

Polyhedral boranes and carboranes are of great interest because of their use as a 10B source in BNCT,[1] as hydrophobic pharmacophores,[2] as weakly coordinating anions,[3] and as ligands for transition metals and other types of metal as well.[4] One of the less explored applications of the polyhedral boranes is one in which they can be used as platforms for the targeted and high payload density delivery of drug molecules and imaging agents.[5] The icosahedral dodecahydro-closo-dodecaborate dianion [closo-B12H12]2−, (Figure 1) is an aromatic species having extensive delocalization of thirteen bonding electron pairs[6] as well as unique properties, such as chemical, hydrolytic and thermal stabilities and low toxicity. Each of the twelve vertices present in [closo-B12H12]2− can be attached to either identical or a variety of substituents to generate attractive molecular construction modules. To date the [closo-B12H12]2− cage has been modified with a variety of functional substituents such as hydroxyl,[7] thiol,[8] thioethers,[9] halogens,[10] amines,[11] alkyl, aryl groups and others[12] to form polyhedral boranes with varying degrees of substitution and reactivity.
Figure 1.
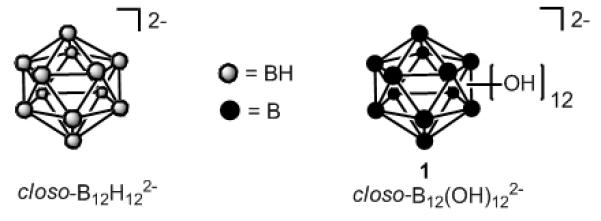
Icosahedral closo-boranes
The discovery of the remarkable B-H hydroxylation reaction with H2O2 has led to a variety of polyhedral borane and carborane structures, such as [closo-B12(OH)12]2− (1), [closo-CHB11(OH)11]− and [closo-1,12-C2H2B10(OH)10][13] that have added new dimensions to the chemistry of polyfunctional molecules. The hydroxylation of all of the B-H vertices of [closo-B12H12]2− using 30% hydrogen peroxide provides 1 in greater than 95% yield.[7b] Icosahedral 1 is stable to hydrolysis, air-oxidation and enzymatic attack while providing a functionalized molecular scaffold that can be used to anchor up to twelve radial arms with desired pendant groups even at generation zero. This is possible due to the fact that the reactivity of the B-OH vertices resembles that of alcohols. Consequently, twelve-fold carboxylate ester[14] and ether[15] derivatives, described by us as “closomers”, are now available.[14a] Closomer derivatives of 1 and similar dendrimers share several chracteristics although basic differences are significant. Closomers are the smaller in size with greater rigidity (dendrimers are more loosely constructed while closomers of the same functionality are more rigidly configured as twelve chains which simultaneously originate at the icosahedral surface in close proximity to each other). Closomers have higher symmetry and consist of a single structure. In addition, a much more compact presentation of functional groups is possible with closomer derivatives of 1 as compared to dendrimer structures having similar twelve-fold functionality. Consequently, the chemistry of 1 provides uniform nanoparticle-size molecular architectures potentially useful for carrying payloads of pharmaceuticals, imaging agents or many other useful substitutents.
Blending click[16] and closomer chemistries would provide increased opportunities for the syntheses of novel therapeutic and diagnostic entities. One obvious addition of click chemistry to that of the icosahedral borane scaffold requires the initial synthesis of twelve-fold azide-substituted closomers that can react with terminal alkynes to generate twelve-fold 1,2,3-triazole rings. Here we report for the first time several examples of this new chemistry which demonstrates the only known methodology capable of producing twelve reaction centers for the click reaction at generation zero.
