normalized to total LRRK2 from four LPS-injected mice and four injection control mice. C, Specificity of the pT1503 antibody is demonstrated by LRRK2 recombinant protein derived from transiently transfected HEK-293FT cells. D, Primary cultures derived from P2 rats were analyzed by Western blot for cell type-specific markers and LRRK2 expression. Lysates were normalized to actin, and ∼20 μg of protein was loaded per lane. E, F, Primary microglia treated with various concentrations of LPS for 12 h and LRRK2 expression evaluated by Western blot (E), with quantification normalized to actin for three independent experiments (F). G, mRNA levels of LRRK2 were determined by relative quantification (ΔΔcT) normalized to TBP. H, Representative immunofluorescence of LRRK2 staining in primary microglia cultures treated with LPS or control (-LPS) for 12 h. I, Human LRRK2 expression characterized by Western blot in human primary microglia cultures compared with human macrophage/monocyte THP-1 cells. J, THP-1 cells treated with 100 ng of LPS for the indicated time and lysates analyzed by Western blot (J), with quantification of LRRK2 levels from three independent experiments (K). *p < 0.01, two-tailed unpaired t test (B); *p<0.01, one-way ANOVA with Tukey–Kramer test (B, F, K), with respect to initial LRRK2 expression. Error bars indicate SEM.
We next determined whether LRRK2 expression is upregulated at the mRNA level but failed to detect any significant differences after 12 h of induction with various doses of LPS, suggesting important post-transcriptional regulation of LRRK2 in microglia (Fig. 3G). LRRK2 distribution by immunofluorescence in primary microglia is consistent with that of previous reports with strong perinuclear staining and nuclear exclusion, both in non-LPS- and LPS-treated microglia (Fig. 3H). LRRK2 expression in human-derived cells of monocytic and microglia origin appears to be conserved (Fig. 3I), with similar LRRK2 upregulation in THP-1 cells treated with LPS. Quantitative PCR analysis for LRRK2 mRNA also revealed no significant changes in LRRK2 levels in these cells, despite strong upregulation of LRRK2 protein levels (Fig. 3K; data not shown).
Inhibition of LRRK2 kinase activity attenuates proinflammatory microglial signaling
A post-transcriptional induction of LRRK2 and enhanced autophosphorylation in purified protein suggests possible involvement of LRRK2 during a proinflammatory response. We first evaluated the inhibitory potential of the two most potent and specific LRRK2 inhibitors yet described, L2in1 and Sunitinib, under common conditions. We derived IC50 values of 40.3 and 126.4 nm under uniform conditions, respectively, demonstrating L2in1 as more potent against LRRK2 kinase activity in vitro (Fig. 4A,B). Sunitinib inhibits with similar potency several receptor tyrosine kinases including the PDGF receptor and VEGF receptor in addition to LRRK2, and L2in1 is highly specific for LRRK2 but is predicted to inhibit several other kinases including MAPK7 and Aurora-A (Dzamko et al., 2010). However, neither small molecule inhibits any known kinases that obviously link to TLR4 receptor signaling when applied at nanomolar or low micromolar concentrations. In addition, both inhibitors are structurally distinct from one another, and there are no common off-targets known.
Figure 4.

LRRK2 kinase inhibition attenuates inflammatory signaling in microglia. A, B, Calculation of the inhibitory potential of the LRRK2 targeted compounds L2in1 (A) and sunitinib (B) using standardized in vitro kinase assays consisting of 30 nm LRRK2 enzyme, 50 μm peptide, and 100 μm ATP with reactions run for 30 min. IC50 values were calculated through nonlinear regression with r2 values of 0.960 and 0.968, respectively, for L2in1 and Sunitinib. C, Graphical depiction of the experimental timeline used to generate lysates and serum from primary microglia analyzed in D–G. D, Quantification of secreted TNFα by ELISA after a 6 h exposure to LPS. Suni, Sunitinib. Drug concentrations are given in micromoles, and mean values are calculated from three independent experiments. E, TNFα mRNA was measured by quantitative PCR (ΔΔcT) normalized to TBP, and mean values are calculated from three independent experiments. F, Representative Western blot analysis of primary microglia lysates. G, Quantification of three independent experiments with levels of iNOS and phospho-p38 levels normalized to VDAC expression. *p < 0.05, **p < 0.01, one-way ANOVA with Tukey–Kramer test, with respect to DMSO (+LPS) conditions. Error bars indicate SEM.
