

Biogenic amine-binding proteins mediate the anti-inflammatory and antihemostatic activities of blood-feeding insect saliva. The structure of the amine-binding protein from R. prolixus reveals the interaction of biogenic amine ligands with the protein.
Keywords: lipocalins, Rhodnius prolixus, Triatominae, serotonin, norepinephrine, tryptamine, nitrophorin
Abstract
Proteins that bind small-molecule mediators of inflammation and hemostasis are essential for blood-feeding by arthropod vectors of infectious disease. In ticks and triatomine insects, the lipocalin protein family is greatly expanded and members have been shown to bind biogenic amines, eicosanoids and ADP. These compounds are potent mediators of platelet activation, inflammation and vascular tone. In this paper, the structure of the amine-binding protein (ABP) from Rhodnius prolixus, a vector of the trypanosome that causes Chagas disease, is described. ABP binds the biogenic amines serotonin and norepinephrine with high affinity. A complex with tryptamine shows the presence of a binding site for a single ligand molecule in the central cavity of the β-barrel structure. The cavity contains significant additional volume, suggesting that this protein may have evolved from the related nitrophorin proteins, which bind a much larger heme ligand in the central cavity.
1. Introduction
In the saliva of blood-feeding arthropods, members of the lipocalin protein family play important roles as anticoagulants, platelet-aggregation inhibitors, vasodilators and anti-inflammatory mediators (Andersen et al., 2003 ▶; Mans & Ribeiro, 2008 ▶; Mans et al., 2008 ▶; Paesen et al., 1999 ▶; Ribeiro & Walker, 1994 ▶). Structurally, lipocalins are composed of an eight-stranded antiparallel β-barrel structure surrounding a central pocket that serves as a site for binding biologically active small-molecule ligands (Andersen et al., 2003 ▶; Assumpção et al., 2010 ▶; Flower, 1996 ▶; Francischetti et al., 2000 ▶; Skerra, 2008 ▶). In the saliva of the kissing bug Rhodnius prolixus, which is a vector of Trypanosoma cruzi, the lipocalin family has been greatly expanded through a process of gene duplication (Ribeiro et al., 2004 ▶). Related species in the genera Triatoma and Dipetalogaster also show an expansion of lipocalin genes, with over 50 distinct forms being present in salivary proteomes (Assumpção et al., 2008 ▶, 2011 ▶).
The functional diversification of lipocalins is made possible by the tolerance of the structure to amino-acid substitution, particularly in the binding-pocket region. This property has been taken advantage of in the development of ‘anticalin’ molecules, where artificial selection of mutant forms has been employed to generate novel binding properties with potential pharmaceutical uses (Skerra, 2008 ▶). In the saliva of R. prolixus and its relatives, the process of natural selection has generated lipocalins that bind heme, ADP, biogenic amines and thromboxane A2 (Andersen et al., 2003 ▶; Andersen & Montfort, 2000 ▶; Assumpção et al., 2010 ▶; Francischetti et al., 2000 ▶). Some lipocalins (and lipocalin-like proteins) have also evolved the ability to participate in surface interactions with important host plasma proteins such as coagulation factor IX(a) and thrombin (Isawa et al., 2000 ▶; Noeske-Jungblut et al., 1995 ▶; Ribeiro et al., 1995 ▶).
In R. prolixus, a group of heme-binding lipocalins given the name nitrophorins (NPs) serve multiple functions as vasodilators, platelet-aggregation inhibitors, antihistamines and inhibitors of coagulation (Montfort et al., 2000 ▶; Ribeiro et al., 1993 ▶, 1995 ▶; Ribeiro & Walker, 1994 ▶). The central pocket of the NP β-barrel binds a single molecule of heme that is tethered to the protein via a histidine residue on the proximal side of the ligand (Andersen et al., 1998 ▶; Roberts et al., 2001 ▶; Weichsel et al., 1998 ▶). Additionally, the distal pocket is lined with hydrophobic and polar residues that provide a stabilizing environment for a nitric oxide (NO) molecule coordinated to the Fe3+ atom of the heme (Roberts et al., 2001 ▶; Weichsel et al., 2000 ▶). Distortions in the normally planar heme conformation that occur as a result of steric interactions in the binding pocket and the effect of nearby ionizable amino-acid side chains act to stabilize the ferric form of the protein (Berry et al., 2009 ▶; Ding et al., 1999 ▶; Roberts et al., 2001 ▶; Shokhireva et al., 2003 ▶). In this state NO binding is readily reversible, facilitating its release into the host blood as a result of dilution, where it acts as a vasodilator and platelet-aggregation inhibitor (Andersen et al., 2000 ▶; Ribeiro et al., 1993 ▶). The NO complex is also stabilized by the low pH (5.5) of the saliva, but the structural reasons for this are not clear (Ribeiro & Walker, 1994 ▶; Soares et al., 2006 ▶). After NO has been released, histamine can bind in its place, giving the NP molecule a secondary anti-inflammatory function (Ribeiro & Walker, 1994 ▶). One member of the group, NP2, has also gained the ability to bind coagulation factor IX and its activated form, factor IXa, and acts to prevent the clotting of blood during feeding. This activity is attributable to surface features of the protein and is not dependent on heme or NO.
