

Abstract
Anthocyanin accumulation is coordinated in plants by a number of conserved transcription factors. In apple (Malus × domestica), an R2R3 MYB transcription factor has been shown to control fruit flesh and foliage anthocyanin pigmentation (MYB10) and fruit skin color (MYB1). However, the pattern of expression and allelic variation at these loci does not explain all anthocyanin-related apple phenotypes. One such example is an open-pollinated seedling of cv Sangrado that has green foliage and develops red flesh in the fruit cortex late in maturity. We used methods that combine plant breeding, molecular biology, and genomics to identify duplicated MYB transcription factors that could control this phenotype. We then demonstrated that the red-flesh cortex phenotype is associated with enhanced expression of MYB110a, a paralog of MYB10. Functional characterization of MYB110a showed that it was able to up-regulate anthocyanin biosynthesis in tobacco (Nicotiana tabacum). The chromosomal location of MYB110a is consistent with a whole-genome duplication event that occurred during the evolution of apple within the Maloideae family. Both MYB10 and MYB110a have conserved function in some cultivars, but they differ in their expression pattern and response to fruit maturity.
Red pigments in plants are thought to be involved in reproductive fitness, since red coloration of the fruit skin indicates to seed-dispersing animals that the fruit is mature and ready to eat (Regan et al., 2001; Stintzing and Carle, 2004). The color of fruit flesh is a less visible characteristic, as it is hidden by the skin and has probably developed from the adaptive strategy to provide nutritional value to the species feeding on the fruit (Cipollini, 2000). One hypothesis is that some fruit species have retained pigments in their flesh as a rich source of antioxidant molecules, rewarding seed dispersers with improved fitness and increased longevity due to a healthier diet, making them more likely to visit these trees and disperse more seeds. Fruit flesh color and its anthocyanin-related antioxidant properties are well documented both scientifically (Hertog et al., 1993; Wolfe et al., 2003; Butelli et al., 2008) and popularly, with some examples receiving considerable public attention, such as the “French paradox” (Renaud and de Lorgeril, 1992; Wang et al., 1997), which is centered on the heart disease-reducing properties of wine grapes.
In the Rosaceae family, Prunus (e.g. peach [Prunus persica], plum [Prunus domestica], apricot [Prunus armeniaca], and cherry [Prunus avium]) and Rubus (raspberry [Rubus idaeus], blackberry [Rubus fruticosus], and boysenberry [Rubus idaeus × Rubus fruticosus]) species have commercial cultivars that are distinguished from each other by variation in fruit flesh anthocyanin concentrations. Both these genera have a long history of coevolution with seed-dispersing animals across their wide natural range (Traveset and Willson, 1998; Jordano et al., 2007) and modern human fruit consumers who also have a preference for red fruit (Jaeger and Harker, 2005). Despite the recognized health benefits from antioxidant pigments, high concentrations of anthocyanin in the apple (Malus × domestica) fruit flesh are a relatively unexplored phenomenon. Two distinct red-fleshed apple phenotypes have been identified (Volz et al., 2006, 2009). The color in both results from the elevation of an anthocyanin compound, cyanidin 3-galactoside (Mazza and Velioglu, 1992). In cultivars displaying the type 1 red-flesh phenotype, cyanidin 3-galactoside is distributed throughout the fruit, from fruit set through maturity, and this phenotype is associated with red leaves, stems, roots, and flowers. The second red-flesh apple phenotype (type 2) is characterized by red pigment only in the fruit cortex and not the core, developing late in fruit development, and is associated with green rather than red leaves (Volz et al., 2009).
Anthocyanins are phenylpropanoids. The genes controlling the plant phenylpropanoid biosynthetic pathway are largely regulated by changes in transcription. In all species studied to date, this regulation is through a complex of MYB transcription factors (TFs), basic helix-loop-helix (bHLH) TFs, and WD-repeat proteins in the MYB-bHLH-WD40 (MBW) complex (Baudry et al., 2004). The MYB members of this complex often appear to be the major determinant of variation in anthocyanin pigmentation (Schwinn et al., 2006; Allan et al., 2008). MYB TFs constitute one of the largest TF gene families in plants, with 126 R2R3 MYB genes identified in Arabidopsis (Arabidopsis thaliana; Stracke et al., 2001). Those associated with up-regulation of the anthocyanin pathway are members of a subclade that includes the PRODUCTION OF ANTHOCYANIN PIGMENT1 (PAP1) gene (AtMYB75; At1g56650), which, when overexpressed in Arabidopsis, results in the accumulation of anthocyanins (Borevitz et al., 2000). Anthocyanin-regulating MYBs have been isolated from many species, including Arabidopsis MYB75 (PAP1), MYB90 (PAP2), MYB113, and MYB114, and all the major Rosaceous fruit crops (Lin-Wang et al., 2010).
In maize (Zea mays), the p1 gene controls red pigmentation in floral organs and corresponds to an R2R3 MYB TF (Grotewold et al., 1991). A second p gene (p2), highly homologous to p1, was identified upstream of p1 and is likely to have arisen from a tandem duplication (Zhang et al., 2000). Both p1 and p2 have differential expression during the development of red phenotypes, due to variations in their 5′ regulatory elements. In Antirrhinum majus, a family of duplicated MYB genes (Rosea1, Rosea2, and Venosa) controls differential floral pigmentation (Schwinn et al., 2006).
In apple, the expression of MYB10 in type 1 red-fleshed apples is strongly correlated with anthocyanin accumulation (Espley et al., 2007). Transgenic cv Royal Gala apples constitutively expressing MYB10 produced highly pigmented shoots. Two more apple MYBs, MYB1 and MYBA, were also reported to regulate genes in the anthocyanin pathway in red-skinned fruit (Takos et al., 2006; Ban et al., 2007). As MYB1 and MYBA share identical sequences, and MYB10 and MYB1 genes are located at very similar positions on linkage group 9, it was concluded that these genes were allelic (Lin-Wang et al., 2010; Zhu et al., 2011). It was shown that the mutation leading to the type 1 red-flesh phenotype lies in the promoter of the MYB10 gene and results in the autoregulation of MYB10 by itself (Espley et al., 2009). In contrast, the type 2 red-flesh apple phenotype does not cosegregate with molecular markers linked to MYB10 (Volz et al., 2006).
Analysis of the assembled whole-genome sequence of domestic apple has shown that Malus spp. experienced a whole-genome duplication approximately 60 to 65 million years ago (Velasco et al., 2010). Pairwise comparison of the sequence of the 17 apple chromosomes has revealed that whole chromosomes, or segments of chromosomes, are colinear (Velasco et al., 2010), and the chromosomal number and genome size of the Pyrineae subtribe (including apple) are double that of the rest of the Rosaceae family. Polyploidy is common as a source of speciation and genomic novelty in plants, and it is believed that most angiosperms have experienced one or more whole-genome duplication events in their evolutionary history (Wendel, 2000; Adams and Wendel, 2005).
We have used genome mapping and quantitative trait locus (QTL) analysis to identify the genetic region associated with type 2 red flesh and map it to linkage group (LG) 17. We have identified the MYB gene within this locus that is expressed in type 2 red-flesh cortex but not in other apple cultivars. If expressed, this gene is able to up-regulate anthocyanin pathway genes, as shown in tobacco (Nicotiana tabacum). We discuss how this gene has arisen due to a whole-genome duplication in apple evolution.
