

Abstract
Background
Human ß-defensins are a family of antimicrobial peptides located at the mucosal surface. Both sequence multi-site variations (MSV) and copy-number variants (CNV) of the defensin-encoding genes are associated with increased risk for various diseases, including cancer and inflammatory conditions such as psoriasis and acute pancreatitis. In a case–control study, we investigated the association between MSV in DEFB104 as well as defensin gene (DEF) cluster copy number (CN), and pancreatic ductal adenocarcinoma (PDAC) and chronic pancreatitis (CP).
Results
Two groups of PDAC (N=70) and CP (N=60) patients were compared to matched healthy control groups CARLA1 (N=232) and CARLA2 (N=160), respectively. Four DEFB104 MSV were haplotyped by PCR, cloning and sequencing. DEF cluster CN was determined by multiplex ligation-dependent probe amplification.
Neither the PDAC nor the CP cohorts show significant differences in the DEFB104 haplotype distribution compared to the respective control groups CARLA1 and CARLA2, respectively.
The diploid DEF cluster CN exhibit a significantly different distribution between PDAC and CARLA1 (Fisher’s exact test P=0.027), but not between CP and CARLA2 (P=0.867).
Conclusion
Different DEF cluster b CN distribution between PDAC patients and healthy controls indicate a potential protective effect of higher CNs against the disease.
Keywords: Defensins, Single nucleotide variants, Copy number variation, Chronic pancreatitis, Pancreatic ductal adenocarcinoma
Background
Pancreatitis, a necroinflammatory condition of the pancreas, has both acute and chronic manifestations. In the recent past, our understanding of the pathogenesis of pancreatic inflammation has improved considerably. Whereas acute pancreatitis is known to be initiated by premature activation of digestive enzymes within the exocrine component of the pancreas, chronic pancreatitis (CP) is characterized by progressive and irreversible damage to both the exocrine and endocrine components of the pancreas. CP is believed to result from repeated overt or silent episodes of acute pancreatitis [1]. The key histopathologic features of CP are pancreatic fibrosis, acinar atrophy, chronic inflammation, and distorted and blocked ducts [2]. The annual incidence of CP in industrialized countries ranges from 3.5 to 10 per 100,000. Alcohol abuse is the major risk factor for CP in Western countries, although other mechanisms such as mutations, pancreatic duct obstruction (caused by strictures), hypertriglyceridemia, hypercalcemia, and autoimmunity also have been implicated [3]. Since patients with CP have an approximately 13-fold higher risk to develop pancreatic cancer than the general population, the identification of disease-related genes is essential for understanding the transformation from benign to malignant disorder, and for developing strategies for early diagnosis [4]. Pancreatic cancer (pancreatic ductal adenocarcinoma, PDAC), is the fourth most common cause of cancer-related death in industrialized countries and characterized by extremely low survival rates [5,6]. Currently, no imaging procedure can reliably differentiate between benign and malignant tumors in CP patients.
Defensins are small cysteine-rich peptides that can be classified as either α-defensin or β-defensin, depending upon the arrangement of six critical cysteine residues. Defensins are synthesized as inactive preproproteins that become post-translationally activated. They are produced in the respiratory, gastrointestinal and genitourinary tracts, the skin, and by circulating blood cells. Defensins are considered a first line of defence against invading pathogens. Of all defensins, the ß-defensins comprise the largest group, with around 40 members encoded in the human genome. Most of the genes are located in defensin (DEF) clusters on chromosomes 8 and 20. In addition to their antimicrobial activity, β-defensins have multifaceted functions in innate and adaptive immunity [7]. The β-defensins are expressed in most epithelial cells and are found to be impaired in many inflammatory diseases, including Crohn's disease, psoriasis, pulmonary inflammation, and periodontal disease [8-13].
Except for DEFB1, all ß-defensin genes (DEFB4, DEFB103109) harbour a high degree of copy-number variation (CNV). Copy numbers (CN) range from 2 to 13 copies per diploid genome and show large inter-genic concordance because the respective genes bunch in a ~200 kb CNV region, called ‘DEF cluster b’ (Figure 1) [14-20]. In principle, both CNV and sequence variation within a given copy can contribute to clinical phenotypes through variation in gene expression [21]. So far, this has only been demonstrated experimentally for DEFB4 but most probably applies to all other DEF genes with variable CN as well [17]. Anyhow, the analysis of genes located in CNV regions poses two methodological challenges: First, conventional genotyping of single nucleotide variations (SNV) cannot resolve whether a variation occurs between paralogs affected by CNV [22]. To acknowledge this problem, we will address SNVs within the copy number variable DEFB cluster as ‘multi-site variation’ (MSV). Second, the assessment of CNs is also complicated by methodological difficulties. Although widely used, qPCR has questionable reliability so that tightly controlled paralog ratio tests or multiplex ligation-dependent probe amplification (MLPA) have been recommended instead [23-28].
Figure 1.