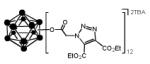
Below, we extend our original work[14] on the synthesis of ester closomers and describe a method for the synthesis of ester-linked azido closomers by the twelve-fold esterification of (TBA)2-1 [TBA = tetran-butylammonium] with an α-haloacetic anhydride followed by the displacement of halide ion with azide (NaN3). The reaction of TBA2-1 and chloroacetic anhydride (5.0 eqv. per vertex) at the reflux temperature in acetonitrile for 5 days gave the twelve-fold chloroacetate closomer 2 after purification using gelfiltration chromatography on a Lipophilic Sephadex® LH-20 column in 91% yield (Scheme 1). The excess, unreacted chloroacetic anhydride was separated and reutilized. The course of the esterification reactions was monitored by mass spectrometric analysis and 11B NMR spectra of crude reaction mixtures. In the 11B NMR spectrum, intermediate stages of esterification were characterized by an array of peaks centered near −17 ppm. Complete reaction was characterized by equivalent B-OCOR vertices and a singlet near −17 ppm (Fig. SI-1 in supporting information). The 1H NMR of the purified product contained a characteristic singlet at δ 4.1 ppm for 24-protons assigned to twelve-Cl-CH2CO2- groups bound to the substituted closo-borane cage.
Scheme 1.
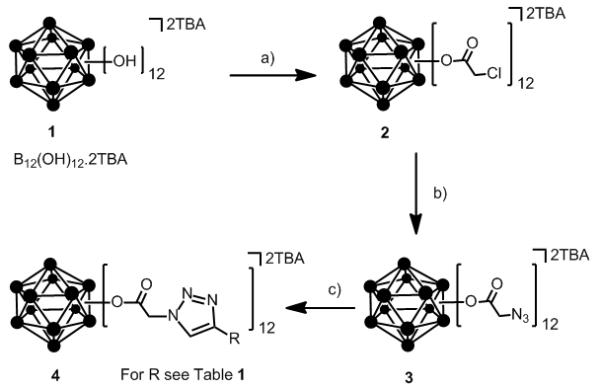
Synthetic route for twelve-fold Click reaction with [closo-B12H12]2−; Conditions, a) chloroacetic anhydride in CH3CN, 5 days, reflux; b) NaN3 in DMF, 2 days, 50°C; c) alkyne, CuI, DIPEA, CH3CN-THF (1:1), 2-3 days, RT. DMF = dimethylformamide, CH3CN = acetonitrile, DIPEA = diisopropylethylamine, THF = tetrahydrofuran.
In a typical procedure for the synthesis of twelve-fold azide terminated closomer 3, closomer 2 was reacted with a 10-fold excess of sodium azide in DMF (dimethylformamide) at 50°C for 2 days to obtain the twelve-fold azido closomer 3 in essentially quantitative yields (Scheme 1). The progress of the reaction was monitored using 1H NMR and mass spectrometric analysis; at this stage the 11B NMR spectrum of the crude reaction mixture had not changed significantly from that of the starting closomer 2. However, in the 1H NMR spectrum, the completion of the reaction was indicated by a complete shift in the characteristic singlet for 24-protons assigned to the twelve Cl-CH2CO2- groups from δ 4.1 ppm to δ 3.7 ppm for the twelve N3-CH2CO2- groups. The IR-spectrum of the product also showed the presence of a characteristic peak at 2109 cm−1 which was attributed to the asymmetrical stretch of the pendant azide-group (Fig. SI-2 in supporting information). Purification of the product was achieved by concentrating the reaction mixture to dryness under reduced pressure, dissolution of the residue in ethyl acetate and filtration using a celite pad to remove unreacted sodium azide and sodium chloride. The filtrate was concentrated to obtain the desired product in quantitative yield. The product could be used in the click reaction with alkyne substrates without further purification.
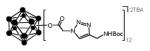
Among various forms of click reactions, the Cu(I)-catalyzed variant of the Hüisgen 1,3-dipolar cycloaddition of azides with alkynes to afford 1,2,3-triazoles has emerged as the most popular for click chemistry due to its reliability, specificity and biocompatibility. In a typical click reaction; twelve-fold azido closomer 3 and acetylenic compounds (5 eq. per vertex), in the presence of copper (I) iodide (1 eq. per vertex) and Hünig’s base (10 eq. per vertex) were reacted for 2-3 days under an argon atmosphere (Scheme 1).
The progress of the click reaction was followed by mass spectrometric analysis and 1H NMR spectra of crude reaction mixtures. Products were purified by size-exclusion column chromatography (Lipophilic Sephadex® LH-20) using acetonitrile as an eluent to yield click products in good to excellent yields (Table 1). All of the click products showed a characteristic singlet between δ 7-8 ppm for twelve protons of alkene-CH of the twelve triazole rings in the 1H NMR spectrum of the purified products. The IR-spectrum of the product also showed the disappearance of the characteristic peak at 2109 cm−1 which was originally attributed to the asymmetric stretching of the azide-group.