RESULTS
Phenotypic Assessment of Type 2 Red Flesh in Apple
Fruit from a seedling population of 328 individual fruiting trees, obtained from a cross between cv Sciros and a red-fleshed cv Sangrado open pollinated (OP) seedling, were sampled at two maturity time points in 2007 and 2008. Both parents of the segregating population have red fruit skin, white fruit core, and green foliage; however, cv Sangrado OP is characterized by anthocyanin expressed in the fruit cortex (type 2 red flesh; Fig. 1A). The progeny segregate for type 2 red flesh. This is phenotypically distinct from type 1 red flesh, where the foliage and the fruit core are also red (Fig. 1B). The frequency distributions for proportions of red-flesh coverage and intensity within the fruit were similar in both years, with 29% of seedlings having completely white flesh and the remainder exhibiting variable amounts of red coloration (Fig. 1C), up to a maximum of 100% cortex area (Fig. 1C), and maximum intensity score of 6 (Supplemental Fig. S1). Cortex coverage showed a bimodal distribution, with a higher proportion of seedlings with 90% to 100% red coverage than those with intermediate coverage (Supplemental Fig. S1). In contrast, there was a higher proportion of red-fleshed seedlings with weaker intensity red-flesh coloration than stronger (Fig. 1C; Supplemental Fig. S1). Three validation families of a total 450 seedlings, with a red-flesh parent (NZSelectionT051) that is an F1 seedling of cv Sangrado OP, had a similar distribution of the type 2 red-flesh phenotype, with 60% of the seedlings having red coloration.
Figure 1.

Comparison of type 1 and type 2 red flesh in apple. A, Type 2 red-flesh apple such as cv Sangrado OP is characterized by red pigmentation in the fruit cortex, white fruit core, and green foliage. B, Type 1 red-flesh apple such as cv Redfield OP is characterized by red pigmentation in the fruit core, fruit cortex, and foliage. C, Phenotype of type 2 red flesh in an F2 population observed in cv Sciros × NZSelectionT051 (cv Sciros × Sangrado OP). Tree numbers are as follows: i, R04T009; ii, NZSelectionT051; iii, R08T068; iv, R03T120; v, R06T062; vi, R08T069; vii, R05T060.
Whole-Genome Scan and QTL Detection
As the phenotypic distributions for red coverage and intensity were strongly nonnormal, indicating the presence of a genomic region associated with a large-effect QTL, a whole-genome scan was performed to identify its location. Thirty-eight markers spanning the genome were screened over extreme individuals. Two markers located on LG 17 (ARGH30 [Baldi et al., 2004] and Hi12f12 [Silfverberg-Dilworth et al., 2006]) cosegregated with the extreme type 2 red-flesh phenotypes. These markers, as well as other simple sequence repeat markers from LG 17 (NZmsEB140474 and NZmsEB137525; Celton et al., 2009), were then screened over 137 fruiting individuals of the segregating population in order to construct a genetic map suitable for use in QTL analysis. A composite interval-mapping analysis detected a significant QTL for weighted cortex intensity (WCI) on LG 17. There was no evidence of additional QTLs on LG 17. The detected QTL explained 31.0% of the phenotypic variation. This finding is consistent with the recent mapping of the self-incompatibility locus (Celton et al., 2009), also linked with red flesh in cv Pink Pearl (Sekido et al., 2010; Umemura et al., 2011) on LG 17 (Maliepaard et al., 1998).
A fine QTL-mapping experiment was then carried out in order to narrow the QTL confidence interval and enable the identification of positional candidate genes using the apple genome sequence. Three validation families (a total of 450 fruiting F1 seedlings) were screened using the apple International RosBREED SNP Consortium Infinium II 8k single-nucleotide polymorphism (SNP) array (Chagné et al., 2012), which yielded 163 polymorphic SNPs on LG 17, corresponding to an average density of one SNP every 164 kb. Thirty-one SNP markers located between genome positions 21.05 and 26.77 Mb of LG 17 had Kruskal-Wallis test statistics significantly associated (P < 0.005) with type 2 red cortex coverage (Fig. 2A). A multiple QTL-mapping analysis revealed that the most significant QTL was within an interval equivalent to 780 kb between SNPs spanning region 24.59 to 25.37 Mb. The SNP marker with the highest log of the odds (LOD) score (GDsnp00341; LOD = 22.1) was at position 24.85 Mb. GDsnp00341 did not have any recombinant between its favorable allele and the presence of red coloration in the fruit cortex.
Figure 2.

Fine-mapping for type 2 red flesh in the QTL validation populations. A, QTL analysis using a Kruskal-Wallis test (K*) using the percentage of red pigmentation in the fruit cortex and 163 SNP markers scored on 450 F1 individuals from the QTL validation population. Significance levels are as follows: P < 0.0001: K* = 23.59 for markers segregating < ab×aa > (marker heterozygous for one parent and homozygous for the other; degrees of freedom [d.f.] = 1) and K* = 19.13 for markers segregating < ab×ab > (marker heterozygous for both parents of the progeny; degrees of freedom = 2). B, Multiple QTL mapping analysis at the bottom of LG 17 in the QTL validation populations for percentage of red coloration in the fruit cortex (solid line) and intensity (dotted line).
Candidate Gene Mapping and Characterization
The location of a QTL associated with type 2 red flesh on LG 17 was used to investigate further the underlying genetic basis of this phenotype. Within the most significant QTL for type 2 red flesh, spanning 24.57 to 25.37 Mb, there were 167 gene models predicted, with 74 nonoverlapping (Supplemental Table S1) in the reference apple genome set (Velasco et al., 2010). Genome coordinates refer to the apple version 1.0 pseudohaplotype (primary assembly). If the genome version 1.0 is used, coordinates were 22.37 to 23.17 Mb (Supplemental Table S1). Only 47 of these gene models had EST evidence (Newcomb et al., 2006; Table I). None of these genes appeared related to anthocyanin biosynthesis. However, two R2R3 MYBs (MYB110a and MYB110b; Table I) showed good homology to MYB10. Apple is an ancient polyploid, with LG 9 and LG 17 shown to be homeologous (i.e. originating from a common ancestral chromosome that was duplicated due to a tetraploidization event; Velasco et al., 2010). As MYB10 (the gene controlling type 1 red flesh) is located at the bottom of LG 9 (Chagné et al., 2007), in a region homeologous to this LG 17 QTL for type 2 red flesh, these MYB gene sequences became lead candidates for the genetic control of this phenotype.
Table I. Candidate genes located in the QTL interval for type 2 red flesh on LG 17.
Gene model identification (Gene ID) and locations on LG 17 (bp) are from Velasco et al. (2010).