A second group of Rhodnius salivary lipocalin genes identified in a cDNA library show a relatively close sequence relationship to the NPs, but the amino-acid position corresponding to the proximal histidine residue is occupied by asparagine (Andersen et al., 2003 ▶). The recombinant protein does not bind heme, but rather binds the biogenic amines serotonin, norepinephrine and epinephrine with high affinity. The protein was shown to act as an inhibitor of biogenic amine-mediated platelet activation and smooth-muscle contraction and was given the name amine-binding protein (ABP).
In this study, we have determined the three-dimensional structure of ABP in the presence and absence of ligands. This allows us to compare the binding-pocket structure of ABP with that of the NPs and to examine the evolution of multiple novel ligand-binding surfaces in two functionally different proteins that are clearly derived from a common ancestor within a single lineage.
2. Materials and methods
2.1. Protein production
A cDNA derived from the full-length ABP sequence (GenBank AAO25746.1) but lacking a signal-peptide sequence (residues 1–21) was cloned into the expression vector pET17b to produce the expression construct described previously (Andersen et al., 2003 ▶). Briefly, PCR mutagenesis was used to remove the signal sequence from the cDNA, replacing it with an initiator methionine codon directly N-terminal to the mature coding sequence of the protein. The 5′ mutagenic primer CGCACCATATGGCATCTGGTTGTTCTACTGTGGATACTG was also used to insert an NdeI restriction site at the position of the start codon that was needed to clone the sequence into the expression vector. The protein was produced as inclusion bodies in Escherichia coli strain BL21 (DE3) pLysS in a manner similar to that described previously (Andersen et al., 2003 ▶). Edman degradation of the refolded protein indicated that the N-terminal methionine residue was largely removed post-translationally. Mutagenesis of ABP was also performed to insert two methionine residues into the structure to facilitate the production of a selenomethionine derivative. The ABP cDNA was mutated using a PCR-based mutagenesis method to replace Ile25 and Leu134 with methionine. The selenomethionine derivative of the I25M/L134M mutant was produced in the E. coli methionine auxotroph B834 (DE3) pLysS using SelenoMet medium (Molecular Dimensions) and was prepared from inclusion bodies as described above.
2.2. Isothermal titration calorimetry
Isothermal titration calorimetry (ITC) was carried out using a VP-ITC calorimeter (Microcal) at 303 K using 10 µl injections. For the tryptamine-binding experiments with ABP, the protein was dialyzed against 20 mM Tris–HCl pH 7.4, 0.15 M NaCl; after dialysis, the ligand was dissolved in the same buffer. All samples were degassed prior to use. After conversion of the heats of injection to enthalpies, the data were fitted to a single-site binding model using the Microcal Origin software package.
2.3. Crystallization and data collection
Both wild-type ABP and the selenomethionine derivative of the I25M/L134M double mutant were crystallized from 28–30% PEG 6000 in 0.1 M Tris–HCl pH 8.2. Data collection was performed on beamlines 22-BM and 22-ID at the Southeast Regional Collaborative Access Team (SER-CAT) and 19-ID of the Structural Biology Center (SBC), Advanced Photon Source (APS), Argonne National Laboratory. The selenomethionine-derivative crystal of the I25M/L134M double mutant diffracted to 3.0 Å resolution in space group P21, with eight molecules in the asymmetric unit (Table 1 ▶). The wild-type protein crystallized under the same crystallization conditions, but in a different P21 crystal form. In this case, the crystal diffracted to 2.3 Å resolution with four monomers in the asymmetric unit (Table 1 ▶). The wild-type protein was also crystallized in the presence of 1.0 mM tryptamine under the same conditions as in the absence of ligands and gave crystals that were isomorphous with the ligand-free wild-type form (Table 1 ▶). The complex crystallized similarly to the wild-type protein in space group P21, with four molecules in the asymmetric unit. The crystals were cooled for data collection using a cryoprotectant solution consisting of the crystallization solution containing 10% glycerol.
Table 1. Phasing and refinement statistics for ABP, the selenomethionine derivative of its mutant and its tryptamine complex.