Table 1.
Click reactions of various terminal alkynes with twelve-fold azido closomer 3 using CuI and DIPEA in CH3CN-THF (1:1).
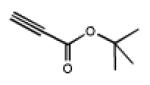
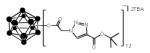
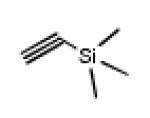
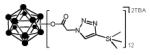
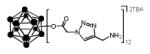
all yields after purification
crude yield
In conclusion, we have established a mild and highly efficient protocol for the synthesis of twelve-chloroacetate esters of (TBA)2-1. The twelve α-chlorine atoms on the ester closomer 2 were readily replaced by an azide functionality, thereby forming a closomer having twelve linker arms with terminal groups, 3, available for the twelve-fold click reaction on a closomer surface. These unique products discrete nano-size molecules carrying multiple copies of diverse functions which serve as model therapeutic and/or diagnostic agents. In addition, the α-chloro and α-azido substituted ester analogues (2 and 3) can be used to perform reactions characteristic of simple alkyl halides and azides, respectively, further expanding the chemical horizons of closo-borane scaffolds. Applications of Hüisgen chemistry have expanded the scope of organic chemistry and at this point we can imagine the potential for closo-borane chemistry amplified with click chemistry.
Experimental Section
Bis(tetrabutylammonium)-closo-dodecachloroacetoxydodecaborate (2): A solution of TBA2-1 (2.00 g, 2.44 mmol) and chloroacetic anhydride (25.1 g, 147 mmol) in 50 mL dry acetonitrile was refluxed for 5 days in an argon atmosphere with vigorous stirring. Progress of the reaction was monitored by 11B NMR. Completion of the reaction was indicated by the appearance of a sharp singlet at −17 ppm. The reaction mixture was then concentrated to dryness and purified using a size-exclusion column (Lipophilic Sephadex® LH-20) with acetonitrile as the eluent. The product was obtained as light brown semi-solid. Yield: 3.85 g (91%). IR (KBr): 3054, 2983, 2305, 1753, 1422, 1330, 1264 and 1231 cm−1. 1H NMR (500 MHz, CDCl3): δ 4.12 (s, 24H), 3.14 (m, 16H), 1.63 (m, 16H), 1.43 (m, 16H), 1.02 (t, 24H, J = 7.5 Hz). 13C NMR (125 MHz, CDCl3): δ 165.18, 58.80, 43.29, 23.76, 19.45 and 13.48. 11B NMR (160 MHz, CDCl3): δ −17.22. HRMS (m/z): Calcd. for C24H24B12Cl12O24 (M2−) 625.8005. Found: 625.9180 and Calcd. for C24H24B12Cl12O24+ C16H36N1− (M+TBA) 1494.0647. Found: 1493.1816.
Bis(tetrabutylammonium)-closo-dodecaazidoacetoxydodecaborate (3): In a 100 mL round bottom flask, Bis(tetrabutylammonium)-dodecachloroacetoxydodecaborate 2 (2.00 g, 1.15 mmol) and sodium azide (9.00 g, 139 mmol) were mixed with 20 mL of dry DMF. This mixture was vigorously stirred at 50°C for 2 days under an argon atmosphere. The progress of the reaction was monitored by 1H NMR. After completion, the reaction mixture was filtered through a celite pad and the filtrate was concentrated to dryness, the residue was re-dissolved in ethyl acetate and filtered again through a celite pad. The filtrate was collected and evaporated to dryness. The product was purified using size-exclusion column chromatography (Lipophilic Sephadex® LH-20) with acetonitrile as eluent. The product was obtained as a light brown semi-solid. Yield: 1.70 g (81%). IR (KBr): 3054, 2984, 2305, 2109, 1742, 1442, 1359 and 1265 cm−1. 1H NMR (500 MHz, CD3CN): δ 3.72 (s, 24H), 3.10 (m, 16H), 1.62 (m, 16H), 1.37 (m, 16H), 0.99 (t, 24H, J = 7.5 Hz). 13C NMR (125 MHz, CD3CN): δ 166.64, 117.24, 58.26, 51.07, 23.22, 19.25 and 12.74. 11B NMR (160 MHz, CD3CN): δ −17.57. HRMS (m/z): Calcd. for C24H2412N36O24 (M2−) 665.2031. Found: 664.9981 and Calcd. for C24H24B12N36O24+ C16H36N1− (M+TBA) 1572.8699. Found: 1573.2446.