| Gene ID | LocationStart | Location End | Identity | GenBank Accession No. | ESTs |
|---|---|---|---|---|---|
| MDP0000634279 | 24,574,539 | 24,577,552 | U-box domain-containing protein4 | CN851564 | 46 |
| MDP0000135567 | 24,587,706 | 24,600,475 | Protease Do-like7 | CN909465 | 98 |
| MDP0000770587 | 24,630,213 | 24,630,824 | Cytochrome d ubiquinol oxidase | CN863894 | 1 |
| MDP0000289236 | 24,662,320 | 24,662,963 | Acyl-CoA oxidase2 | CN916535 | 6 |
| MDP0000188426 | 24,691,178 | 24,692,853 | Tropinone reductase | None | 4 |
| MDP0000202749 | 24,694,496 | 24,701,436 | THO complex subunit 4 | GO536973 | 68 |
| MDP0000202750 | 24,704,968 | 24,710,954 | DNA repair protein | CN863361 | 7 |
| MDP0000202751 | 24,720,470 | 24,721,780 | HMG-Y-related protein A | EB131982 | 8 |
| MDP0000185817 | 24,732,289 | 24,736,196 | Histone-Lys N-methyltransferase MLL5 | CN889116 | 82 |
| MDP0000544957 | 24,746,893 | 24,750,819 | Valyl-tRNA synthetase | None | 7 |
| MDP0000286576 | 24,768,518 | 24,771,172 | Zinc finger RNA-binding protein | CN911169 | 6 |
| MDP0000186929 | 24,764,954 | 24,766,577 | Cytochrome P450 71A24 | EG631270 | 1 |
| MDP0000889179 | 24,790,952 | 24,792,196 | Valyl-tRNA synthetase | CN495735 | 9 |
| MDP0000182767 | 24,795,628 | 24,798,332 | Calcium-binding protein CML27 | DT041281 | 8 |
| MDP0000123700 | 24,804,329 | 24807,923 | Ribosomal RNA-processing protein UTP23 homolog | DY255624 | 1 |
| MDP0000296152 | 24,825,987 | 24,823,517 | ADP-ribosylation factor-like protein8A | GO539178 | 4 |
| MDP0000669449 | 24,867,322 | 24,864,246 | Copia protease | None | 2 |
| MDP0000669451 | 24,852,646 | 24,854,400 | MdSCL33 DELLA protein GAI | EB131978 | 16 |
| MDP0000206691 | 24,876,543 | 24,878,401 | ADP-ribosylation factor GTPase | GO562351 | 4 |
| MDP0000295218 | 24,890,449 | 24,896,521 | MYB110a AtMYB114-like | EB710109 | 2 |
| MDP0000317257 | 24,960,842 | 24,961,396 | MYB110b At1g14600-like | CN993940 | 5 |
| MDP0000169910 | 24,985,536 | 24,988,079 | G2-like KANADI | CV657790 | 2 |
| MDP0000499017 | 25,017,563 | 25,020,991 | Cucumisin | CO865728 | 1 |
| MDP0000290729 | 25,037,965 | 25,042,561 | Importin5 | CN872280 | 5 |
| MDP0000290730 | 25,032,159 | 25,037,044 | Replication protein A | None | 6 |
| MDP0000243352 | 25,054,084 | 25,054,851 | MdC2H2L1 zinc finger protein4 | EB125198 | 3 |
| MDP0000129445 | 25,061,661 | 25,063,001 | U-box domain-containing protein21 | DT003420 | 9 |
| MDP0000435842 | 25,063,491 | 25,070,283 | Probable receptor protein kinase TMK1 | EB112902 | 6 |
| MDP0000146581 | 25,096,920 | 25,097,180 | Hypothetical protein | None | 1 |
| MDP0000548690 | 25,087,549 | 25,091,369 | Protein LURP-one-related7 | GO524670 | 1 |
| MDP0000248694 | 25,113,792 | 25,120,391 | Potassium channel SKOR | CN897708 | 30 |
| MDP0000182939 | 25,174,843 | 25,179,327 | Protein Mpv17 | GO518583 | 6 |
| MDP0000283631 | 25,168,103 | 25,173,514 | SPRY domain-containing protein | DT043639 | 31 |
| MDP0000550915 | 25,162,503 | 25,164,638 | Poly(A) polymerase | CN910697 | 3 |
| MDP0000213300 | 25,230,420 | 25,235,382 | T-complex protein1 subunit θ | CN913102 | 121 |
| MDP0000213302 | 25,210,522 | 25,214,797 | Chaperone protein dnaJ | GO558627 | 14 |
| MDP0000306514 | 25,202,770 | 25,209,354 | Ras-related protein Rab11D | CN914745 | 13 |
| MDP0000766705 | 25,252,993 | 25,253,643 | Vacuolar-processing enzyme (VPE) | CN921691 | 1 |
| MDP0000131537 | 25,250,844 | 25,252,523 | Glc-6-P 1-epimerase | EB151188 | 32 |
| MDP0000244968 | 25,242,969 | 25,249,684 | TCP domain class TF | CN930481 | 121 |
| MDP0000354348 | 25,246,250 | 25,246,370 | ABC transporter C | CN923992 | 1 |
| MDP0000616695 | 25,240,031 | 25,241,873 | 60S ribosomal protein L11-1 | CN866368 | 12 |
| MDP0000194307 | 25,281,950 | 25,291,315 | Shaggy-related protein kinase ζ | CN916950 | 109 |
| MDP0000292164 | 25,274,885 | 25,280,715 | Asp aminotransferase | CN887154 | 32 |
| MDP0000629636 | 25,313,338 | 25,325,872 | Peroxidase52 | CN855852 | 1 |
| MDP0000629637 | 25,326,103 | 25,327,960 | Ser/Thr protein kinase | GO538010 | 16 |
| MDP0000822752 | 25,358,724 | 25,359,977 | Triacylglycerol lipase | CN916378 | 1 |
| MDP0000363881 | 25,372,981 | 25,374,449 | Multiprotein-bridging factor 1b | CN925492 | 35 |
When the sequence of MYB10 (accession no. DQ267896) was used to BLAST the whole-genome assembly of cv Golden Delicious (Velasco et al., 2010), these two MYBs were the best matches (E > 1e-50). For this reason, we termed the two genes on LG 17 MYB110a (MDC013323_290; GenBank accession no. JN711473) and MYB110b (MDC035405_20; GenBank accession no. JN711474). Like MYB10, both MYB110a and MYB110b have three exons separated by two introns (Fig. 3). MYB110b has a large second intron of 5.7 kb, and the third exon in MYB110b appears to have nucleotide changes that would lead to a premature stop codon (marked with black dots in Fig. 3) compared with MYB10 and MYB110a and other related MYB genes. Following this premature stop codon in exon 3, MYB110b has a 272-bp insertion compared with both MYB10 and MYB110a. In the current genome assembly, MYB110a and MYB110b are oriented in opposing directions with adjacent start codons (Fig. 3).
Figure 3.

Schematic representation of the three R2R3 MYB anthocyanin-activating homeologous genes in the apple genome. MYB10 controlling type 1 red flesh and skin color is located at the bottom of chromosome 9 in contig MDC13323_319, and MYB110a (GenBank accession no. JN711473) and MYB110b (GenBank accession no. JN711474) are located on homeologous apple chromosome 17 on contigs MDC013323_290 and MDC035405_20, respectively. According to the apple genome primary assembly, MYB110a and MYB110b are in opposite orientations, with their putative start codons facing each other at an estimated distance of 60 kb. The MYB110b second intron is larger than the second intron of MYB10 and MYB110a and is truncated in its third exon (premature stop codons shown as black dots), based on the protein translation. A 272-bp insertion is also present in exon 3.
A phylogenetic analysis of predicted protein sequences placed MYB110a and MYB110b close to MYB10 and indicates that they are in the same family as other anthocyanin-activating MYB genes, such as A. majus Rosea1 and Rosea2, maize p1 and C1, and Arabidopsis PAP1 (Fig. 4). Although truncated, MYB110b has DNA-binding motifs, predicted interaction domains for bHLH partners, and a C-terminal consensus sequence, indicating that it could activate anthocyanin synthesis.
Figure 4.

Sequence characterization of MYB10, MYB110a, and MYB110b. A, Phylogenetic analysis of anthocyanin-related MYBs in plant species. B, Amino acid sequence alignment of MYB110a, MYB110b, MYB10, and MYB1. The amino acid sequence of MYB110a was translated from sequences obtained from the apple cv Sangrado OP (SG), Royal Gala (RG; parent of cv Sciros), and Golden Delicious (GD). The R2 and R3 MYB motifs are indicated. Arrowheads indicate residues needed for the interaction with the bHLH partner, box A is a domain characteristic of anthocyanin-regulating MYBs, and box B indicates a conserved C-terminal motif.
The genome assembly contigs containing MYB110a and MYB110b are physically located within the QTL interval for type 2 red flesh on LG 17. The SNP marker with the highest LOD score for red cortex coloration (GDsnp00341) is located 45 kb upstream of MYB110a. A high-resolution melting (HRM)-based genetic marker developed from the second exon of MYB110a mapped to LG 17 of cv Sangrado OP and in the QTL detection population. All seedlings with the MYB110a HRM marker red allele had red-colored cortex with coverage greater than 80%.