Values in parentheses are for the highest resolution shell.
| Selenomethionine (peak) | Selenomethionine (inflection) | Ligand-free | Tryptamine complex | |
|---|---|---|---|---|
| PDB code | 4hfo | 4get | 4ge1 | |
| Resolution (Å) | 50.0–3.00 | 50.0–3.00 | 32.9–2.24 | 25.3–2.15 |
| Beamline | 19-ID | 19-ID | 22-BM | 22-ID |
| Wavelength (Å) | 0.97910 | 0.97926 | 1.0000 | 0.92309 |
| Completeness (%) | 100 (100) | 99.2 (96.0) | 98.9 (90.6) | 98.7 (90.3) |
| Average multiplicity | 8.2 (8.4) | 8.1 (7.2) | 5.6 (4.6) | 5.2 (4.6) |
| R merge † (%) | 10.7 (41.6) | 13.1 (61.8) | 8.1 (36.3) | 7.6 (61.5) |
| 〈I/σ(I)〉 | 8.7 (6.2) | 7.3 (3.6) | 9.1 (3.0) | 11.3 (2.2) |
| Observed reflections | 253864 | 251429 | 206343 | 226752 |
| Unique reflections | 29175 | 28761 | 37046 | 43654 |
| Space group | P21 | P21 | P21 | P21 |
| Unit-cell parameters | ||||
| a (Å) | 102.24 | 102.43 | 68.73 | 69.25 |
| b (Å) | 70.71 | 70.80 | 72.33 | 73.09 |
| c (Å) | 108.42 | 108.46 | 79.49 | 80.93 |
| α = γ (°) | 90 | 90 | 90 | 90 |
| β (°) | 99.15 | 99.09 | 90.04 | 91.46 |
| Phasing statistics | ||||
| No. of sites | 14 | |||
| FOM (RESOLVE) | 0.61 | |||
| R.m.s. deviations | ||||
| Bond lengths (Å) | 0.005 | 0.004 | 0.005 | |
| Bond angles (°) | 0.912 | 0.83 | 0.89 | |
| Ramachandran plot (MolProbity ‡) | ||||
| Favored (%) | 97.1 | 97.1 | 97.4 | |
| Allowed (%) | 99.9 | 99.9 | 99.9 | |
| MolProbity ‡ clash score (%) | 15.04 | 14.06 | 10.99 | |
| MolProbity ‡ rotamer outliers (%) | 1.6 | 1.6 | 1.8 | |
| Wilson temperature factor (Å2) | 54.4 | 30.4 | 40.4 | |
| R cryst/R free § (%) | 32.1/34.9 | 18.5/25.4 | 19.2/24.9 | |
R
merge = 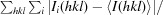
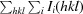 , where 〈I(hkl)〉 is the mean intensity of all symmetry-related reflections I
i(hkl).
, where 〈I(hkl)〉 is the mean intensity of all symmetry-related reflections I
i(hkl).
Chen et al. (2010 ▶).
R
cryst = 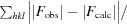
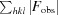 . R
free is calculated as for R
cryst but using a 5% random subset of the data for calculation.
. R
free is calculated as for R
cryst but using a 5% random subset of the data for calculation.
2.4. Structure solution and refinement
The structure of ABP was determined by multiple anomalous diffraction (MAD) methods using the selenomethionine derivative. Data collected at the selenium peak and inflection wavelengths were indexed, integrated and scaled using HKL-3000 (Minor et al., 2006 ▶). Selenium sites were located with SHELXD (Sheldrick, 2008 ▶) implemented in HKL-3000 and phasing/density modification was performed with SHELXE (Sheldrick, 2008 ▶), MLPHARE (Otwinowski, 1991 ▶), DM (Cowtan & Main, 1998 ▶) and RESOLVE (Terwilliger, 2003 ▶), also implemented in HKL-3000. The initial experimental map allowed the tracing of some of the β-strand regions, but no side chains were visible. Using the structure of NP2 as a guide, much of the peptide backbone of the β-barrel structure of a single molecule could be traced using Coot (Emsley & Cowtan, 2004 ▶). The coordinates of this single molecule were then used as a molecular-replacement search model in Phaser (McCoy et al., 2007 ▶), resulting in the positioning of all eight monomers. Noncrystallographic symmetry averaging of the experimental map in RESOLVE using the symmetry operators generated from the molecular-replacement solution significantly improved the quality of the experimental map. Backbone traces could then be completed for all eight molecules in the asymmetric unit.
After obtaining a high-quality data set from a crystal of the wild-type protein, a single-molecule model from the selenomethionine-derivative solution was used as a search model in Phaser. All four molecules of the asymmetric unit of the P21 unit cell were located in this manner. Side chains were added to the model, numerous cycles of rebuilding and refinement using Coot and REFMAC5 (Winn et al., 2011 ▶; Murshudov et al., 2011 ▶) were performed and NCS restraints were applied. As the model neared completion, refinement was carried out using phenix.refine (Afonine et al., 2012 ▶) without NCS restraints and with a TLS model applied. Validation calculations for the refined structures were performed using the MolProbity web server (Chen et al., 2010 ▶). The refined model of the wild-type structure was then used to generate the octomeric contents of the mutant selenomethionine-derivative asymmetric unit and this model was refined using phenix.refine. The structures of the ligand-free wild-type protein, its tryptamine complex and the mutant selenomethionine-derivative protein have been deposited in the RCSB Protein Data Bank as entries 4get, 4ge1 and 4hfo, respectively.