Secretion of the proinflammatory cytokine TNFα by microglia may represent a major modifier of neurotoxicity in models of Parkinson's disease (Harms et al., 2011). Pretreatment of microglia with nanomolar concentrations of either L2in1 or Sunitinib significantly attenuated the release and transcriptional induction of TNFα in primary cultured microglia (Fig. 4D,E). iNOS and p38 are both critical targets of TNFα and effectors of continued TNFα release in autocrine signaling. In agreement with an attenuated inflammatory response to TLR4 stimulation, significantly reduced levels of iNOS induction and phosphorylated p38 were observed 6 h post-LPS treatment (Fig. 5F,G). To our knowledge, this is the first demonstration of anti-inflammatory activity for either small-molecule LRRK2 inhibitor.
Figure 5.

LRRK2 knockdown attenuates inflammatory signaling in microglia. A, Graphical depiction of the experimental timeline used to generate lysates and serum from primary microglia cultures. B, A total of 5 × 105 primary microglia per condition were exposed to the indicated copies of purified lentivirus encoding EGFP (no RNAi control), a noncoding control shRNA (NC-shRNA), or LRRK2 shRNAs (A, B), in 0.5 ml in culture. C, Serum from primary microglia treated with 2 × 107 lentiviral copies/ml of the respective lentivirus for 7 d were analyzed for TNFα secretion 6 h post-LPS (100 ng/ml) or control exposures. Results are calculated from three independent experiments. D, Primary microglia exposed to LPS for 6 h as indicated were lysed in SDS buffer and analyzed by Western blot. Two micrograms of total protein was loaded per lane and analyzed with the indicated antibody. VDAC is used as a loading control. Blots shown are representative of three independent experiments. E, Quantification of iNOS levels normalized to VDAC expression. *p < 0.05, **p < 0.01 by one-way ANOVA with Tukey–Kramer, with respect to LV-NC-shRNA (+LPS) conditions. Error bars indicate SEM.
To determine whether removal of total LRRK2 protein produces effects comparable to acute kinase inhibition, we developed lentivirus capable of knocking down LRRK2 expression in primary microglia (Fig. 5B). Lentivirus has previously been described as an effective method to genetically modify primary microglia without adverse effects on inflammatory responses (Balcaitis et al., 2005). To knock down LRRK2 by RNAi in microglia, highly purified lentivirus was applied in increasing concentrations to determine the minimum amount of virus required to knock down ∼90% of LRRK2 protein. Control shRNA (NC) lentivirus was also applied at identical concentrations, and no adverse toxic or inflammatory effects on the microglia were observed. Knockdown of LRRK2 was sufficient to reduce TNFα secretion similar to LRRK2 kinase inhibition via small-molecule inhibitor exposure (Fig. 5C). LRRK2 knockdown also inhibited TLR4-mediated iNOS induction (Fig. 6D,E). Phosphorylation of p38 was unchanged by LRRK2 RNAi, although basal levels of phospho-p38 rose to the levels of post-LPS phospho-p38 as a consequence of the extended in vitro culture period required for LRRK2 protein knockdown (Fig. 6D). Collectively, these results suggest that LRRK2 kinase activity and expression are required for a full inflammatory response in microglia.
Figure 6.