Only MYB110a Is Expressed in the Fruit Cortex of Type 2 Red-Fleshed Apples
Chemical composition and gene expression were measured at harvest in mature type 1 (completely red-fleshed apple; cv Redfield OP) and type 2 apples (Fig. 5). Anthocyanin concentrations in red-fleshed apples generally increased with increasing red color coverage and intensity, with the dominant anthocyanin in type 2 being mainly that of cyanidin 3-galactoside (Fig. 5B). At this harvest point, the expression of genes using real-time quantitative PCR (qPCR) showed that Leucoanthocyanidin dioxygenase (LDOX) was only expressed in red-fleshed tissue, and generally, levels correlated with anthocyanin concentration (Fig. 6). The expression of MYB110a correlated with LDOX expression and anthocyanin levels in the flesh of type 2 material, and expression was absent in type 1 apples and cv Sciros peel (Fig. 6, A and B). In contrast, MYB10 is highly expressed in type 1 red-fleshed cv Redfield OP and cv Sciros peel but is absent in type 2 genotypes (Fig. 6C). No expression could be detected for MYB110b in any of the samples tested. This was despite the use of different gene-specific primers designed to amplify different regions across the MYB110b gene, from the 5′ untranslated region (UTR) to the exon1/exon2 boundary and also in exon 3, prior to the premature stop codon (Supplemental Table S2).
Figure 5.

HPLC analysis of anthocyanin profiles in the cortex of progeny of an F2 cross of cv Sciros × Sangrado OP. A, Total anthocyanin (µg g−1 fresh weight [FW]) for progeny (same trees as numbered in Fig. 1C) and cv Redfield cortex (RFc), cv Sciros cortex (ScRc), and cv Sciros peel (ScRp). B, Liquid chromatography-quadrupole-time of flight-mass spectrometry traces from pooled fruit of three representative progeny, i, ii, and iii (as in Fig. 1). Letters besides peaks represent different cyanidin glycosides: a, cyanidin 3-galactoside; b, cyanidin 3-arabinoside; c, cyanidin 3-pentoside1; d, cyanidin 3-pentoside2. [See online article for color version of this figure.]
Figure 6.

qPCR analysis of transcript abundance of LDOX (A), MYB110a (B), and MYB10 (C) in the cortex of progeny of an F2 cross of cv Sciros × NZSelectionT051 (numbered as in Fig. 1), Redfield cortex (RFc), Sciros cortex (ScRc), and Sciros peel (ScRp). Ratio is relative to three apple reference genes, Act, EF1α, and GAPDH. NZSelectionT051 (ii) was set as a calibrator for LDOX and MYB110a, and cv Sciros peel (ScRp) was set as a calibrator for MYB10. Primers are shown in Supplemental Table S2. Data are presented as means ± se of four biological replicates.
MYB110a Codes for a Functional Protein That Regulates the Synthesis of Anthocyanin
Analysis of the derived polypeptide alignment of MYB110a, aligned with its paralogs (MYB110b and MYB10/MYB1), reveals that all three genes have motifs suggestive of MYBs that activate anthocyanin biosynthesis (Fig. 4B). Both MYB110a and MYB110b have residues that indicate a requirement for a bHLH TF partner (Fig. 4B, arrowheads) and a C-terminal motif (Fig. 4B, box B) identified for PAP1-like MYBs (Stracke et al., 2001; Zimmermann et al., 2004). However, MYB110b was not expressed in any apple tissue examined, and there are nucleotide changes that predict a premature stop in the C-terminal region. The expression of MYB110a in the flesh of cv Sangrado OP and its progeny (Fig. 6) suggests that this gene is active in this tissue concomitant with the production of anthocyanin pigments in the fruit cortex. A transient assay was performed to validate the functionality of MYB110a. The promoter sequence of apple Chalcone synthase (CHS; AB074485) was fused to the luciferase reporter and infiltrated into the leaves of tobacco with members of the MBW complex, each under the control of the 35S cauliflower mosaic virus promoter. MYB110a was able to transactivate the CHS promoter, particularly when coinfiltrated with other candidate members of the MBW complex, bHLH3 (Espley et al., 2007) and the WD40-like gene from apple, TTG1 (Brueggemann et al., 2010; Fig. 7A). MYB110a gave more transactivation of the CHS promoter than MYB10 when coinfiltrated with bHLH3 and WD40 expression constructs. In addition, anthocyanic regions were observed in tobacco leaves 7 d after infiltration with MYB110a (Fig. 7B).
Figure 7.

Transient assays demonstrate the function of MYB110a as a regulator of anthocyanin biosynthesis. A, Leaves of N. benthamiana were infiltrated with the apple CHS promoter-LUC fusions on their own or coinfiltrated with 35S:MYB110a, with or without 35S:bHLH3 and 35S:WD40. As a positive control, 35S:MYB10 was similarly infiltrated. Luminescence of luciferase and renilla was measured 3 d later and expressed as a ratio of luciferase to renilla. Data are presented as means of four replicate reactions. B, Red coloration around the infiltration site in the leaves of tobacco 8 d after transient transformation with 35S:MYB110a showing the adaxial leaf surface (i), 35S:MYB110a abaxial surface (ii), and 35S:MYB10 adaxial leaf surface (iii). All three patches were coinfiltrated with 35S:bHLH3. No coloration was observed with infiltration of 35S:bHLH3 alone.
Sequence Polymorphisms in the MYB110a Region
While there are differences in the expression profile of MYB110a between cv Sangrado OP and its progeny and white-fleshed apples, there is only one predicted amino acid change between MYB110a of cv Sangrado OP and Golden Delicious (Fig. 4B). However, this Cys/Tyr change is also found in MYB10 and so, therefore, is unlikely to affect activity. Hence, an examination of the promoter sequences of MYB110a from 12 apple cultivars was made (Fig. 8) for potential cis-regulatory motifs using the PLACE program (Higo et al., 1999). One BELL homeodomain-binding site and one MYBCORE site were identified within a 930-bp region upstream of the MYB110a START codon. In addition, two sequences conserved with the Arabidopsis PAP1 promoter and the R1 motif found in the promoter of apple MYB10 were identified. Seven SNPs and one insertion-deletion were detected in this region. Interestingly, the R1-like motif (Espley et al., 2009) is altered by a SNP in the promoter of cv Sangrado OP. However, both alleles of this SNP are found in the promoter of white-fleshed cultivars, suggesting that it is not causative for the type 2 red-flesh phenotype.
Figure 8.

Nucleotide sequence of the 5′ UTR and proximal promoter region of MYB110a. The positions of seven SNPs (black triangles) and one insertion-deletion (black diamond), cis-regulatory motifs, and the start codon (boxed) are indicated.
To identify repetitive elements, such as retrotransposons, the program RepeatMasker was employed using apple-specific settings (Smit et al., 1996) from the Repbase database (Jurka et al., 2005). Many repetitive elements were revealed in the interval for type 2 red flesh (Fig. 3). These include a Gypsy-like transposon element 835 bp from the translational start site of MYB110a and a Copia element downstream of MYB110a (Figs. 3 and 8). Another Copia element appears to have caused the truncation of MYB110b. In addition, a hAT element has increased the size of intron 2 of MYB110b. The Gypsy element in the promoter of MYB110a is 5,800 nucleotides in length but does not encode the Gag and Pol proteins required for the reverse transcription of the element and integration into the host genome, so it is likely to be an incomplete inactive retrotransposon.