Genomic organization of the human defensin (DEF) gene clusters on chromosome 8p23.1. DEF cluster a is essentially single-copy, except a CNV region encompassing DEFA1A3 and DEFT1p (2 to 13 copies per diploid genome), whereas DEF cluster b as a whole is CN-variable (2 to 12 copies). Black triangle (zoomed): DEFB104 gene with the four multi site variations (MSV) investigated in this study. A: DEFA, α-defensin; B: DEFB, ß-defensin; ex = exon, p = pseudogene.
Disease association studies of the CN-variable ß-defensin genes are scarce. Hitherto reported associations mostly either lack replication or are conflicting [21,29-32]. No CN association, but a skewed distribution of MSV haplotypes, was identified in two prostate cancer groups [33]. The corresponding haplotypes comprise four MSV (rs17843871, rs2680507, rs17843872 and rs4259430) around exon 1 of DEFB104. While haplotypes GGGC and CAAT were significantly under-represented among patients, GAAT and GAAC were significantly over-represented. Moreover, high CNs of the ß-defensin cluster (≥9 copies per diploid genome) were found to be less frequent among prostate cancer patients than among healthy controls.
The aim of the present study was to search for associations between MSV-based DEFB104 haplotypes and DEF cluster b CNs on the one hand, and pancreatic ductal adenocarcinoma and chronic pancreatitis on the other.
Results
DEFB104 haplotypes are not associated with PDAC or CP
Two independent cohorts of patients with pancreatic ductal adenocarcinoma (PDAC) and chronic pancreatitis (CP) were investigated in comparison with complementary age- and sex-matched healthy control groups [34] named CARLA1 and CARLA2, respectively (Table 1 and Methods section).
Table 1.
Description of study groups
| Group | PDAC | CARLA1 | CP | CARLA2 | |
|---|---|---|---|---|---|
| Number (%) |
male |
37 (53%) |
123 (53%) |
49 (82%) |
133 (83%) |
| |
female |
33 (47%) |
109 (47%) |
11 (18%) |
27 (17%) |
| |
total |
70 |
232 |
60 |
160 |
| Age (years) |
minimum |
41 |
46 |
36 |
45 |
| |
maximum |
77 |
77 |
74 |
74 |
| |
mean |
63.8 |
65.0 |
50.2 |
52.5 |
| median | 67.0 | 66.8 | 49.0 | 50.0 | |
PDAC: pancreatic ductal adenocarcinoma; CP: chronic pancreatitis; CARLA1, CARLA2: healthy control groups which were age- and sex-matched to PDAC and CP, respectively.
The haplotypes of four exon 1 MSV in DEFB104 (Figure 1) were determined by PCR on the genomic DNAs from the four cohorts as well as from a commercially available pool of ~100 anonymous human DNAs. The PCR products were pooled by cohorts in equimolar amounts and cloned. Subsequently, clones were sequenced, haplotypes were inferred from the sequence traces and the haplotype fractions within cohorts were calculated (Table 2). Since these fractions do not take into account the effects of post-PCR pooling, however, they cannot be compared directly between patients and controls using standard statistical tests. Instead, the expected haplotype distribution under the null hypothesis had to be simulated as previously described [33]. An omnibus χ2 test based upon these simulations yielded a p value of 0.239 for the comparison of PDAC and CARLA1, and of 0.129 for CP and CARLA2, respectively, suggesting that there were no significant differences between the DEFB104 haplotype distributions among cases and controls.