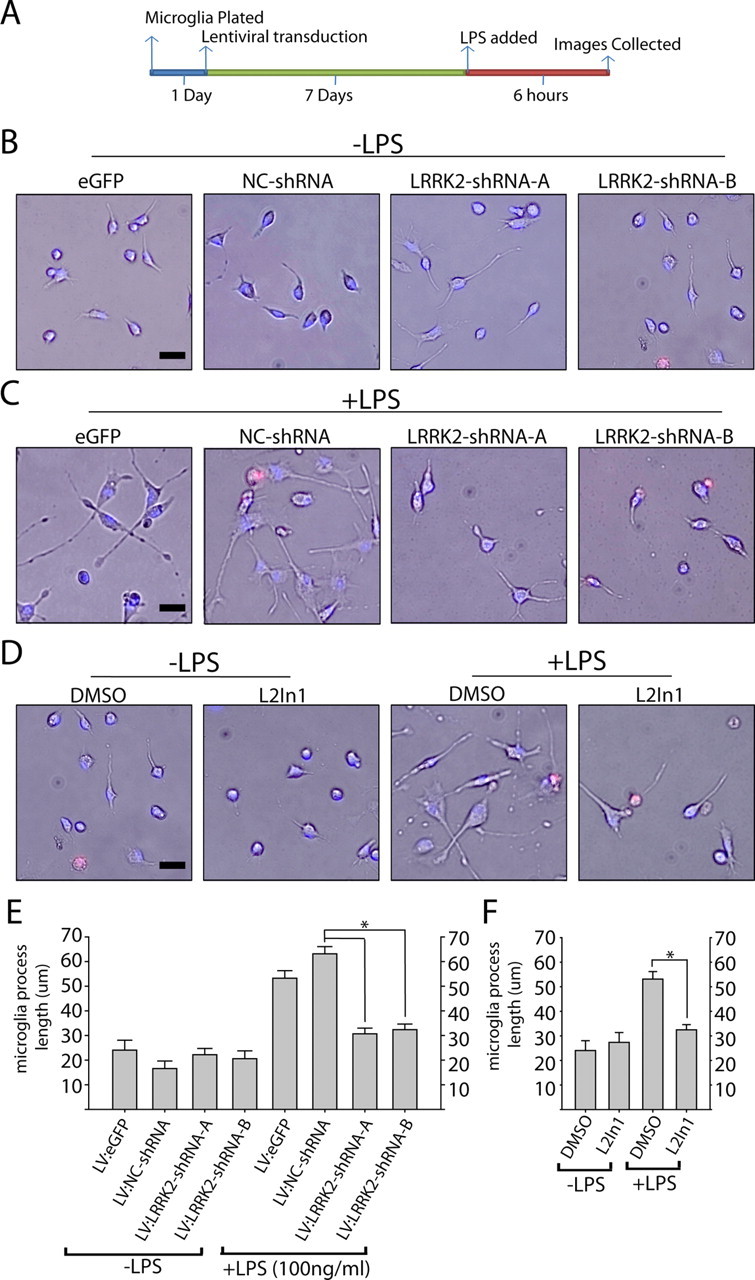
LRRK2 controls TLR4-responsive process outgrowth. A, Graphical depiction of the experimental timeline. B–D, Representative phase-contrast images with Hoechst stain (blue) and propidium iodide (red) stain overlay. Scale bars, 40 μm. E, F, Quantification of average microglia process length calculated from >150 microglia analyzed from three experiments per condition. L2in1 was used at 1 μm concentration with a 1 h pretreatment before LPS addition. Round cells with healthy nuclear staining but no process extensions were counted as 0 for process length determination, and propidium iodide-positive cells or cells with abnormal Hoechst staining (<10% of cells in every condition) were excluded from analysis. *p < 0.01 compared with LV:NC-shRNA/+LPS (E) or DMSO/+LPS (F) as determined by one-way ANOVA with Tukey–Kramer test. Error bars are SEM.
LRRK2 kinase activity is required for microglial morphological remodeling during activation
Microglia initially respond to LPS and other proinflammatory factors by extending processes into the environment (Sheng et al., 2011), with eventual (>12 h) retraction into a classic activated amoeboid morphology as autocrine effects contribute to a positive feedback loop in pure microglial cultures. We noticed that in the presence of LRRK2 inhibitors or LRRK2 shRNA, LPS treatment failed to cause any significant morphological differences with non-LPS-treated controls (Fig. 6). This failure to morphologically respond to LPS was not due to an enhanced rate of cell death since Hoechst and propidium iodide staining revealed comparable results across conditions.
Microglial dynamic fine process extension have been suggested to play a critical role in mediating responses to local brain injury and stressors (Davalos et al., 2005). Because of migroglial dependence on LRRK2 for mediating a morphological response to LPS, we hypothesized that a chemotactic response to the potent microglial chemoattractant ADP may likewise be compromised in the absence of LRRK2 activity. Microglial cells immediately plated onto transwell-permeable membranes after removal from astrocyte cultures demonstrated reduced migration to the bottom well in the presence of LRRK2 inhibitors (Fig. 7). An actin inhibitor, cytochalasin D, nearly abolished migration of microglia through the membrane, demonstrating the importance of cytoskeleton architecture in this process.
Figure 7.