We also examined the genomic region surrounding MYB110a and MYB110b using second-generation sequencing. We resequenced the entire genome of five apple accessions varying in their flesh color phenotype and compared these sequences with the reference genome of cv Golden Delicious. Our detection criteria successfully identified 64,206 SNPs on 27 Mb of LG 17, corresponding to an average of one SNP every 423 bp. We focused our SNP search in the region surrounding MYB110a and MYB110b (Table II). In total, 33 SNPs were located between the two stop codons of MYB110a and MYB110b. Three of the 33 SNPs had an allele found to be present in cv Sangrado OP and absent from the white flesh (cv Golden Delicious, Cox Orange Pippin, and Granny Smith) and type 1 red flesh (cv Geneva). No SNP was detected in the MYB110a exons and introns. Twenty-four SNPs were in the 60 kb upstream of the MYB110a start codon. Eight SNPs were found in MYB110b in the large second intron, including two SNPs with an allele only found in cv Sangrado OP.
Table II. SNPs detected by second-generation sequencing around MYB110a and MYB110b.
Five cultivars were used: Redfield OP (both type 1 and type 2 red flesh), Sangrado OP (type 2), Geneva (type 1), Granny Smith, and Cox Orange Pippin (white flesh). Three SNPs have an allele (boldface and underlined) only found in the type 2 red-flesh parent cv Sangrado OP. x, Missing data.
| Position (bp) on LG 17 | cv Redfield OP | cv Sangrado OP | cv Geneva | cv Granny Smith | cv Cox Orange Pippin | Position Relative to MYB110a and MYB110b | Distance to MYB110a Start |
|---|---|---|---|---|---|---|---|
| 24,904,168 | CC | AC | AC | AC | CC | In front of MYB110a start codon | −7,648 |
| 24,904,176 | AA | AG | AG | AG | AA | In front of MYB110a start codon | −7,656 |
| 24,904,807 | CC | CC | AC | AC | AC | In front of MYB110a start codon | −8,287 |
| 24,904,854 | TT | CT | CT | CT | CT | In front of MYB110a start codon | −8,334 |
| 24,904,964 | AA | AA | AA | AG | AA | In front of MYB110a start codon | −8,444 |
| 24,904,977 | TT | CT | TT | TT | TT | In front of MYB110a start codon | −8,457 |
| 24,913,900 | TT | CT | CT | CT | CT | In front of MYB110a start codon | −17,380 |
| 24,914,139 | AA | AC | CC | AC | AC | In front of MYB110a start codon | −17,619 |
| 24,914,161 | CC | CC | CT | CT | CT | In front of MYB110a start codon | −17,641 |
| 24,914,569 | CC | CT | CT | CT | CT | In front of MYB110a start codon | −18,049 |
| 24,914,621 | CC | CC | CC | CC | CT | In front of MYB110a start codon | −18,101 |
| 24,914,657 | GG | GG | AG | GG | GG | In front of MYB110a start codon | −18,137 |
| 24,914,783 | CC | CC | CT | CT | CT | In front of MYB110a start codon | −18,263 |
| 24,944,789 | CT | CT | CT | CT | CC | In front of MYB110a start codon | −48,269 |
| 24,944,792 | GG | GG | CG | CG | CC | In front of MYB110a start codon | −48,272 |
| 24,947,468 | CT | CT | CT | CT | CT | In front of MYB110a start codon | −50,948 |
| 24,950,313 | AA | AC | AC | AC | AC | In front of MYB110a start codon | −53,793 |
| 24,950,314 | TT | CT | CT | CT | CT | In front of MYB110a start codon | −53,794 |
| 24,954,654 | GG | GG | GT | GT | GT | In front of MYB110a start codon | −58,134 |
| 24,954,749 | CC | CC | AC | AC | AC | In front of MYB110a start codon | −58,229 |
| 24,954,753 | AA | AG | AG | AG | AG | In front of MYB110a start codon | −58,233 |
| 24,954,775 | CC | CC | CT | CT | CT | In front of MYB110a start codon | −58,255 |
| 24,956,590 | TT | CT | CT | CT | CT | In front of MYB110a start codon | −60,070 |
| 24,956,593 | GG | GG | AG | GG | AG | In front of MYB110a start codon | −60,073 |
| 24,963,058 | TT | CT | TT | TT | TT | MYB110b second intron | |
| 24,964,981 | CC | CT | CC | CC | CC | MYB110b second intron | |
| 24,965,139 | TT | CT | CT | CT | CT | MYB110b second intron | |
| 24,966,427 | CT | CC | CC | CC | CT | MYB110b second intron | |
| 24,969,165 | GG | GG | GG | GG | GT | MYB110b second intron | |
| 24,969,234 | TT | CT | CT | CT | CT | MYB110b second intron | |
| 24,969,403 | CC | CC | AC | AC | AC | MYB110b second intron | |
| 24,969,660 | CT | CT | CT | CT | CT | MYB110b second intron | |
| 24,969,911 | AG | GG | AG | x | AG | MYB110b second intron |
DISCUSSION
Transcriptional Control of Apple Anthocyanin Levels
Anthocyanins and proanthocyanidins are a class of phenylpropanoid whose accumulation is important for the expression of plant traits, such as color and astringency, which govern interactions with seed dispersers, as well as plant responses to biotic and abiotic stress. Hence, the control elements for the phenylpropanoid pathway are key factors in the evolution of plants, and their elucidation is of considerable interest. It is well known that R2R3 MYB TFs are responsible for controlling different aspects of the phenylpropanoid pathway in a wide range of different plant species. In apple, three MYB genes mapping to the same locus, MYB10, MYB1, and MYBA, have already been independently isolated (Takos et al., 2006; Ban et al., 2007; Espley et al., 2007). All of these control anthocyanin concentrations and exhibit a very high degree of sequence similarity. It has been suggested that these three genes are alleles arising from the different varieties from which they were cloned (Lin-Wang et al., 2010).
We have identified genes homologous to MYB10 that colocate with a QTL for type 2 red flesh on LG 17. According to analysis of the cv Golden Delicious sequence (Velasco et al., 2010), LG 9 and LG 17 are highly colinear and arose from whole-genome duplication. Therefore, it is likely that MYB110a and MYB110b are homeologous to MYB10 and control a similar trait (red coloration of the fruit). Although these genes have similar exon-intron structure to MYB10, MYB110b appears to code for a truncated protein. Real-time qPCR indicated that MYB110b does not accumulate transcript, suggesting that the predicted truncated protein is not responsible for type 2 red flesh. Transcript profiling of the expression of the functional homeologous genes MYB10 and MYB110a in the fruit cortex and colocation of MYB110a with a type 2 red-flesh QTL on LG 17 suggest that MYB110a specifically controls the type 2 red-flesh phenotype. Transient assays confirmed that MYB110a is able to regulate the anthocyanin pathway genes in a model plant system, analogous to previously characterized apple MYB genes.
MYB10 and MYB110: Insight into the Maloideae Genome Evolution
Several evolutionary models have been proposed to elucidate how gene copies are retained within duplicated regions of genomes (Force et al., 1999). Pseudogenization, which can be defined as a silencing or loss of function of one of the gene copies due to deleterious sequence mutations, does not seem to fully apply to MYB10 (LG 9) and MYB110a (LG 17), as our findings suggest that both homeologs were retained following the Maloideae whole-genome duplication and that both genes control a phenotypic variation. Possible hypotheses for the evolution of MYB10 and MYB110a genes could be that they are either subfunctionalized or neofunctionalized. Subfunctionalization assumes that each of the duplicated genes has specialized to give rise to functions that are complementary to each other and jointly match the function of an ancestral gene. When the publicly available apple EST data sets are analyzed (Sanzol, 2010), extensive subfunctionalization appears to have occurred in the apple genome, meaning that a large number of gene copies from the WGD were retained. This hypothesis could apply to the evolution of MYB10 and MYB110a, as both MYB10 and MYB110a are capable of activating the anthocyanin pathway genes, although their tissue specificity differs. MYB10 possesses alleles coding for the presence or absence of skin, flesh, and foliage (type 1) coloration (Takos et al., 2006; Ban et al., 2007; Chagné et al., 2007; Espley et al., 2007, 2009), and these alleles are expressed in these tissues. MYB110a may be controlling type 2 red flesh coloration, which is only expressed in the fruit cortex and late during fruit development. The expression of MYB110a is shown in fruit cortex at the end of the season, coinciding with pigmentation and the expression of pathway genes. The protein sequences of MYB10 and MYB110a are very similar, suggesting that the different expression of the two genes, and therefore their subfunctionalization, might be due to differences in the regulatory regions. Another plausible hypothesis is that both genes evolved through neofunctionalization, which assumes that their sequences independently accumulate mutations until one of the copies generates a novel character and the other retains the original function. In this case, we could hypothesize that MYB10 is very similar to the ancestral gene, with different alleles coding for the presence or absence of skin, flesh, and foliage (type 1) coloration. Cortex coloration would be a novel trait that neofunctionalized during the evolution of the ancestral gene into MYB110a.