Click reaction (Compounds 4a-4f)
In a 50 mL oven-dry round bottom flask, the azide-functionalized closomer 3 (1eq.), alkyne (5 eq. per vertex, total 60 eq.) and copper (I) iodide (1 eq. per vertex, total 12 eq.) were dissolved in a 50:50 mixture of tetrahydrofuran and acetonitrile (15 mL). To this mixture, diisopropylethylamine (10 eq. per vertex, total 120 eq.) was added and the reaction mixture was vigorously stirred at room temperature for 3 days under an argon atmosphere. After completion, the reaction mixture was concentrated to dryness, redissolved in ethyl acetate and filtered through a celite pad. The filtrate was concentrated and purification via size-exclusion column chromatography (Lipophilic Sephadex® LH-20) using acetonitrile as eluent to afford the pure product. Using the general strategy described above, 4a was synthesized from azide functionalized closomer 3 (100 mg, 0.055 mmol), phenylacetylene (337 mg, 3.30 mmol), copper (I) iodide (126 mg, 0.660 mmol) and diisopropylethylamine (853 mg, 6.60 mmol). Yield: 120 mg (72%). IR (KBr): 3054, 2985, 2305, 1742, 1422, 1369 and 1265 cm−1. 1H NMR (500 MHz, CD3CN): δ 7.97 (s, 12H), 7.83 (m, 24H), 7.31 (m, 24H), 7.23 (m, 12H), 4.87 (s, 24H), 3.05 (m, 16H), 1.57 (m, 16H), 1.32 (m, 16H), 0.94 (t, 24H, J = 7.5 Hz). 13C NMR (125 MHz, CD3CN): δ 165.50, 146.93, 130.48, 128.74, 127.70, 125.39, 121.95, 58.19, 51.92, 23.17, 19.20 and 12.74. 11B NMR (96 MHz, CD3CN): δ −17.00. HRMS (m/z): Calcd. for C120H96B12N36O24 (M2−) 1278.42575. Found: 1278.4110.
Supplementary Material
Footnotes
This research was funded by National Cancer Institute-NIH grant no. R21 CA114090
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
References
- [1].Hawthorne MF. Angew. Chem. 1993;105:997–1033. [Google Scholar]; Angew. Chem. Int. Ed. Engl. 1993;32:950–984. [Google Scholar]; (b) Soloway AH, Tjarks W, Barnum BA, Rong F-G, Barth RF, Codogni IM, Wilson JG. Chem. Rev. 1998;98:1515–1562. doi: 10.1021/cr980493e. [DOI] [PubMed] [Google Scholar]
- [2].Julius RL, Farha OK, Chiang J, Perry LJ, Hawthorne MF. Proc. Natl. Acad. Sci. USA. 2007;104:4808–4813. doi: 10.1073/pnas.0700316104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Reed CA. Acc. Chem. Res. 1998;31:133–139. [Google Scholar]
- [4] (a).Hawthorne MF, Dunks GB. Science. 1972;178:462–471. doi: 10.1126/science.178.4060.462. [DOI] [PubMed] [Google Scholar]; (b) Hawthorne MF, Zink JI, Skelton JM, Bayer MJ, Liu C, Livshits E, Baer R, Neuhauser D. Science. 2004;303:1849–1851. doi: 10.1126/science.1093846. [DOI] [PubMed] [Google Scholar]
- [5] (a).Hawthorne MF, Maderna A. Chem. Rev. 1999;99:3421–3434. doi: 10.1021/cr000001+. [DOI] [PubMed] [Google Scholar]; (b) Hawthorne MF, Farha OK, Julius R, Ma L, Jalisatgi SS, Li T, Bayer MJ. (in ACS Symposium Series).Modern Aspects of Main Group Chemistry. 2006;917:312–324. [Google Scholar]