LRRK2 inhibition impairs microglial chemotaxis. A, Graphical depiction of experimental approach. The bottom chamber is supplemented with 100 μm ADP, and microglia are allowed to migrate through the 8 μm pore membrane over a 30 h period of time to the bottom chamber. B–E, Representative depiction of a 0.5 mm2 area of the bottom chamber with cells stained with Hoechst dye. Suni, Sunitinib; Cyto-D, cytochalasin D. F, Relative quantification of the number of microglia migrating to the bottom chamber, calculated from three independent experiments. **p < 0.01 as determined by one-way ANOVA with Tukey–Kramer test. Error bars indicate SEM.
Since inhibition of LRRK2 has been shown to modify intracellular cytoskeleton architecture and process outgrowth in neurons (MacLeod et al., 2006), we reasoned that LRRK2 inhibition of process outgrowth and chemotaxis may link LRRK2 action, albeit indirectly, to anti-inflammatory activities. To test this hypothesis, we applied the actin inhibitor cytochalasin D to repress LPS-induced morphological remodeling. A 2 μm concentration of cytochalasin D was sufficient to phenocopy LRRK2 in preventing LPS-induced process extension, although, as opposed to LRRK2 inhibition, cytochalasin D also reduced normal process morphology in non-LPS-treated microglia (Fig. 8B). Surprisingly, cytochalasin D failed to demonstrate any anti-inflammatory activity; rather, TNFα secretion was positively regulated, although we were not able to detect an overall change in iNOS induction (Fig. 8C,D). We conclude that LRRK2-dependent morphological changes are a result of upstream LRRK2-dependent anti-inflammatory signaling. Disruption of LRRK2 activity thus prevents a full inflammatory response in these cells that is reflected both morphologically, by TNFα secretion, and by iNOS induction.
Figure 8.
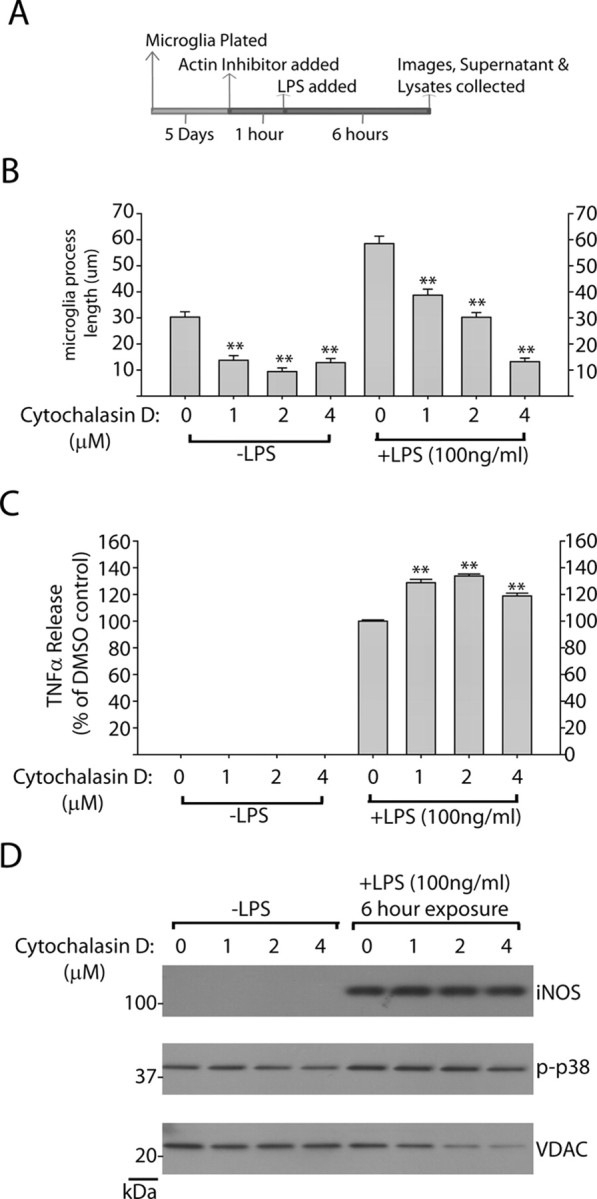
Inhibition of microglial process outgrowth does not affect proinflammatory signaling. A, Graphical depiction of experimental approach. B, Quantification of average microglia process length in response to the indicated concentration of cytochalasin D. Mean lengths were calculated from >150 microglia analyzed from three independent experiments per condition. Round cells with healthy nuclear staining but no process extensions were counted as 0 for process length determination, and propidium iodide-positive cells or cells with abnormal Hoechst staining (<10% of cells in every condition) were excluded from analysis. C, Quantification of secreted TNFα by ELISA after a 6 h exposure to LPS in the presence of the indicated concentration of cytochalsin D. Mean values were calculated from three independent experiments. D, Representative Western blots demonstrating levels of iNOS and phospho-p38 levels in response to LPS addition in the presence of cytochalsin D or control. No significant differences in iNOS or phospho-p38 levels could be detected over three independent experiments. **p < 0.01 as determined by one-way ANOVA with Tukey–Kramer test. Error bars indicate SEM.