Toward the Identification of Mutations Responsible for Type 2 Red Flesh in Apple
Upstream of MYB110a, there are regulatory regions that may be associated with the elevation of MYB110a expression in certain cultivars to control type 2 red flesh. In the case of type 1 red flesh, a minisatellite containing a MYB-binding domain was found to be responsible for MYB10 inducing its own expression (Espley et al., 2009). However, such a mutation is not responsible for type 2 red flesh. Using second-generation resequencing of five apple cultivars, we identified 33 SNPs in the 60-kb region surrounding MYB110a and MYB110b, of which three are found only in the type 2 red-flesh parent cv Sangrado OP. In addition, there are many transposons and repetitive elements in this region. It will be necessary to functionally assay these sequence polymorphisms as well as methylation patterns to determine the potential causative mutations for type 2 red flesh. In blood oranges (Citrus sinensis), an insertion of a Copia-like retrotransposon within the promoter adjacent to the MYB Ruby activates anthocyanin production (Butelli et al., 2012). Expression is elicited by cold, because the long terminal repeats contain cold-responsive elements. However, in the case of orange, such insertions are much closer to the ATG start site than that seen in apple (Fig. 8).
The Copia and Gypsy elements seen within the type 2 red-flesh QTL interval are both class I long terminal repeat retrotransposons that replicate through an RNA intermediate that is reverse transcribed into a DNA copy that can insert elsewhere in the genome (Kumar and Bennetzen, 1999). Such elements are not uncommon in the apple genome (with 425K repeats estimated; Velasco et al., 2010). These may attract both genetic and epigenetic changes to activate or deactivate genes. In Arabidopsis, for example, epigenetic regulation of transposable elements and tandem repeats contribute to the regulation of neighboring genes (Lippman et al., 2004; Hollister and Gaut, 2009) and divergence between Arabidopsis species (Pereira, 2004). Such activity could well be the genetic or epigenetic cause of the red-fleshed phenotype. In grape (Vitis vinifera), insertion of the Gypsy-like retrotransposon, Gret1, suppresses the expression of MYBA1, resulting in the development of white-skinned berries (Kobayashi et al., 2004).
CONCLUSION
We have shown how a duplicated TF in the apple genome, retained as two functional copies that evolved independently due to the recent whole-genome duplication, could have given rise to a novel character. This scenario of the evolution of two duplicated genes is likely to be found widely within duplicated plant genomes. The assembly and annotation of the whole-genome sequence for apple have indicated that the overall number of genes within the apple genome is higher than in other plant species so far sequenced (Velasco et al., 2010). It has been hypothesized that evolution of the pome, a fruit found only in the Pyreae tribe (Potter et al., 2007), was enabled by the Maloideae whole-genome duplication (Velasco et al., 2010). Our results support this hypothesis, providing a good visual demonstration of how pairs of recently duplicated genes can evolve to define novel fruit phenotypes in an apple-specific tissue. Additionally, we have generated a highly specific gene-based marker linked to type 2 red flesh in apple for use by apple breeders.
MATERIALS AND METHODS
Plant Material
A seedling family produced from a cross in 1998 between the white-fleshed commercial apple (Malus × domestica) line cv Sciros and an OP red-fleshed selection derived from cv Sangrado was used as a QTL detection population. A total of 442 own-rooted seedlings were planted in four rows at 0.5-m × 3.0-m spacing in a Plant and Food Research orchard block in 2001. Three additional seedling families from crosses made in 2005 between one red-fleshed individual (NZSelectionT051) from the QTL detection population and three white-fleshed cultivars (NZSelectionT153 [229 trees], NZSelectionT179 [40 trees], and Sciros [285 trees]; Kumar et al., 2012) were used for QTL validation and fine-mapping. The three QTL validation families were grafted on M.9 rootstock and planted in a Plant and Food Research orchard in 2008. All trees were sprayed with pesticides according to commercial guidelines and otherwise received minimal management (pruning and thinning).
Fruit Phenotyping
In 2007 and 2008, each fruiting tree in the QTL detection family was harvested once when fruit on each tree were judged to be eating ripe. Six ripe fruit were randomly sampled within the tree canopy, and then each fruit was cut in transverse cross-section through its equator. The six fruit were positioned on a glass plate with the cut surface facing downward; the glass plate was placed on the bed of a color scanner (HP Scanjet 5400c) and scanned at 150 dots per inch. The proportion of red color and the intensity of the red (on a 0–9 scale using a reference chart) in the cross-sectional area of each fruit image were later estimated visually for cortex and core separately by a single trained assessor.
For the QTL validation of seedling populations, fruit were harvested in 2010 at midmaturity and at eating ripe, stored for 10 weeks at 0.5°C plus 1 week at 20°C, and then assessed for red color intensity and coverage in the cortex as described above by two trained assessors.
A derived variable, WCI, was calculated for each fruit as cortex coverage × cortex intensity/100 for both the QTL detection and validation populations, as it gave a more continuous distribution than either cortex coverage or intensity taken alone (Supplemental Fig. S1).
Genome-Scan Analysis
Extreme phenotypes were selected from the segregating population (i.e. intensely red fruit flesh with greater than 80% cortex coverage or complete absence of red flesh). We used 38 markers covering the whole apple genome, including microsatellite (Silfverberg-Dilworth et al., 2006; Celton et al., 2009), sequence-characterized amplified region (Baldi et al., 2004), and SNP markers (Chagné et al., 2008), to screen DNA extracted from leaves of both parents, three high-cortex-coverage red-fleshed and three white-fleshed individuals. The PCR conditions for each class of marker were as described in their original publication. PCR fragments for simple sequence repeat markers were separated using a CePRO 9600 capillary electrophoresis system (Advance Analytical Technologies). Sequence-characterized amplified region and SNP markers were PCR amplified on a LightCycler 480 (Roche), and the polymorphisms were detected using the HRM technique (Liew et al., 2004) as described by Chagné et al. (2008).
The mapping strategy used was the double pseudo-test-cross (Grattapaglia and Sederoff, 1994). Genetic linkage analysis was performed using Joinmap version 3.0 (Van Ooijen and Voorrips, 2001) using the Kosambi mapping function with a LOD score of 6 for grouping.
Candidate Gene Mapping
DNA polymorphism detection and candidate gene marker genotyping were with the HRM technique (Liew et al., 2004; Chagné et al., 2008) on a LightCycler 480 (Roche). PCR primers for the MYB110a HRM marker were as follows: forward (5′-GAAGACCTTGTTAGAAGACGACGATATA-3′) and reverse (5′-TATGATCTTGCCGACTGTTGCATAT-3′).
Candidate apple anthocyanin-activating MYBs were identified by BLAST search against the apple genome (Velasco et al., 2010). A phylogenetic tree of R2R3 MYBs was built with PhyML (Guindon et al., 2005), based on the alignment of the MYBs with Mafft (Katoh et al., 2002), using default parameters and the LG substitution method along with a maximum likelihood with 100 bootstrap (Le and Gascuel, 2008).