3. Results and discussion
3.1. Structure of ABP
The structure of ABP was determined by multiple anomalous diffraction methods using a selenomethionine derivative crystallizing in space group P21 with eight molecules in the asymmetric unit (Table 1 ▶). Since the ABP sequence contains no internal methionine residues, two methionine codons were introduced by site-directed mutagenesis of Ile25 and Leu134. Subsequently, the structure of the wild-type protein was determined by molecular replacement using a single-molecule model from the selenomethionine-derivative mutant protein structure. The wild-type ligand-free and tryptamine-bound proteins crystallized in a second P21 crystal form with four molecules in the asymmetric unit (Table 1 ▶). Serotonin (Fig. 1 ▶ a), rather than tryptamine, is a presumed natural ligand for ABP, but we found that it oxidized in the crystallization drops, resulting in discolored crystals that exhibited poor diffraction properties. Because tryptamine lacks a phenolic hydroxyl group, it did not oxidize and was suitable as a replacement for serotonin. Tryptamine binding was measured using ITC, yielding a dissociation constant (K d) of 370 nM, compared with the published value of 102 nM for serotonin (Fig. 1 ▶ b; Andersen et al., 2003 ▶).
Figure 1.

Structures of ligands of R. prolixus lipocalins and the results of ITC analysis of tryptamine binding to ABP. (a) Structures of ligands known to bind to ABP. (b) Results of ITC analysis of tryptamine binding to ABP. Top, measured heats as a function of time. Bottom, individual binding enthalpies calculated by integration of raw data as a function of ligand:protein molar ratio. Squares represent the enthalpy per injection, while the solid line represents the result of fitting to a single-site binding model. Parameters: ΔH = −12.0 ± 0.2 kcal mol−1, K assoc = (2.7 ± 0.3) × 106 M −1, TΔS = −3.06 kcal mol−1, N = 1.12 ± 0.01 molecules ligand bound per protein molecule. 1 cal = 4.186 J.
ABP forms an eight-stranded antiparallel β-barrel that is characteristic of the lipocalin protein family and similar in structure to the NPs (Fig. 2 ▶ a). The molecule is stabilized by two disulfide bonds that occupy positions equivalent to those found in the NPs (Figs. 2 ▶ a and 2 ▶ b). One disulfide bond links Cys4 in the N-terminal coil region of the protein with Cys125 in β-strand H of the β-barrel, while the second disulfide bond links Cys42 in β-strand B with Cys176 in the C-terminal coil (Figs. 2 ▶ a and 2 ▶ b). An α-helix (α1) typical of the lipocalin family lies against the side of the β-barrel in the region immediately C-terminal to β-strand H, and a second α-helix (α2) is present in the region of the C-terminal coil (Fig. 2 ▶ a). The end of the β-barrel that serves as the entry point to the ligand-binding pocket is surrounded by a series of flexible loops connecting β-strands A–B, C–D, E–F and G–H. As in the NPs, the ω-loop A–B is larger than the others. The N-terminal coil region passes over the opposite end of the barrel and closes it to the solvent (Fig. 2 ▶ a). Superposition of ABP with models of NPs produced r.m.s.d.s of 2.08 Å (138 Cα positions) for NP2 and 2.38 Å (121 Cα positions) for NP4.
Figure 2.

The structure of ABP. (a) Ribbon representation of the ABP structure. The component β-strands of the β-barrel are labelled A–H and the loops surrounding the binding pocket are labelled A–B to G–H. The two helices occurring ‘downstream’ of the β-barrel are labelled α1 and α2. The positions of the two disulfide bonds are shown as stick diagrams and the positions of the C- and N-termini are indicated. The right-hand figure was obtained by rotation of the left-hand figure around the axis shown. (b) Amino-acid sequence alignment of the ABP-coding sequence produced as recombinant protein with the related proteins NP2 and NP4. The N-terminal methionine is largely removed post-translationally, giving the mature polypeptide shown in the alignment. Amino-acid identities are marked in red. The boundaries of the important secondary-structure elements from Fig. 2 ▶(a) are marked below the alignment. The numbering at the right end of each sequence line indicates the amino-acid position for that sequence. The numbering above the sequences marks every ten positions of the alignment.
3.2. Relationship between the wild-type and the selenomethionine-derivative mutant crystal forms
The P21 unit cell of the selenomethionine-derivative mutant crystal is approximately twice the volume of the wild-type native unit cell and contains eight molecules in the asymmetric unit (Table 1 ▶). The two unit cells share a common b edge and the larger cell is related to the smaller by the transformation a′ ≃ a + c, c′ ≃ a − c. The octomer in the asymmetric unit of the large unit cell is composed of two tetramers related by noncrystallographic twofold symmetry, with each tetramer being equivalent to the asymmetric unit contents of the small unit cell (Fig. 3 ▶). In this arrangement a pseudo-translation vector ½a′ + ½c′ ≃ a is present, producing a large native Patterson peak at u = ½, v = 0, w = ½. The packing of protein molecules in the two crystal forms is essentially identical, with an r.m.s. deviation of 1.36 Å for the selenomethionine-derivative mutant protein octomer superimposed with a model consisting of two symmetry-related tetramers from the wild-type native structure (Fig. 3 ▶).