Discussion
Genetic studies unambiguously tie LRRK2 to several human diseases. Most notably, high frequencies of causative pathological mutations have been identified in late-onset PD. Whole genome association studies pinpoint LRRK2 as one of a few genes where common genetic variability underlies susceptibility to leprosy and Crohn's disease, highlighting a potential immunologic function for LRRK2. Other evidence also suggests that immune function is involved in the pathogenesis of PD, since genetic variation in the HLA region associates with PD (Hamza et al., 2010), and numerous pathologic studies describe microglia activation in PD. LRRK2 appears to be expressed in cells of the innate but not adaptive immune system. A recent study demonstrated that the expression of LRRK2 in cultured bone marrow-derived macrophages from mice is upregulated in response to LPS (Hakimi et al., 2011). Another recent study demonstrated that LRRK2 expression is also stimulated by the IFN-γ response in peripheral blood mononuclear cells (Gardet et al., 2010). Thus, it has been hypothesized that LRRK2 may mediate some aspect of signaling or differentiation in innate immune cells.
The recent development of highly specific and sensitive rabbit monoclonal LRRK2 antibodies and LRRK2 knock-out mice enables immunohistochemistry studies with a higher degree of confidence. Our results show that in brain tissue challenged with the potent TLR4 agonist LPS, LRRK2 expression is induced in activated microglia. Immunoprecipitation of LRRK2 protein from brains treated with LPS revealed enhanced levels of autophosphorylation, which implies greater LRRK2 activity. Direct demonstration of enhanced LRRK2 enzyme activity in neuroinflammation awaits the identification of bone fide LRRK2 kinase substrates. Interestingly, we found that the accumulation of LRRK2 protein, which occurs during inflammatory signaling in primary microglia, is not accompanied by significant changes in mRNA levels, suggesting important post-transcriptional regulation.
We find that inhibition of LRRK2, either by small-molecule kinase inhibitors or RNAi knockdown, attenuates proinflammatory signaling in response to TLR4 activation. LRRK2 is hierarchically clustered in the tyrosine kinase-like superfamily nearby kinases important for inflammatory signaling in immune activation, such as the interleukin-1 receptor-associated kinase family and the mixed-lineage kinase family (West et al., 2007). Thus, we hypothesized that LRRK2 may function as a stress response kinase during a neuroinflammatory stimulus in the brain by facilitating signal transduction pathways in affected cells. We used two approaches to dissect the role of LRRK2 in TLR4-mediated inflammatory responses in primary microglia: small-molecule inhibition and RNAi knockdown of total protein. While a single inhibitory molecule may be confounded by off-target effects, we find agreement with multiple effective molecules that are unlikely to have overlapping off-targets. In cultured rat microglia, peak TNFα release was observed 6 h after LPS exposure and reduced by 12 h, and, in the context of LRRK2 inhibition, peak induction was reduced by >20% by either LRRK2 small-molecule inhibition or RNAi. Likewise, reduced levels of the TNFα target iNOS were detected by Western blot. Although this effect is modest in the overall proinflammatory response, a cumulative effect over time may link this modifying effect with susceptibility to neurodegeneration since both TNFα and iNOS have been implicated as critical determinants of neurotoxicity.