Table 2.
Haplotype frequencies (fh) as derived for DEFB104 MSV rs17843871, rs2680507, rs17843872 and rs4259430 by pooled PCR/cloning and Sanger sequencing
|
Haplotype |
Frequency as determined by pooled PCR/sequencing |
Group comparison |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
PDAC |
CARLA1 |
CP |
CARLA2 |
Human genomic pool |
PDAC vs CARLA1 |
CP vs CARLA2 |
|||||
| No. clones | fh | No. clones | fh | No. clones | fh | No. clones | fh | No.clones | fh | P* | P* | |
| GAGC |
224 |
0.40 |
286 |
0.34 |
310 |
0.41 |
391 |
0.34 |
413 |
0.38 |
0.160 |
0.061 |
| GGGC |
152 |
0.27 |
207 |
0.25 |
200 |
0.27 |
277 |
0.24 |
233 |
0.21 |
0.540 |
0.444 |
| GAAT |
39 |
0.07 |
92 |
0.11 |
92 |
0.12 |
153 |
0.13 |
106 |
0.10 |
0.109 |
0.747 |
| CAGT |
60 |
0.11 |
65 |
0.08 |
63 |
0.08 |
91 |
0.08 |
62 |
0.06 |
0.232 |
0.813 |
| CAAT |
29 |
0.05 |
48 |
0.06 |
27 |
0.04 |
76 |
0.07 |
47 |
0.04 |
0.773 |
0.123 |
| CAGC |
15 |
0.03 |
36 |
0.04 |
23 |
0.03 |
56 |
0.05 |
59 |
0.05 |
0.316 |
0.298 |
| GAGT |
22 |
0.04 |
28 |
0.03 |
9 |
0.01 |
41 |
0.04 |
54 |
0.05 |
0.721 |
0.079 |
| GAAC |
5 |
0.01 |
22 |
0.03 |
7 |
0.01 |
28 |
0.02 |
12 |
0.01 |
0.136 |
0.194 |
| GGAT |
3 |
0.01 |
22 |
0.03 |
2 |
<0.01 |
14 |
0.01 |
27 |
0.02 |
0.059 |
0.217 |
| GGGT |
7 |
0.01 |
10 |
0.01 |
2 |
<0.01 |
9 |
0.01 |
25 |
0.02 |
0.961 |
0.430 |
| CGGC |
6 |
0.01 |
5 |
0.01 |
2 |
<0.01 |
12 |
0.01 |
22 |
0.02 |
0.547 |
0.297 |
| CGGT |
0 |
0.00 |
5 |
0.01 |
1 |
<0.01 |
3 |
<0.01 |
0 |
0.00 |
0.163 |
0.720 |
| GGAC |
0 |
0.00 |
4 |
<0.01 |
0 |
0.00 |
0 |
0.00 |
6 |
0.01 |
0.222 |
n.a. |
| CAAC |
1 |
<0.01 |
2 |
<0.01 |
0 |
0.00 |
2 |
<0.01 |
0 |
0.00 |
0.781 |
0.318 |
| CGAT |
1 |
<0.01 |
2 |
<0.01 |
0 |
0.00 |
0 |
0.00 |
4 |
<0.01 |
0.783 |
n.a |
| Total | 564 | 834 | 738 | 1.153 | 1.070 | 0.239 | 0.129 | |||||
* P values for the comparison of case and control haplotype frequencies were based upon simulations as previously described [33].
DEF cluster b CN distribution differs between PDAC and controls
Diploid DEF cluster b CNs were determined by MLPA for 65, 232, 63 and 161 individuals from the PDAC, CARLA1, CP and CARLA2 groups, respectively (Additional files 1, 2, 3, 4 and 5). The median CN was 4 copies per diploid genome for all groups. CNs ranged from 2 to 7 in the PDAC group (mean: 4.22) and from 3 to 7 in the CP group (mean: 4.57). In both control groups, CNs were between 2 and 8 copies (mean CARLA1: 4.42, CARLA2: 4.55). Differences in mean CN between cases and control groups were not statistically significant (PDAC vs CARLA1: 0.20, P=0.151; CP vs CARLA2: 0.02, P=0.915) (Additional file 6).
The diploid CN distributions within the four cohorts are depicted in Figure 2. Application of Fisher’s exact test revealed that these distributions differed significantly between PDAC and CARLA1 (P=0.027), but not between CP and CARLA2 (P=0.867). The two control groups also did not differ significantly from each other (P=0.580).
Figure 2.
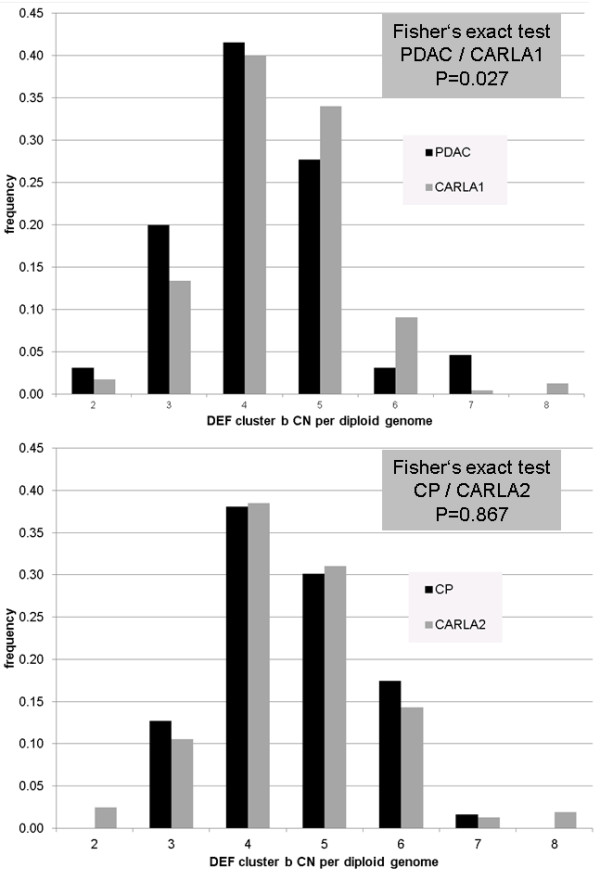
Distribution of DEF cluster b CN per diploid genome in the PDAC and CARLA1 (top) and CP and CARLA2 (bottom) cohorts.