Figure 3.

Crystal packing in the wild-type and the selenomethionine-derivative mutant structures reveals a nearly identical molecular arrangement. (a) The tetrameric contents of the asymmetric unit of the wild-type native crystals are shown as a red ribbon diagram, with a tetramer related by rotational symmetry shown in blue. (b) The octameric contents of the selenomethionine-derivative mutant asymmetric unit are shown in red.
3.3. Binding pocket of ABP and structure of the tryptamine complex
The central binding pocket of ABP is large despite the small size of its ligand. In the absence of ligands, the pocket volume was calculated to be 784 Å3 using CASTp (Dundas et al., 2006 ▶). A mixture of polar and hydrophobic side chains line the interior of the pocket, with the backbone atoms making little contribution to the binding surface (Fig. 4 ▶). A single well ordered ligand molecule is bound in the tryptamine complex at a site located towards the side of the main cavity. In the presence of ligand, the pocket contains substantial unoccupied space which is accessible to solvent (Figs. 4 ▶ a and 4 ▶ d). Although some nonprotein electron density is present in this part of the pocket, there is no indication that a second tryptamine molecule is bound. Nevertheless, the possibility of simultaneous binding of other biologically relevant ligands has not been ruled out. The observed binding stoichiometry in the complex was corroborated by thermodynamic analysis of tryptamine binding using ITC, which clearly indicated that a single molecule of ligand is bound per molecule of APB (Fig. 1 ▶ b).
Figure 4.

The biogenic amine-binding pocket of ABP. (a) Stick diagram showing the side-chain structure of the binding pocket with the tryptamine ligand bound. Protein C atoms are shown in white, O atoms in red and N atoms in blue. Ligand C atoms are shown in green and N atoms in blue. Water molecules are shown as red spheres and hydrogen bonds are shown as red dashed lines. (b) Stick diagram showing the hydrogen-bonding network that acts to stabilize the aliphatic amino group of tryptamine. The color scheme is the same as in (a). (c) Left, space-filling diagram of cation–π and π–π interactions involved in ligand binding. Right, the same view as a stick diagram with electron density shown (2mF o − DF c contoured at 1.0σ). (d) Space-filling model showing tryptamine bound in a portion of the ABP central cavity. Protein atoms are colored as in (a), while tryptamine is colored yellow.
A number of hydrophobic amino-acid side chains surround the indole nucleus of the bound tryptamine ligand, including those of Phe45, Phe59, Leu107, Val109, Val124, Val122 and Leu134 (Fig. 3 ▶ a). A hydrogen bond is also formed between the indole N atom of the ligand and the side chain of Gln138 (Fig. 4 ▶ a). Tryptamine lacks the C-5 hydroxyl group that is present in serotonin, but the side chain of Ser74 is positioned such that it could form a hydrogen bond to the C-5 hydroxyl group of a similarly placed serotonin molecule (Fig. 4 ▶ a). A model hydroxyl group placed at C-5 of tryptamine lies at a distance of 3.4 Å from the hydroxyl of Ser74 in the tryptamine complex structure. The higher binding affinity for serotonin (Andersen et al., 2003 ▶) suggests that interactions with the C-5 hydroxyl add significantly to the stability of that complex.
The ammonium group of tryptamine interacts extensively with binding-pocket amino-acid residues in ABP. It is stabilized by hydrogen-bond and salt-bridge interactions with the side chain of Asp72, as well as with the side chain of Asn61 through an intervening water molecule (Fig. 4 ▶ a). The side chains of Thr43, Asn61, Asp72, Asn92 and Arg105 are all directed towards the ligand and participate in a hydrogen-bonding network that positions Asp72 and Asn61 for interaction with the ligand (Fig. 4 ▶ b). In addition to electrostatic and hydrogen-bonding interactions, the amino group of tryptamine participates in a cation–π interaction with the aromatic ring of Phe59 (Figs. 4 ▶ a and 4 ▶ c; Gallivan & Dougherty, 1999 ▶). The amino N atom is positioned 3.1 Å from the ring centroid of Phe59 and the dihedral angle of the interaction deviates from perpendicular to the ring by approximately 4°. Binding of the amino group of biogenic amine ligands by a mixture of cation–π and hydrogen-bonding interactions is also a feature of serotonin- and histamine-binding proteins from the saliva of hard and soft ticks (Mans et al., 2008 ▶). These proteins have evolved independently in these two highly divergent groups, making this an interesting example of convergent evolution.
Comparison of the ligand-free structure of ABP with the tryptamine complex shows that no large conformational changes are induced as a result of binding. However, in the four molecules making up the asymmetric unit of both structures Tyr132 (in loop G–H) is disordered to varying degrees, with the side chain appearing to face predominantly outwards towards the solvent in some molecules and inwards towards the bound ligand in others. Based on comparison of the wild-type and tryptamine-complex structures, this disorder does not appear to influence tryptamine binding.