We also observed an effect of LRRK2 inhibition on the morphological response of microglia to LPS. Exposure of microglia to LPS in the context of inhibited LRRK2 prevents the normal morphological response of fine process extension and cytoskeleton remodeling. The functional role for microglial fine process extension during initial phases of inflammatory signaling are not clear, but may involve physical sequestration of infiltrating pathogens in vivo in damaged areas by providing a network blockage (Davalos et al., 2005). We hypothesized that inhibition of fine process extension during LPS stimulation may itself have an anti-inflammatory effect due to negative effects on receptor clustering and lipid raft shuttling. Although actin inhibition nearly abolished fine process extension, there were no anti-inflammatory effects observed. We therefore conclude that a full proinflammatory response mediates process extension, but process extension itself does not mediate proinflammatory signaling. Thus, LRRK2 inhibition may prevent a full inflammatory response required for fine-process extension, placing LRRK2 as an upstream stress-responsive kinase to TLR4 activation.
Although the current data would suggest an overall proinflammatory role for LRRK2, given the complexity of neuroinflammation and the limitations of interpreting microglial action in purified cultures in vitro, it is possible that an overall anti-inflammatory role for LRRK2 may be likewise envisaged. Indeed, we find that LRRK2 inhibitors quell a chemotaxic response to the potent microglial chemoattractant ADP. However, we interpret these results with some caution since we were not able to perform comparable experiments under conditions of LRRK2 RNAi. In these experiments, it was not possible to remove microglia already transduced with lentiviral LRRK2 shRNA molecules, incubated for the time required to knock down LRRK2 protein, without killing the microglia cells. In addition to microglia, LRRK2 may also be expressed in anti-inflammatory IL-10-producing macrophages in the brain, and inhibition of LRRK2 in these cells may mitigate anti-inflammatory signaling. Transgenic and LRRK2 knock-out mice can be used in future studies to clarify overall effects of LRRK2 in neuroinflammation.
In this study, we contribute to a growing body of evidence that suggests a possible modifying role for the immune system and inflammation in PD by demonstrating LRRK2 activation in microglia and critical function in proinflammatory responses. It seems hypothetically possible that activating mutations in LRRK2, such as the G2019S missense mutation in the kinase activation loop, may serve to exaggerate neuroinflammatory responses that predispose to neurodegeneration susceptibility in PD. Further studies with in vivo models of neuroinflammation and associated neurodegeneration are warranted and will be critical to address the pathophysiological function of LRRK2.
Footnotes
This work was supported by the Michael J. Fox Foundation for Parkinson's Research, the American Parkinson's Disease Association, National Institutes of Health Grant R01-NS064934, UAB Neuroscience Core Center Grant NS047466, and the benevolence of John A. and Ruth R. Jurenko. M.S.M. is supported by NIH/NINDS Grant T32 NS061788. We thank Dario Alessi for L2in1 compound; Bassel Sawaya for purified human microglia cells; and Heather Melrose and Matthew Farrer for LRRK2 knock-out mice.
References
- Alegre-Abarrategui J, Christian H, Lufino MM, Mutihac R, Venda LL, Ansorge O, Wade-Martins R. LRRK2 regulates autophagic activity and localizes to specific membrane microdomains in a novel human genomic reporter cellular model. Hum Mol Genet. 2009;18:4022–4034. doi: 10.1093/hmg/ddp346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balcaitis S, Weinstein JR, Li S, Chamberlain JS, Möller T. Lentiviral transduction of microglial cells. Glia. 2005;50:48–55. doi: 10.1002/glia.20146. [DOI] [PubMed] [Google Scholar]