Genetic Map Construction for Fine-QTL Mapping
DNA from 450 F1 individuals of the three QTL validation families was extracted using the Nucleospin kit (Macherey-Nagel). Genomic DNA was amplified using the Genomi-φ kit and then screened using the Apple International RosBREED SNP Consortium Infinium II 8k SNP array (Chagné et al., 2012). SNP data were analyzed using GenomeStudio (Illumina). SNPs were evaluated based on their GenTrain score: SNPs with GenTrain score greater than 0.6 were retained, and SNPs with GenTrain score between 0.3 and 0.6 were visually checked for accuracy of the SNP calling. Clusters were manually edited when the parent-child segregation was not correct or when the number of missing genotypes was too great.
Genetic markers segregating in a backcross manner were selected for parental map construction following the double pseudo-test-cross mapping strategy (Grattapaglia and Sederoff, 1994). Joinmap 3.0 (van Oiijen 2004) was used for genetic map construction using a LOD threshold of 6 for grouping and the Kosambi function for genetic distance calculation. A framework genetic map with genetic markers evenly distributed at regular intervals was developed to allow the use of interval-mapping analysis during the QTL analysis.
QTL Analysis of Phenotype Data
For the QTL detection population, a mean WCI was calculated from measurements taken in 2007 and 2008. The unequal precision of the data were handled by using a BLUP analysis to obtain predictions of the seedling red phenotype. This ensures that seedlings with fewer replicates (year × harvest × fruit) are shrunk toward the average. The mixed model is:
where the seedling gi and the rep rij effects are random variables with corresponding variances 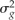 and
and 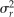 , respectively, and
, respectively, and 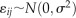 , whose residual variance includes variation between fruit and any variation due to uncertainty in the measurement. The BLUP analysis was carried out using SAS (SAS Institute).
, whose residual variance includes variation between fruit and any variation due to uncertainty in the measurement. The BLUP analysis was carried out using SAS (SAS Institute).
For the QTL validation population, no BLUP value could be calculated, as only 1 year of phenoptyping was performed. Therefore, QTL analysis was performed using raw phenotypic data for fruit cortex coverage and intensity transformed to WCI. QTL analysis was done using MapQTL version 5 (www.kyazma.nl). For the Kruskal-Wallis test, the entire set of polymorphic SNPs on LG 17 was used after calculating the marker phase using Joinmap 3.0. The polymorphic markers were sorted by their physical locations on LG 17 based on the apple genome assembly (Velasco et al., 2010). For the interval-mapping analysis, a reduced framework map was developed due to computing limitations. The LOD threshold for the significance of a QTL was calculated at the genome level using 1,000 permutations, and QTLs significant at 99% genome wide were retained. The most significant marker for each QTL was then used as a cofactor for a multiple QTL analysis for detecting minor QTLs that were hidden by the major QTL in the previous interval-mapping analysis.
Real-Time qPCR Expression Analysis
For qPCR analysis, samples were extracted from four replicates, each replicate from three pooled mature fruit of the progeny of type 2 phenotype red flesh from the cv Sciros × NZSelectionT051 segregating population. The individual samples were labeled i to vii and refer to individual trees, numbered as follows: i, R04T009; iii, R08T068; iv, R03T120; v, R06T062; vi, R08T069; and vii, R05T060. For the same analysis, three pooled samples of each replicate were also extracted from the cortical flesh of NZSelectionT051 (ii), a representative type 1 red-flesh apple, cv Redfield OP, and from the peel of the representative commercial cv Sciros. For the apple fruit development series, samples were also collected from the cortical flesh of NZSelectionT051, Redfield OP, and Sciros at five time points: stage 1, 40 d after full bloom (DAFB); stage 2, 67 DAFB; stage 3, 102 DAFB; stage 4, 130 DAFB; stage 5, 146 DAFB (eating ripe). All trees were planted at the Plant and Food Research orchard in Hawke’s Bay, New Zealand.
RNA was isolated from fruit flesh using a method adapted from Chang et al. (1993). To remove genomic DNA contamination, all of the RNA samples were DNase treated (DNA-free; Ambion). First-strand complementary DNA synthesis was carried out using oligo(dT) according to the manufacturer’s instructions (SuperScript III; Invitrogen). To distinguish the expression of MYB110a from MYB10, primers for MYB110a were designed in the region from exon 3 into the 3′ UTR, where the sequence of MYB110a is significantly different from the sequence of MYB10. Ten qPCR amplicons for MYB110a were sequenced to confirm that neither MYB110b nor MYB10 was coamplified with the primers specific to MYB110a. The apple reference genes Actin (CN932644), EF1α (CN887003), and GAPDH (Schaffer et al., 2007) were selected due to their consistent transcript levels throughout fruit samples, with crossing threshold values changing by less than 2.
Real-time qPCR DNA amplification and analysis were carried out using the LightCycler 480 Real-Time PCR System (Roche), with LightCycler 480 software version 1.5. The LightCycler 480 SYBR Green I Master Mix (Roche) was used following the manufacturer’s method. The qPCR conditions were 5 min at 95°C, followed by 40 cycles of 5 s at 95°C, 5 s at 60°C, and 10 s at 72°C, followed by 65°C to 95°C melting-curve detection. The qPCR efficiency of each gene was obtained by analyzing the standard curve of a cDNA serial dilution of that gene.
Liquid Chromatography-Quadrupole-Time of Flight-Mass Spectrometry Analysis of Anthocyanins
For the analysis of anthocyanins, 1.5 g of fruit sample was flash frozen powdered, and the pigments were extracted in 10 mL of ethanol:water:formic acid (80:20:1, v/v/v). Cyandin 3-galactoside and cyanidin 3-glucoside were purchased from Exsynthese. Formic acid and methanol (liquid chromatography-mass spectrometry grade) were obtained from Thermo Scientific. The liquid chromatography-mass spectrometry system used was composed of a Dionex Ultimate 3000 Rapid Separation liquid chromatography system and a micrOTOF II mass spectrometer (Bruker Daltonics) fitted with an electrospray source operating in positive mode. The liquid chromatography system contained the following components: SRD-3400 solvent rack/degasser, HPR-3400RS binary pump, WPS-3000RS thermostatted autosampler, and a TCC-3000RS thermostatted column compartment. The analytical column used was a Zorbax SB-C18, 2.1 × 100, 1.8 µm (Agilent), maintained at 50°C and operated in gradient mode. Solvents were as follows: A = methanol:water (90:10); B = 0.5% formic acid at a flow of 400 µL min−1. The gradient was as follows: 5% A/95% B, 0 to 0.5 min; linear gradient to 40% A/60% B, 0.5 to 8 min; linear gradient to 75% A/25% B, 8 to 11 min; linear gradient to 100% A, 11 to 12 min; composition held at 100% A, 12 to 14 min; linear gradient to 5% A/95% B, 14 to 14.2 min; then return to the initial conditions before another sample injection at 16.5 min. The injection volume for samples and standards was 2 μL. The micrOTOF II source parameters were as follows: temperature, 200°C; drying N2 flow, 8 L min−1; nebulizer N2, 4 bars; end plate offset, −500 V; capillary voltage, −4,000 V; mass range, 100 to 1,500 D, acquired at 2 scans s−1. Postacquisition internal mass calibration used sodium formate clusters with the sodium formate delivered by a syringe pump at the start of each chromatographic analysis.