Discussion
Defensins are expressed in the pancreas although it is not entirely clear which cells actually produce and secrete which of these diverse peptides for which purpose. In pancreatic juice, only HNP-3 (α-defensin 3, encoded by DEFA3) has been detected but mRNA expression of ß-defensins has also been demonstrated in pancreatic tissue [35]. The important role of defensins in the innate immune system due to their antimicrobial, chemotactic and regulatory functions, and their involvement in inflammatory processes vindicates the assumption that defensins are also involved in the pathogenesis of pancreatitis and pancreatic cancer.
Pancreatitis may develop as a chronic disease after long-term alcohol abuse. Chronic pancreatitis is a strong risk factor for pancreatic cancer but alcohol does not appear to be an independent causative agent for the disease [36,37]. Interestingly, acute and chronic pancreatic inflammation occurs as an extra-intestinal co-morbidity of inflammatory bowel disease for which an involvement of defensins is also discussed [27,38,39]. Furthermore, in view of the microbicidal properties of defensins, it appears noteworthy that a link between infectious diseases and pancreatic cancer has been drawn both for viral diseases (mumps, HBV infections) and bacterial infections (Helicobacter pylori) [37,40].
In the present study, both sequence variants in a ß-defensin gene and CN variants of the cluster containing this gene were investigated for a putative association with PDAC and CP. As haplotyping of the CN-variable DEFB104 gene was performed in pools, haplotypes cannot be assigned to the individual diploid CN for DEF cluster b. Respectively, both features had to be tested for association independently. All data from the patient groups were compared to age- and sex-matched healthy controls (CARLA1 and CARLA2).
Earlier, we have demonstrated association between DEFB104 haplotypes and sporadic prostate cancer as well as under-representation of high diploid DEF cluster CN in patients with this disease [33]. In the present study, no statistical support was found for an association between DEFB104 haplotypes and either PDAC or CP. However, analysis of the diploid CN distributions revealed a statistically significant difference between PDAC and CARLA1 that was due mainly to a paucity of 5- and 6-copy samples and an excess of 3-copy ones in the PDAC cohort.
Recently, under-representation of higher diploid DEFB4 CNs (>4) was reported in patients with acute pancreatitis (AP) and severe acute pancreatitis (SAP) [41]. Since DEFB4 is part of DEF cluster b and concordance for the CN of all genes within cluster b has been shown [16], this result is in agreement with our findings in the PDAC cohort. However, for CP, known to increase the risk for developing pancreatic cancer by 10 to 20-fold and a possible outcome of AP, we and others [42] did not observe significant associations with genetic features of the DEF cluster b, potentially pinpointing different roles of defensins in the etiopathogenesis of pancreatic diseases.
Although the functional consequences of the lower DEF cluster b CN observed for PDAC and AP are not yet resolved, lower CNs are rather associated with lower defensin expression [17]. In the light of inflammation as key feature of AP and the established link between inflammation and cancer [43], a low CN would be consistent with an anti-inflammatory effect of defensins described recently [44]. Assuming instead defensins to exert a pro-inflammatory effect [45] would favor a role of perturbed antimicrobial barrier defense in the etiopathogenesis of PDAC and AP. Further studies are necessary to find these missing functional links and to clarify which genetic variants may serve as reliable and feasible markers in the diagnosis and prognosis of pancreatic diseases.
Conclusion
Different DEF cluster b CN distribution between PDAC patients and healthy controls indicate a potential protective effect of higher CNs against the disease. Replication of the study with larger sample numbers are needed to confirm the result and to draw definitive conclusions thereof.
Methods
Patients, DNA samples and Oligonucleotides
All individuals were of European origin. Cases of PDAC and CP were taken from two cohorts of patients with PDAC and CP who previously had undergone pylorus-preserving pancreatico-duodenectomy. They were complemented by age- and sex-matched healthy control groups sampled from the CARLA Study, a prospective cohort study of the general elderly population [34]. The sampled controls from CARLA were free from heart disease, cancer or any other severe chronic disease, and without intake of antiphlogistic medication (Anatomical Therapeutic Chemical Classification System (ATC) code A07). A description of the age- and sex-distribution of the groups is given in Table 1.
Genomic DNA was obtained from peripheral blood collected in EDTA tubes (QIAamp DNA Mini Kit). The studies were approved by the ethics committees of the Universities of Dresden (Vote No. EK96042007) and Halle (Vote No. 1983-01/07). Written informed consent was obtained from all participants. The funding sources of the study played no role in the study design, data collection, data analysis, data interpretation or writing of the report. A human genomic DNA pool derived from ~100 anonymous individuals (Roche Diagnostics, Cat.No. 1691112) served as an additional control. All primers were synthesized by Metabion AG (Martinsried, Germany).