- Biskup S, Moore DJ, Celsi F, Higashi S, West AB, Andrabi SA, Kurkinen K, Yu SW, Savitt JM, Waldvogel HJ, Faull RL, Emson PC, Torp R, Ottersen OP, Dawson TM, Dawson VL. Localization of LRRK2 to membranous and vesicular structures in mammalian brain. Ann Neurol. 2006;60:557–569. doi: 10.1002/ana.21019. [DOI] [PubMed] [Google Scholar]
- Bosgraaf L, Van Haastert PJ. Roc, a Ras/GTPase domain in complex proteins. Biochim Biophys Acta. 2003;1643:5–10. doi: 10.1016/j.bbamcr.2003.08.008. [DOI] [PubMed] [Google Scholar]
- Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, Littman DR, Dustin ML, Gan WB. ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci. 2005;8:752–758. doi: 10.1038/nn1472. [DOI] [PubMed] [Google Scholar]
- Dzamko N, Deak M, Hentati F, Reith AD, Prescott AR, Alessi DR, Nichols RJ. Inhibition of LRRK2 kinase activity leads to dephosphorylation of Ser(910)/Ser(935), disruption of 14-3-3 binding and altered cytoplasmic localization. Biochem J. 2010;430:405–413. doi: 10.1042/BJ20100784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardet A, Benita Y, Li C, Sands BE, Ballester I, Stevens C, Korzenik JR, Rioux JD, Daly MJ, Xavier RJ, Podolsky DK. LRRK2 is involved in the IFN-gamma response and host response to pathogens. J Immunol. 2010;185:5577–5585. doi: 10.4049/jimmunol.1000548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillardon F. Leucine-rich repeat kinase 2 phosphorylates brain tubulin-beta isoforms and modulates microtubule stability—a point of convergence in parkinsonian neurodegeneration? J Neurochem. 2009;110:1514–1522. doi: 10.1111/j.1471-4159.2009.06235.x. [DOI] [PubMed] [Google Scholar]
- Gloeckner CJ, Schumacher A, Boldt K, Ueffing M. The Parkinson disease-associated protein kinase LRRK2 exhibits MAPKKK activity and phosphorylates MKK3/6 and MKK4/7, in vitro. J Neurochem. 2009;109:959–968. doi: 10.1111/j.1471-4159.2009.06024.x. [DOI] [PubMed] [Google Scholar]
- Goldwurm S, Zini M, Mariani L, Tesei S, Miceli R, Sironi F, Clementi M, Bonifati V, Pezzoli G. Evaluation of LRRK2 G2019S penetrance: relevance for genetic counseling in Parkinson disease. Neurology. 2007;68:1141–1143. doi: 10.1212/01.wnl.0000254483.19854.ef. [DOI] [PubMed] [Google Scholar]
- Hakimi M, Selvanantham T, Swinton E, Padmore RF, Tong Y, Kabbach G, Venderova K, Girardin SE, Bulman DE, Scherzer CR, LaVoie MJ, Gris D, Park DS, Angel JB, Shen J, Philpott DJ, Schlossmacher MG. Parkinson's disease-linked LRRK2 is expressed in circulating and tissue immune cells and upregulated following recognition of microbial structures. J Neural Transm. 2011;118:795–808. doi: 10.1007/s00702-011-0653-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamza TH, Zabetian CP, Tenesa A, Laederach A, Montimurro J, Yearout D, Kay DM, Doheny KF, Paschall J, Pugh E, Kusel VI, Collura R, Roberts J, Griffith A, Samii A, Scott WK, Nutt J, Factor SA, Payami H. Common genetic variation in the HLA region is associated with late-onset sporadic Parkinson's disease. Nat Genet. 2010;42:781–785. doi: 10.1038/ng.642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harms AS, Barnum CJ, Ruhn KA, Varghese S, Treviño I, Blesch A, Tansey MG. Delayed dominant-negative TNF gene therapy halts progressive loss of nigral dopaminergic neurons in a rat model of Parkinson's disease. Mol Ther. 2011;19:46–52. doi: 10.1038/mt.2010.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLeod D, Dowman J, Hammond R, Leete T, Inoue K, Abeliovich A. The familial Parkinsonism gene LRRK2 regulates neurite process morphology. Neuron. 2006;52:587–593. doi: 10.1016/j.neuron.2006.10.008. [DOI] [PubMed] [Google Scholar]
- McGeer PL, Itagaki S, Boyes BE, McGeer EG. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson's and Alzheimer's disease brains. Neurology. 1988;38:1285–1291. doi: 10.1212/wnl.38.8.1285. [DOI] [PubMed] [Google Scholar]