3.4. Similarities in the binding-pocket structures of NPs and ABP
The large size and similarity of the binding-pocket shape in ABP and the NPs suggests that both are derived from an ancestral lipocalin form having a large binding pocket. One possibility would be that the ABP has evolved directly from a heme-binding NP progenitor. Amino-acid sequence identities between ABP and NPs calculated from a multiple sequence alignment are 29% for NP2 and 27% for NP4 (Fig. 2 ▶ b). NP1, NP3 and NP7 also show similar degrees of similarity (data not shown). The ABP-binding pocket is roughly similar in volume to those of the NPs and contains considerable unoccupied space in the tryptamine complex (Figs. 4 ▶ d, 5 ▶ a and 5 ▶ b). The biogenic amine-binding site of ABP is not the result of a major collapse or filling of an NP-like pocket to create a site corresponding in size to the small biogenic amine ligand. Rather, a binding site for the biogenic amine is formed by modifying part of the cavity, while the remaining volume is left unoccupied by the ligand. It may be that this extra space is important for accommodating other known ABP binding partners, particularly the catecholamine norepinephrine, which contains a dihydroxyphenyl moiety in place of the indole and also contains a hydroxyl group at the β-carbon of the alkylamine chain (Fig. 1 ▶ a). However, previous studies have shown that in the presence of a saturating concentration of serotonin, binding of norepinephrine is abrogated, indicating that the sites for serotonin and the catecholamines are at least partially coincident (Andersen et al., 2003 ▶).
Figure 5.

Comparison of the structures of ABP and NP2. (a) Ribbon diagram of ABP showing the molecular surface of the central cavity in magenta. The bound tryptamine is shown in blue. The cavity surface was calculated using CASTp (Dundas et al., 2006 ▶). (b) Ribbon diagram of NP2 (PDB entry 1euo; Andersen & Montfort, 2000 ▶) showing the molecular surface of the central cavity in magenta. The bound heme is shown in red. (c) Stereoview of the superimposed binding-pocket regions of ABP and NP2. For ABP, C atoms are colored white, O atoms red and N atoms blue. For NP2, C atoms are colored cyan, O atoms red and N atoms blue. Tryptamine of ABP is colored yellow, with its N atoms blue, and heme from NP2 is colored red, with its N atoms blue.
The heme moiety of the NPs is held in place by the proximal histidine ligand as well as a large number of interactions with aliphatic and aromatic amino-acid side chains (Roberts et al., 2001 ▶; Weichsel et al., 1998 ▶). In ABP, a number of structurally equivalent residues also play a major role in ligand binding, with most of the notable interactions being performed by positions equivalent to those in the proximal pocket of the NPs. His57 is the proximal ligand for the ferric iron of NP2 and is located on β-strand C (Fig. 5 ▶ c). This residue is stabilized by hydrogen bonding to Asn68 through an intervening water molecule. His57 has been substituted by Asn61 in ABP, while Asn68 of NP2 has been substituted by Asp72 (Fig. 5 ▶ c). These residues make up part of the hydrogen-bonding network described above that stabilizes the amino group of tryptamine (Fig. 4 ▶ b).
In NP2, three aromatic residues, Phe27, Phe42 and Tyr85, are situated such that their faces make contact with the porphyrin edge at pyrrole rings A, B and C, respectively, and appear to play a role in positioning the heme moiety in the pocket (Fig. 5 ▶ c). Tyr104 also faces the porphyrin in a similar manner and is situated between pyrrole rings B and C, while the side chain of Phe66 lies close to the γ-meso C atom in the proximal pocket (Fig. 5 ▶ c). The phenolic ring of Tyr38 also lies on the proximal side of the porphyrin and has recently been shown to form a radical species in an efficient peroxidase reaction performed by NP2 (Singh et al., 2010 ▶). It is notable that norepinephrine is efficiently oxidized as substrate in this reaction. It may be that the NPs and ABP act together as an enzyme/scavenger system for elimination of norepinephrine during blood feeding.
In addition to the aromatic residues of the NP2 binding pocket, the hydrophobic residues Ile120, Leu122 and Leu132 of NP2 contact the distal heme face near the γ-meso position and pyrrole ring A and clearly play a role in inducing the ruffled conformation of the porphyrin. An additional leucine residue, Leu55, contacts the heme at the β-meso C atom and near pyrrole ring B, and Val24 contacts the heme near the vinyl group of pyrrole ring A (Fig. 5 ▶ c).
Many of the heme-binding positions in NP2 have also taken on important roles in biogenic amine binding by ABP (Figs. 2 ▶ b and 5 ▶ c). Leu55 of NP2 is substituted by Phe59 in ABP, which interacts with the amino group through a cation–π interaction. Tyr85 has been substituted by Asn92 in ABP, which forms a hydrogen bond to Asp72, in turn stabilizing the amino group of the ligand. Phe42 in NP2 is conserved in ABP (Phe45) and participates in a stacking interaction with Phe59, while Phe27 of NP2 is also conserved as tyrosine in ABP (Tyr28) but does not interact significantly with bound tryptamine. Leu120, Leu132 and Leu106 from the distal pocket of NP2 show conservation in ABP as Val124, Leu134 and Val109 in ABP. All of these contact the indole portion of tryptamine. Tyr104 of NP2 is substituted by Leu107, which also acts to position the indole ring (Fig. 5 ▶ c).