DEFB104 haplotyping
Amplification from individual genomic DNAs was carried out using primers 5'-TTCTGTAGCCCCAACACCTC-3' and 5'-GGTGCCAAGGACATCTAGGA-3', resulting in a 500 bp PCR product spanning four MSV (rs17843871, rs2680507, rs17843872, rs4259430) around exon 1 of DEFB104 (GenBank Refseq NM_080389.2). PCR reactions were performed as described with the following cycling conditions: 95°C for 1 min; 5 cycles at 95°C for 30 s, 56°C for 30 s, and 72°C for 60 s, 27 cycles at 95°C for 30 s, 58°C for 30 s, and 72°C for 60 s, with a final extension at 72°C for 5 min [33]. The concentrations of PCR products were measured by use of a Nanodrop device and equal amounts were pooled per cohort. Pooled DNAs were cloned into pCR2.1-TOPO (Invitrogen) according to the manufacturer’s instructions and transformed into E.coli by electroporation. Well-isolated colonies were transferred and grown in LB broth supplemented with ampicillin. Plasmid DNA was isolated from the cultures by BioRobot 8000 and MagAttract 96 Miniprep Core Kit (Qiagen) and inserts were sequenced in both directions using M13 universal primers. Haplotypes were called by visual inspection of the sequence traces.
DEF cluster copy numbers
For all individuals of the PDAC, CP, CARLA1 and CARLA2 cohorts, CNs of DEF cluster b (including DEFB104, Figure 1) were determined by multiplex ligation-dependent probe amplification (MLPA), using the P139 kit (MRC Holland), as previously described [16]. The MLPA probe set consists of 43 probes of which 10 are hybridizing to genes/pseudogenes within DEF cluster a, 10 to genes within DEF cluster b and 23 to bona fide single-copy genes flanking the defensin clusters as well as on other chromosomes, respectively. Peak areas were normalized against the summed peak areas of the “five nearest neighbor” (5nn) reference probes for each individual sample, relative locus doses were calculated and the diploid copy numbers were inferred. As internal quality control, four DNAs (NA18552, NA15324, NA12760, NA18858) with known CN (2, 4, 6 and 8, respectively) from commercially available lymphoblastoid cell lines (Coriell Cell repository http://www.coriell.org/) were used as copy number standards. Reliable copy number details from these samples are from independent, methodologically different determinations from different laboratories (see Table 2 in Groth et al. [2008] and references therein).
Statistics
Following our PCR and sequencing approach, DEFB104 haplotype determination is hampered by the fact that post-PCR pooling leads to an over-representation of alleles derived from individuals with low DEF cluster CN whilst alleles from genomes with high CN are under-represented. Furthermore, the number of sequenced clones differed considerably between the groups, ranging from 564 to 1153. This implies that haplotypes as called from sequence traces do not represent statistically independent observations and do not reflect the truly underlying haplotype distribution. Therefore, a haplotype-wise χ2 test could not be applied in the case–control comparisons. Instead, we simulated genotypes with respect to individual CN under the null hypothesis (i.e. no difference between cases and controls) and derived reference haplotype distributions for statistical testing from these simulated data, as previously described [33].
Differences in DEF cluster b CNs between cases and controls were first assessed by a comparison of the group-specific mean diploid CNs. Since the mean may not be a sufficient statistic for the underlying genotype distribution, diploid CN was also treated as a qualitative variable and gauged for statistically significant differences between groups using Fisher’s exact test as implemented in the SAS statistical analysis package V9.2 (SAS Inc., Cary, NY).
Competing interests
The authors declare that they have no competing interests.
Author’s contributions
GG, CP and RG collected the disease cohort’s samples and provided the patient’s data. KHG, KW, AK and OK conceived the CARLA study, did the cohort matching and provided the control samples. They also performed the statistical analyses with the support of MK, AW and MN. ST and MG carried out the molecular genetic studies with support by PR. ST, KH and MP drafted the manuscript. All authors read and approved the final manuscript.
Supplementary Material
Integer DEF cluster b copy numbers per diploid genome determined by MLPA, PDAC cohort.
Integer DEF cluster b copy numbers per diploid genome determined by MLPA, CP cohort.
Integer DEF cluster b copy numbers per diploid genome determined by MLPA, CARLA1 cohort.
Integer DEF cluster b copy numbers per diploid genome determined by MLPA, CARLA2 cohort.
Summary of DEF cluster b CN distribution.
Statistics for comparisons of DEF cluster b CN distribution.
Contributor Information
Stefan Taudien, Email: stau@fli-leibniz.de.
Gabor Gäbel, Email: Gabor.Gaebel@uniklinikum-dresden.de.
Oliver Kuss, Email: oliver.kuss@medizin.uni-halle.de.
Marco Groth, Email: mgroth@fli-leibniz.de.
Robert Grützmann, Email: Robert.Gruetzmann@uniklinikum-dresden.de.
Klaus Huse, Email: khuse@fli-leibniz.de.
Alexander Kluttig, Email: alexander.kluttig@medizin.uni-halle.de.
Andreas Wolf, Email: wolf@medinfo.uni-kiel.de.
Michael Nothnagel, Email: nothnagel@medinfo.uni-kiel.de.
Philip Rosenstiel, Email: p.rosenstiel@mucosa.de.
Karin Halina Greiser, Email: h.greiser@dkfz-heidelberg.de.