- Paisán-Ruíz C, Jain S, Evans EW, Gilks WP, Simón J, van der Brug M, López de Munain A, Aparicio S, Gil AM, Khan N, Johnson J, Martinez JR, Nicholl D, Carrera IM, Pena AS, de Silva R, Lees A, Martí-Massó JF, Pérez-Tur J, Wood NW, et al. Cloning of the gene containing mutations that cause PARK8-linked Parkinson's disease. Neuron. 2004;44:595–600. doi: 10.1016/j.neuron.2004.10.023. [DOI] [PubMed] [Google Scholar]
- Parisiadou L, Xie C, Cho HJ, Lin X, Gu XL, Long CX, Lobbestael E, Baekelandt V, Taymans JM, Sun L, Cai H. Phosphorylation of ezrin/radixin/moesin proteins by LRRK2 promotes the rearrangement of actin cytoskeleton in neuronal morphogenesis. J Neurosci. 2009;29:13971–13980. doi: 10.1523/JNEUROSCI.3799-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccoli G, Condliffe SB, Bauer M, Giesert F, Boldt K, De Astis S, Meixner A, Sarioglu H, Vogt-Weisenhorn DM, Wurst W, Gloeckner CJ, Matteoli M, Sala C, Ueffing M. LRRK2 controls synaptic vesicle storage and mobilization within the recycling pool. J Neurosci. 2011;31:2225–2237. doi: 10.1523/JNEUROSCI.3730-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen S, Webber PJ, West AB. Dependence of leucine-rich repeat kinase 2 (LRRK2) kinase activity on dimerization. J Biol Chem. 2009;284:36346–36356. doi: 10.1074/jbc.M109.025437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng W, Zong Y, Mohammad A, Ajit D, Cui J, Han D, Hamilton JL, Simonyi A, Sun AY, Gu Z, Hong JS, Weisman GA, Sun GY. Pro-inflammatory cytokines and lipopolysaccharide induce changes in cell morphology, and upregulation of ERK1/2, iNOS and sPLA2-IIA expression in astrocytes and microglia. J Neuroinflammation. 2011;8:121. doi: 10.1186/1742-2094-8-121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thévenet J, Pescini Gobert R, Hooft van Huijsduijnen R, Wiessner C, Sagot YJ. Regulation of LRRK2 Expression Points to a Functional Role in Human Monocyte Maturation. PLoS One. 2011;6:e21519. doi: 10.1371/journal.pone.0021519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomlinson RL. Optimized production of lentivirus using FuGENE HD transfection reagent. Biochemica. 2008;(3):17–19. [Google Scholar]
- Umeno J, Asano K, Matsushita T, Matsumoto T, Kiyohara Y, Iida M, Nakamura Y, Kamatani N, Kubo M. Meta-analysis of published studies identified eight additional common susceptibility loci for Crohn's disease and ulcerative colitis. Inflamm Bowel Dis. 2011;17:2407–2715. doi: 10.1002/ibd.21651. [DOI] [PubMed] [Google Scholar]
- Webber PJ, Smith AD, Sen S, Renfrow MB, Mobley JA, West AB. Autophosphorylation in the leucine-rich repeat kinase 2 (LRRK2) GTPase domain modifies kinase and GTP-binding activities. J Mol Biol. 2011;412:94–110. doi: 10.1016/j.jmb.2011.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AB, Moore DJ, Biskup S, Bugayenko A, Smith WW, Ross CA, Dawson VL, Dawson TM. Parkinson's disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc Natl Acad Sci U S A. 2005;102:16842–16847. doi: 10.1073/pnas.0507360102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AB, Moore DJ, Choi C, Andrabi SA, Li X, Dikeman D, Biskup S, Zhang Z, Lim KL, Dawson VL, Dawson TM. Parkinson's disease-associated mutations in LRRK2 link enhanced GTP-binding and kinase activities to neuronal toxicity. Hum Mol Genet. 2007;16:223–232. doi: 10.1093/hmg/ddl471. [DOI] [PubMed] [Google Scholar]
- Zhang FR, Huang W, Chen SM, Sun LD, Liu H, Li Y, Cui Y, Yan XX, Yang HT, Yang RD, Chu TS, Zhang C, Zhang L, Han JW, Yu GQ, Quan C, Yu YX, Zhang Z, Shi BQ, Zhang LH, et al. Genomewide association study of leprosy. N Engl J Med. 2009;361:2609–2618. doi: 10.1056/NEJMoa0903753. [DOI] [PubMed] [Google Scholar]
- Zimprich A, Biskup S, Leitner P, Farrer M, Kachergus J, Hulihan M, Uitti RJ, Calne DB, Stoessel AJ, Pfeiffer RF, Patenge N, Carbajal IC, Vieregge P, Asmus F, Müller-Myhsok B, Dickson DW, Meitinger T, Strom TM, Wszolek ZK, Gasser T. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. 2004;44:601–607. doi: 10.1016/j.neuron.2004.11.005. [DOI] [PubMed] [Google Scholar]