4. Conclusions
In many cases, salivary proteins of blood-feeding arthropods are encoded by related multigene families that have arisen through rounds of tandem duplications over evolutionary time. The maintenance of these duplications has apparently allowed the acquisition of new functions by related protein forms. Here, we examine the structures of related ligand-binding members of the lipocalin family in R. prolixus that have diverged to give drastically different ligand-binding selectivities. We find that large changes in the overall architecture of the protein or in the dimensions of the binding pocket have not occurred. Rather, the identities of many of the key residues for binding heme in the NPs have been changed and play equally important roles in the binding of biogenic amines.
Supplementary Material
PDB reference: amine-binding protein, 4get
PDB reference: complex with tryptamine, 4ge1
PDB reference: I25M/L134M mutant, 4hfo
Acknowledgments
The authors thank D. Garboczi and A. Gittis for discussions and R. Hearn for technical assistance. We also thank the staffs of the Southeast Regional Collaborative Access Team and the Structural Biology Center, Advanced Photon Source, Argonne National Laboratory for assistance with X-ray data collection. Use of the Advanced Photon Source beamlines was supported by the US Department of Energy, Office of Science, Office of Basic Energy Sciences under contract No. W-31-109-Eng-38. This work was supported by the intramural research program of the NIAID, National Institutes of Health.
References
- Afonine, P. V., Grosse-Kunstleve, R. W., Echols, N., Headd, J. J., Moriarty, N. W., Mustyakimov, M., Terwilliger, T. C., Urzhumtsev, A., Zwart, P. H. & Adams, P. D. (2012). Acta Cryst. D68, 352–367. [DOI] [PMC free article] [PubMed]
- Andersen, J. F., Ding, X. D., Balfour, C., Shokhireva, T. K., Champagne, D. E., Walker, F. A. & Montfort, W. R. (2000). Biochemistry, 39, 10118–10131. [DOI] [PubMed]
- Andersen, J. F., Francischetti, I. M., Valenzuela, J. G., Schuck, P. & Ribeiro, J. M. (2003). J. Biol. Chem. 278, 4611–4617. [DOI] [PubMed]
- Andersen, J. F. & Montfort, W. R. (2000). J. Biol. Chem. 275, 30496–30503. [DOI] [PubMed]
- Andersen, J. F., Weichsel, A., Balfour, C. A., Champagne, D. E. & Montfort, W. R. (1998). Structure, 6, 1315–1327. [DOI] [PubMed]
- Assumpção, T. C., Alvarenga, P. H., Ribeiro, J. M., Andersen, J. F. & Francischetti, I. M. (2010). J. Biol. Chem. 285, 39001–39012. [DOI] [PMC free article] [PubMed]
- Assumpção, T. C., Charneau, S., Santiago, P. B., Francischetti, I. M., Meng, Z., Araújo, C. N., Pham, V. M., Queiroz, R. M., de Castro, C. N., Ricart, C. A., Santana, J. M. & Ribeiro, J. M. (2011). J. Proteome Res. 10, 669–679. [DOI] [PMC free article] [PubMed]
- Assumpção, T. C., Francischetti, I. M., Andersen, J. F., Schwarz, A., Santana, J. M. & Ribeiro, J. M. (2008). Insect Biochem. Mol. Biol. 38, 213–232. [DOI] [PMC free article] [PubMed]
- Berry, R. E., Shokhirev, M. N., Ho, A. Y., Yang, F., Shokhireva, T. K., Zhang, H., Weichsel, A., Montfort, W. R. & Walker, F. A. (2009). J. Am. Chem. Soc. 131, 2313–2327. [DOI] [PMC free article] [PubMed]
- Chen, V. B., Arendall, W. B., Headd, J. J., Keedy, D. A., Immormino, R. M., Kapral, G. J., Murray, L. W., Richardson, J. S. & Richardson, D. C. (2010). Acta Cryst. D66, 12–21. [DOI] [PMC free article] [PubMed]
- Cowtan, K. & Main, P. (1998). Acta Cryst. D54, 487–493. [DOI] [PubMed]
- Ding, X. D., Weichsel, A., Andersen, J. F., Shokhireva, T. K., Balfour, C., Pierik, A. J., Averill, B. A., Montfort, W. R. & Walker, F. A. (1999). J. Am. Chem. Soc. 121, 128–138.