Karl Werdan, Email: karl.werdan@medizin.uni-halle.de.
Michael Krawczak, Email: krawczak@medinfo.uni-kiel.de.
Christian Pilarsky, Email: Christian.Pilarsky@uniklinikum-dresden.de.
Matthias Platzer, Email: mplatzer@fli-leibniz.de.
Acknowledgements
We thank Ivonne Heinze, Ivonne Görlich, Kathleen Seitz, Beate Szafranski and Daniela Werler (FLI Jena) for skilful technical assistance. The PDAC and CP studies were financially supported by grants from the Bundesministerium für Bildung und Forschung for the National Genome Research Network of Environmental Disorders (01GS0809), the Deutsche Forschungsgemeinschaft (DFG) and from the Technical University of Dresden (“MedDrive der Medizinischen Fakultät Dresden”). The CARLA study was funded by a DFG grant as part of the Collaborative Research Center 598 “Heart failure in the elderly – cellular mechanisms and therapy” at the Medical Faculty of the Martin Luther University Halle-Wittenberg; by a grant of the Wilhelm Roux Programme of the Martin Luther University Halle-Wittenberg; and by the Federal Employment Office.
References
- Skipworth JR, Shankar A, Pereira SP. Managing acute and chronic pancreatitis. Practitioner. 2010;254:23–27. 22. [PubMed] [Google Scholar]
- Kloppel G, Maillet B. Development of chronic pancreatitis from acute pancreatitis: a pathogenetic concept. Zentralbl Chir. 1995;120:274–277. [PubMed] [Google Scholar]
- Ahmed SA, Wray C, Rilo HL, Choe KA, Gelrud A, Howington JA, Lowy AM, Matthews JB. Chronic pancreatitis: recent advances and ongoing challenges. Curr Probl Surg. 2006;43:127–238. doi: 10.1067/j.cpsurg.2005.12.004. [DOI] [PubMed] [Google Scholar]
- Raimondi S, Lowenfels AB, Morselli-Labate AM, Maisonneuve P, Pezzilli R. Pancreatic cancer in chronic pancreatitis; aetiology, incidence, and early detection. Best Pract Res Clin Gastroenterol. 2010;24:349–358. doi: 10.1016/j.bpg.2010.02.007. [DOI] [PubMed] [Google Scholar]
- Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- Raptis DA, Fessas C, Belasyse-Smith P, Kurzawinski TR. Clinical presentation and waiting time targets do not affect prognosis in patients with pancreatic cancer. Surgeon. 2010;8:239–246. doi: 10.1016/j.surge.2010.03.001. [DOI] [PubMed] [Google Scholar]
- Lehrer RI. Immunology: Peptide gets in shape for self-defence. Nature. 2011;469:309–310. doi: 10.1038/469309a. [DOI] [PubMed] [Google Scholar]
- Beisswenger C, Bals R. Antimicrobial peptides in lung inflammation. Chem Immunol Allergy. 2005;86:55–71. doi: 10.1159/000086651. [DOI] [PubMed] [Google Scholar]
- Diamond G, Ryan L. Beta-defensins: what are they REALLY doing in the oral cavity? Oral Dis. 2011;17(7):628–635. doi: 10.1111/j.1601-0825.2011.01799.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guani-Guerra E, Santos-Mendoza T, Lugo-Reyes SO, Teran LM. Antimicrobial peptides: general overview and clinical implications in human health and disease. Clin Immunol. 2010;135:1–11. doi: 10.1016/j.clim.2009.12.004. [DOI] [PubMed] [Google Scholar]
- Herr C, Shaykhiev R, Bals R. The role of cathelicidin and defensins in pulmonary inflammatory diseases. Expert Opin Biol Ther. 2007;7:1449–1461. doi: 10.1517/14712598.7.9.1449. [DOI] [PubMed] [Google Scholar]
- Hollox EJ. Copy number variation of beta-defensins and relevance to disease. Cytogenet Genome Res. 2008;123:148–155. doi: 10.1159/000184702. [DOI] [PubMed] [Google Scholar]
- Paris S, Wolgin M, Kielbassa AM, Pries A, Zakrzewicz A. Gene expression of human beta-defensins in healthy and inflamed human dental pulps. J Endod. 2009;35:520–523. doi: 10.1016/j.joen.2008.12.015. [DOI] [PubMed] [Google Scholar]
- Hollox EJ, Armour JA, Barber JC. Extensive normal copy number variation of a beta-defensin antimicrobial-gene cluster. Am J Hum Genet. 2003;73:591–600. doi: 10.1086/378157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taudien S, Galgoczy P, Huse K, Reichwald K, Schilhabel M, Szafranski K, Shimizu A, Asakawa S, Frankish A, Loncarevic IF. et al. Polymorphic segmental duplications at 8p23.1 challenge the determination of individual defensin gene repertoires and the assembly of a contiguous human reference sequence. BMC Genomics. 2004;5:92. doi: 10.1186/1471-2164-5-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groth M, Szafranski K, Taudien S, Huse K, Mueller O, Rosenstiel P, Nygren AO, Schreiber S, Birkenmeier G, Platzer M. High-resolution mapping of the 8p23.1 beta-defensin cluster reveals strictly concordant copy number variation of all genes. Hum Mutat. 2008;29:1247–1254. doi: 10.1002/humu.20751. [DOI] [PubMed] [Google Scholar]