- [6] (a).Pitochelli AR, Hawthorne MF. J. Am. Chem. Soc. 1960;82:3228–3229. [Google Scholar]; (b) Wunderlich JA, Lipscomb WN. J. Am. Chem. Soc. 1960;82:4427–4428. [Google Scholar]; (c) Moore JEB, Lohr JLL, Lipscomb WN. J. Chem. Phys. 1961;35:1329–1334. [Google Scholar]; (d) Miller HC, Miller NE, Muetterties EL. J. Am. Chem. Soc. 1963;85:3885–3886. [Google Scholar]
- [7] (a).Knoth WH, Sauer JC, England DC, Hertler WR, Muetterties EL. J. Am. Chem. Soc. 1964;86:3973–3983. [Google Scholar]; (b) Bayer MJ, Hawthorne MF. Inorg. Chem. 2004;43:2018–2020. doi: 10.1021/ic030289w. [DOI] [PubMed] [Google Scholar]; (c) Peymann T, Knobler CB, Hawthorne MF. Inorg. Chem. 2000;39:1163–1170. doi: 10.1021/ic991105+. [DOI] [PubMed] [Google Scholar]
- [8].Tolpin EI, Wellum GR, Berley SA. Inorg. Chem. 1978;17:2867–2873. [Google Scholar]
- [9].Gabel D, Moller D, Harfst S, Roesler J, Ketz H. Inorg. Chem. 1993;32:2276–2278. [Google Scholar]
- [10] (a).Knoth WH, Miller HC, Sauer JC, Balthis JH, Chia YT, Muetterties EL. Inorg. Chem. 1964;3:159–167. [Google Scholar]; (b) Srebny HG, Preetz W. Anorg. Allg. Chem. 1984;513:7–14. [Google Scholar]; (c) Srebny HG, Preetz W, Marsmann H. Z. Naturfosch. 1984;39B:189–196. [Google Scholar]
- [11] (a).Miller HC, Miller NE, Muetterties EL. Inorg. Chem. 1964;3:1456–1463. [Google Scholar]; (b) Peymann T, Lork E, Schmidt M, Nöth H, Gabel D. Chem. Ber. 1997;130:795–799. [Google Scholar]
- [12].Peymann T, Knobler CB, Hawthorne MF. Inorg. Chem. 1998;37:1544–1548. [Google Scholar]
- [13] (a).Peymann T, Herzog A, Knobler CB, Hawthorne MF. Angew. Chem. 1999;111:1129–1132. doi: 10.1002/(SICI)1521-3773(19990419)38:8<1061::AID-ANIE1061>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. Engl. 1999;38:1062–1064. [Google Scholar]; (b) Peymann T, Knobler CB, Khan SI, Hawthorne MF. J. Am. Chem. Soc. 2001;123:2182–2185. doi: 10.1021/ja0014887. [DOI] [PubMed] [Google Scholar]
- [14] (a).Maderna A, Knobler CB, Hawthorne MF. Angew. Chem. Angew. Chem. Int. Ed. Engl. 2001;2001;11340:1709–1712. 1662–1664. [Google Scholar]; (c) Li T, Jalisatgi SS, Bayer MJ, Maderna A, Khan SI, Hawthorne MF. J. Am. Chem. Soc. 2005;127:17832–17841. doi: 10.1021/ja055226m. [DOI] [PubMed] [Google Scholar]
- [15] (a).Farha OK, Julius RL, Lee MW, Huertas RE, Knobler CB, Hawthorne MF. J. Am. Chem. Soc. 2005;127:18243–18251. doi: 10.1021/ja0556373. [DOI] [PubMed] [Google Scholar]; (b) Lee MW, Farha OK, Hawthorne MF, Hansch C. Angew. Chem. 2007;119:3078–3082. doi: 10.1002/anie.200605126. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2007;46:3018–3022. doi: 10.1002/anie.200605126. [DOI] [PubMed] [Google Scholar]
- [16].Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. Angew. Chem. 2002;113:1713–1715. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2002;41:2596–2599. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.