- Dundas, J., Ouyang, Z., Tseng, J., Binkowski, A., Turpaz, Y. & Liang, J. (2006). Nucleic Acids Res. 34, W116–W118. [DOI] [PMC free article] [PubMed]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Flower, D. R. (1996). Biochem. J. 318, 1–14. [DOI] [PMC free article] [PubMed]
- Francischetti, I. M., Ribeiro, J. M., Champagne, D. & Andersen, J. (2000). J. Biol. Chem. 275, 12639–12650. [DOI] [PubMed]
- Gallivan, J. P. & Dougherty, D. A. (1999). Proc. Natl Acad. Sci. USA, 96, 9459–9464. [DOI] [PMC free article] [PubMed]
- Isawa, H., Yuda, M., Yoneda, K. & Chinzei, Y. (2000). J. Biol. Chem. 275, 6636–6641. [DOI] [PubMed]
- Mans, B. J. & Ribeiro, J. M. (2008). Insect Biochem. Mol. Biol. 38, 862–870. [DOI] [PMC free article] [PubMed]
- Mans, B. J., Ribeiro, J. M. & Andersen, J. F. (2008). J. Biol. Chem. 283, 18721–18733. [DOI] [PMC free article] [PubMed]
- McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C. & Read, R. J. (2007). J. Appl. Cryst. 40, 658–674. [DOI] [PMC free article] [PubMed]
- Minor, W., Cymborowski, M., Otwinowski, Z. & Chruszcz, M. (2006). Acta Cryst. D62, 859–866. [DOI] [PubMed]
- Montfort, W. R., Weichsel, A. & Andersen, J. F. (2000). Biochim. Biophys. Acta, 1482, 110–118. [DOI] [PubMed]
- Murshudov, G. N., Skubák, P., Lebedev, A. A., Pannu, N. S., Steiner, R. A., Nicholls, R. A., Winn, M. D., Long, F. & Vagin, A. A. (2011). Acta Cryst. D67, 355–367. [DOI] [PMC free article] [PubMed]
- Noeske-Jungblut, C., Haendler, B., Donner, P., Alagon, A., Possani, L. & Schleuning, W.-D. (1995). J. Biol. Chem. 270, 28629–28634. [DOI] [PubMed]
- Otwinowski, Z. (1991). Proceedings of the CCP4 Study Weekend. Isomorphous Replacement and Anomalous Scattering, edited by W. Wolf, P. R. Evans & A. G. W. Leslie, pp. 80–86. Warrington: Daresbury Laboratory.
- Paesen, G. C., Adams, P. L., Harlos, K., Nuttall, P. A. & Stuart, D. I. (1999). Mol. Cell, 3, 661–671. [DOI] [PubMed]
- Ribeiro, J. M., Andersen, J., Silva-Neto, M. A., Pham, V. M., Garfield, M. K. & Valenzuela, J. G. (2004). Insect Biochem. Mol. Biol. 34, 61–79. [DOI] [PubMed]
- Ribeiro, J. M., Hazzard, J. M., Nussenzveig, R. H., Champagne, D. E. & Walker, F. A. (1993). Science, 260, 539–541. [DOI] [PubMed]
- Ribeiro, J. M., Schneider, M. & Guimarães, J. A. (1995). Biochem. J. 308, 243–249. [DOI] [PMC free article] [PubMed]
- Ribeiro, J. M. & Walker, F. A. (1994). J. Exp. Med. 180, 2251–2257. [DOI] [PMC free article] [PubMed]
- Roberts, S. A., Weichsel, A., Qiu, Y., Shelnutt, J. A., Walker, F. A. & Montfort, W. R. (2001). Biochemistry, 40, 11327–11337. [DOI] [PubMed]
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Shokhireva, T. K, Berry, R. E., Uno, E., Balfour, C. A., Zhang, H. & Walker, F. A. (2003). Proc. Natl Acad. Sci. USA, 100, 3778–3783. [DOI] [PMC free article] [PubMed]
- Singh, R., Berry, R. E., Yang, F., Zhang, H., Walker, F. A. & Ivancich, A. (2010). Biochemistry, 49, 8857–8872. [DOI] [PubMed]
- Skerra, A. (2008). FEBS J. 275, 2677–2683. [DOI] [PubMed]
- Soares, A. C., Carvalho-Tavares, J., Gontijo, N. de F., dos Santos, V. C., Teixeira, M. M. & Pereira, M. H. (2006). J. Insect Physiol. 52, 468–472. [DOI] [PubMed]
- Terwilliger, T. C. (2003). Methods Enzymol. 374, 22–37. [DOI] [PubMed]
- Weichsel, A., Andersen, J. F., Champagne, D. E., Walker, F. A. & Montfort, W. R. (1998). Nature Struct. Biol. 5, 304–309. [DOI] [PubMed]
- Weichsel, A., Andersen, J. F., Roberts, S. A. & Montfort, W. R. (2000). Nature Struct. Biol. 7, 551–554. [DOI] [PubMed]
- Winn, M. D. et al. (2011). Acta Cryst. D67, 235–242.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: amine-binding protein, 4get
PDB reference: complex with tryptamine, 4ge1
PDB reference: I25M/L134M mutant, 4hfo