- Groth M, Wiegand C, Szafranski K, Huse K, Kramer M, Rosenstiel P, Schreiber S, Norgauer J, Platzer M. Both copy number and sequence variations affect expression of human DEFB4. Genes Immun. 2010;11:458–466. doi: 10.1038/gene.2010.19. [DOI] [PubMed] [Google Scholar]
- Hardwick RJ, Machado LR, Zuccherato LW, Antolinos S, Xue Y, Shawa N, Gilman RH, Cabrera L, Berg DE, Tyler-Smith C. et al. A worldwide analysis of beta-defensin copy number variation suggests recent selection of a high-expressing DEFB103 gene copy in East Asia. Hum Mutat. 2011;32:743–750. doi: 10.1002/humu.21491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taudien S, Groth M, Huse K, Petzold A, Szafranski K, Hampe J, Rosenstiel P, Schreiber S, Platzer M. Haplotyping and copy number estimation of the highly polymorphic human beta-defensin locus on 8p23 by 454 amplicon sequencing. BMC Genomics. 2010;11:252. doi: 10.1186/1471-2164-11-252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taudien S, Szafranski K, Felder M, Groth M, Huse K, Raffaelli F, Petzold A, Zhang X, Rosenstiel P, Hampe J. et al. Comprehensive assessment of sequence variation within the copy number variable defensin cluster on 8p23 by target enriched in-depth 454 sequencing. BMC Genomics. 2011;12:243. doi: 10.1186/1471-2164-12-243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollox EJ, Huffmeier U, Zeeuwen PL, Palla R, Lascorz J, Rodijk-Olthuis D, van de Kerkhof PC, Traupe H, de Jongh G, den Heijer M. et al. Psoriasis is associated with increased beta-defensin genomic copy number. Nat Genet. 2008;40:23–25. doi: 10.1038/ng.2007.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredman D, White SJ, Potter S, Eichler EE, Den Dunnen JT, Brookes AJ. Complex SNP-related sequence variation in segmental genome duplications. Nat Genet. 2004;36:861–866. doi: 10.1038/ng1401. [DOI] [PubMed] [Google Scholar]
- Armour JA, Palla R, Zeeuwen PL, den Heijer M, Schalkwijk J, Hollox EJ. Accurate, high-throughput typing of copy number variation using paralogue ratios from dispersed repeats. Nucleic Acids Res. 2007;35:e19. doi: 10.1093/nar/gkl1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deutsch S, Choudhury U, Merla G, Howald C, Sylvan A, Antonarakis SE. Detection of aneuploidies by paralogous sequence quantification. J Med Genet. 2004;41:908–915. doi: 10.1136/jmg.2004.023184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HH, Chang JG, Lin SP, Chao HT, Yang ML, Ng HT. Rapid detection of trisomy 21 by homologous gene quantitative PCR (HGQ-PCR) Hum Genet. 1997;99:364–367. doi: 10.1007/s004390050373. [DOI] [PubMed] [Google Scholar]
- Schouten JP, McElgunn CJ, Waaijer R, Zwijnenburg D, Diepvens F, Pals G. Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res. 2002;30:e57. doi: 10.1093/nar/gnf056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldhous MC, Abu Bakar S, Prescott NJ, Palla R, Soo K, Mansfield JC, Mathew CG, Satsangi J, Armour JA. Measurement methods and accuracy in copy number variation: failure to replicate associations of beta-defensin copy number with Crohn's disease. Hum Mol Genet. 2010;19:4930–4938. doi: 10.1093/hmg/ddq411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perne A, Zhang X, Lehmann L, Groth M, Stuber F, Book M. Comparison of multiplex ligation-dependent probe amplification and real-time PCR accuracy for gene copy number quantification using the beta-defensin locus. Biotechniques. 2009;47:1023–1028. doi: 10.2144/000113300. [DOI] [PubMed] [Google Scholar]
- Chen Q, Hakimi M, Wu S, Jin Y, Cheng B, Wang H, Xie G, Ganz T, Linzmeier RM, Fang X. Increased genomic copy number of DEFA1/DEFA3 is associated with susceptibility to severe sepsis in Chinese Han population. Anesthesiology. 2010;112:1428–1434. doi: 10.1097/ALN.0b013e3181d968eb. [DOI] [PubMed] [Google Scholar]
- Aldhous MC, Noble CL, Satsangi J. Dysregulation of human beta-defensin-2 protein in inflammatory bowel disease. PLoS One. 2009;4:e6285. doi: 10.1371/journal.pone.0006285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentley RW, Pearson J, Gearry RB, Barclay ML, McKinney C, Merriman TR, Roberts RL. Association of Higher DEFB4 Genomic Copy Number With Crohn's Disease. Am J Gastroenterol. 2009;105(2):354–359. doi: 10.1038/ajg.2009.582. [DOI] [PubMed] [Google Scholar]
- Fellermann K, Stange DE, Schaeffeler E, Schmalzl H, Wehkamp J, Bevins CL, Reinisch W, Teml A, Schwab M, Lichter P. et al. A chromosome 8 gene-cluster polymorphism with low human beta-defensin 2 gene copy number predisposes to Crohn disease of the colon. Am J Hum Genet. 2006;79:439–448. doi: 10.1086/505915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huse K, Taudien S, Groth M, Rosenstiel P, Szafranski K, Hiller M, Hampe J, Junker K, Schubert J, Schreiber S. et al. Genetic variants of the copy number polymorphic beta-defensin locus are associated with sporadic prostate cancer. Tumour Biol. 2008;29:83–92. doi: 10.1159/000135688. [DOI] [PubMed] [Google Scholar]
- Greiser KH, Kluttig A, Schumann B, Kors JA, Swenne CA, Kuss O, Werdan K, Haerting J. Cardiovascular disease, risk factors and heart rate variability in the elderly general population: design and objectives of the CARdiovascular disease, Living and Ageing in Halle (CARLA) Study. BMC Cardiovasc Disord. 2005;5:33. doi: 10.1186/1471-2261-5-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gronborg M, Bunkenborg J, Kristiansen TZ, Jensen ON, Yeo CJ, Hruban RH, Maitra A, Goggins MG, Pandey A. Comprehensive proteomic analysis of human pancreatic juice. J Proteome Res. 2004;3:1042–1055. doi: 10.1021/pr0499085. [DOI] [PubMed] [Google Scholar]
- Lowenfels AB, Maisonneuve P, Cavallini G, Ammann RW, Lankisch PG, Andersen JR, Dimagno EP, Andren-Sandberg A, Domellof L. Pancreatitis and the risk of pancreatic cancer. International Pancreatitis Study Group. N Engl J Med. 1993;328:1433–1437. doi: 10.1056/NEJM199305203282001. [DOI] [PubMed] [Google Scholar]
- Raimondi S, Maisonneuve P, Lowenfels AB. Epidemiology of pancreatic cancer: an overview. Nat Rev Gastroenterol Hepatol. 2009;6:699–708. doi: 10.1038/nrgastro.2009.177. [DOI] [PubMed] [Google Scholar]
- Pitchumoni CS, Rubin A, Das K. Pancreatitis in inflammatory bowel diseases. J Clin Gastroenterol. 2010;44:246–253. doi: 10.1097/MCG.0b013e3181cadbe1. [DOI] [PubMed] [Google Scholar]
- Hollox EJ. Beta-defensins and Crohn's disease: confusion from counting copies. Am J Gastroenterol. 2010;105:360–362. doi: 10.1038/ajg.2009.573. [DOI] [PubMed] [Google Scholar]
- Jerrells TR, Vidlak D, Strachota JM. Alcoholic pancreatitis: mechanisms of viral infections as cofactors in the development of acute and chronic pancreatitis and fibrosis. J Leukoc Biol. 2007;81:430–439. doi: 10.1189/jlb.1004622. [DOI] [PubMed] [Google Scholar]
- Tiszlavicz Z, Szabolcs A, Takacs T, Farkas G, Kovacs-Nagy R, Szantai E, Sasvari-Szekely M, Mandi Y. Polymorphisms of beta defensins are associated with the risk of severe acute pancreatitis. Pancreatology. 2010;10:483–490. doi: 10.1159/000276987. [DOI] [PubMed] [Google Scholar]
- Farrow B, Sugiyama Y, Chen A, Uffort E, Nealon W, Mark Evers B. Inflammatory mechanisms contributing to pancreatic cancer development. Ann Surg. 2004;239:763–769. doi: 10.1097/01.sla.0000128681.76786.07. discussion 769–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–867. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohlgraf KG, Pingel LC, Dietrich DE, Brogden KA. Defensins as anti-inflammatory compounds and mucosal adjuvants. Future Microbiol. 2010;5:99–113. doi: 10.2217/fmb.09.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganz T. Defensins: antimicrobial peptides of innate immunity. Nat Rev Immunol. 2003;3:710–720. doi: 10.1038/nri1180. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Integer DEF cluster b copy numbers per diploid genome determined by MLPA, PDAC cohort.
Integer DEF cluster b copy numbers per diploid genome determined by MLPA, CP cohort.
Integer DEF cluster b copy numbers per diploid genome determined by MLPA, CARLA1 cohort.
Integer DEF cluster b copy numbers per diploid genome determined by MLPA, CARLA2 cohort.
Summary of DEF cluster b CN distribution.
Statistics for comparisons of DEF cluster b CN distribution.