Dual Luciferase Assay of Transiently Transformed Nicotiana benthamiana Leaves
A fragment containing 1,300 bp upstream of the ATG of an apple CHS gene (AB074485) was isolated, inserted into the cloning site of pGreenII 0800-LUC (Hellens et al., 2005), and modified to introduce an NcoI site at the 3′ end of the sequence. This allowed the promoter to be cloned as a transcriptional fusion with the firefly Luciferase gene (LUC). The promoter-LUC fusion in pGreenII 0800-LUC was used in transient transformation by mixing 100 µL of Agrobacterium tumefaciens strain GV3101(MP90) transformed with the reporter cassette with 300 µL each of three other A. tumefaciens cultures. These three cultures had been transformed with cassettes containing a cDNA of MYB TF gene, bHLH TF gene, or WD40 gene fused to the 35S promoter, respectively, in either pART27 (Gleave, 1992) or pHex2 (Hellens et al., 2005). N. benthamiana plant growing conditions, A. tumefaciens infiltration processes, and luminescence measurements were as described by Hellens et al. (2005).
Induction of Anthocyanins by Transiently Transformed Tobacco Leaves
Two A. tumefaciens cultures containing the MYB110a or MYB10 TF gene and the bHLH3 TF gene fused to the 35S promoter were mixed (500 µL each) and infiltrated into the abaxial leaf surface of tobacco (Nicotiana tabacum) as for the dual luciferase assay described by Espley et al. (2007). The level of pigmentation increased throughout the experimental period, and digital photographs were taken 8 d after infiltration.
Second-Generation Whole-Genome Sequencing, and SNP and Repetitive Element Detection
Ten micrograms of pure genomic DNA was extracted using the Qiagen Plant mini kit (Qiagen) for five apple cultivars: Sangrado (OP), Redfield (OP), Geneva, Granny Smith, and Cox Orange Pippin. One library was prepared for each genomic DNA sample. Each library was then sequenced using one lane of the Illumina Genome Analyzer II at 75-bp pair-end reads. Sequences were retrieved as a FASTQ file and mapped to the cv Golden Delicious genome using the Short Oligonucleotide Analysis Package (soap2.20; http://soap.genomics.org.cn). SNPs were detected as described by Wang et al. (2008) using the following criteria: reference genome base must be A, C, G, or T (nonambiguous); base quality score was greater than 20; the overall sequencing depth was less than the average depth plus three times the sd of the sequence depth; the average copy number of the surrounding region was less than 2; the minimum distance between any two sequential SNPs was 5 bp or greater; 150 flanking bases must be present on either side of the SNP position (excludes SNPs called in the first or last 151 bases of the reference sequence); the left flanking 150-base reference genome region must not start with one or more unknown nucleotides; the right flanking 150-base reference genome region must terminate in one or more unknown nucleotides. Filtering was performed using a in-house Perl script.
Repetitive elements were identified using RepeatMasker with apple-specific settings (Smit et al., 1996) from the Repbase database (Jurka et al., 2005).
Sequence data from this article can be found in the GenBank/EMBL data libraries under accession numbers JN711473 and JN711474.
Supplemental Data
The following materials are available in the online version of this article.
Supplemental Figure S1. Phenotypic variation of type 2 red flesh in the cv Sciros × Sangrado OP segregating population.
Supplemental Figure S2. Nucleotide sequence alignment of MYB10, MYB110a and MYB110b.
Supplemental Table S1. Candidate genes located in the QTL interval for type 2 red flesh on LG 17.
Supplemental Table S2. Oligonucleotide primer sequences used for qPCR analysis.
Acknowledgments
We thank Kim Green and Mukarram Mohammed (Plant & Food Research, Palmerston North) for their technical help with the genetic map construction and Slipstream Automation for assistance with DNA purification. We also thank Prof. Amy Iezzoni, Dr. Nahla Bassil, Dr. Jeff Landgraf, and Shari Tjugum-Holland (Michigan State University) for providing whole-genome resequencing service; Dianne Hyndman, Paul Fisher, Rosemary Rickman, and Matt Bixley (AgResearch Invermay) for providing Illumina genotyping service; and Drs. Kathy Schwinn and Kevin Davies (Plant & Food Research, Palmerston North) for their useful comments on the manuscript.
Glossary
- TF
transcription factor
- bHLH
basic helix-loop-helix
- QTL
quantitative trait locus
- LG
linkage group
- OP
open pollinated
- WCI
weighted cortex intensity
- SNP
single-nucleotide polymorphism
- LOD
log of the odds
- HRM
high-resolution melting
- qPCR
quantitative PCR
- UTR
untranslated region
- MBW
MYB-bHLH-WD40
- DAFB
days after full bloom
References
- Adams KL, Wendel JF. (2005) Polyploidy and genome evolution in plants. Curr Opin Plant Biol 8: 135–141 [DOI] [PubMed] [Google Scholar]
- Allan AC, Hellens RP, Laing WA. (2008) MYB transcription factors that colour our fruit. Trends Plant Sci 13: 99–102 [DOI] [PubMed] [Google Scholar]
- Baldi P, Patocchi A, Zini E, Toller C, Velasco R, Komjanc M. (2004) Cloning and linkage mapping of resistance gene homologues in apple. Theor Appl Genet 109: 231–239 [DOI] [PubMed] [Google Scholar]
- Ban Y, Honda C, Hatsuyama Y, Igarashi M, Bessho H, Moriguchi T. (2007) Isolation and functional analysis of a MYB transcription factor gene that is a key regulator for the development of red coloration in apple skin. Plant Cell Physiol 48: 958–970 [DOI] [PubMed] [Google Scholar]
- Baudry A, Heim MA, Dubreucq B, Caboche M, Weisshaar B, Lepiniec L. (2004) TT2, TT8, and TTG1 synergistically specify the expression of BANYULS and proanthocyanidin biosynthesis in Arabidopsis thaliana. Plant J 39: 366–380 [DOI] [PubMed] [Google Scholar]
- Borevitz JO, Xia YJ, Blount J, Dixon RA, Lamb C. (2000) Activation tagging identifies a conserved MYB regulator of phenylpropanoid biosynthesis. Plant Cell 12: 2383–2394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brueggemann J, Weisshaar B, Sagasser M. (2010) A WD40-repeat gene from Malus × domestica is a functional homologue of Arabidopsis thaliana TRANSPARENT TESTA GLABRA1. Plant Cell Rep 29: 285–294 [DOI] [PubMed] [Google Scholar]
- Butelli E, Licciardello C, Zhang Y, Liu J, Mackay S, Bailey P, Reforgiato-Recupero G, Martin C. (2012) Retrotransposons control fruit-specific, cold-dependent accumulation of anthocyanins in blood oranges. Plant Cell 24: 1242–1255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butelli E, Titta L, Giorgio M, Mock HP, Matros A, Peterek S, Schijlen EG, Hall RD, Bovy AG, Luo J, et al. (2008) Enrichment of tomato fruit with health-promoting anthocyanins by expression of select transcription factors. Nat Biotechnol 26: 1301–1308 [DOI] [PubMed] [Google Scholar]
- Celton JM, Tustin DS, Chagné D, Gardiner SE. (2009) Construction of a dense genetic linkage map for apple rootstocks using SSRs developed from Malus ESTs and Pyrus genomic sequences. Tree Genet Genomes 5: 93–107 [Google Scholar]
- Chagné D, Carlisle CM, Blond C, Volz RK, Whitworth CJ, Oraguzie NC, Crowhurst RN, Allan AC, Espley RV, Hellens RP, et al. (2007) Mapping a candidate gene (MdMYB10) for red flesh and foliage colour in apple. BMC Genomics 8: 212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chagné D, Crowhurst RN, Troggio M, Davey MW, Gilmore B, Lawley C, Vanderzande S, Hellens RP, Kumar S, Cestaro A, et al. (2012) Genome-wide SNP detection, validation, and development of an 8K SNP array for apple. PLoS ONE 7: e31745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chagné D, Gasic K, Crowhurst RN, Han Y, Bassett HC, Bowatte DR, Lawrence TJ, Rikkerink EHA, Gardiner SE, Korban SS. (2008) Development of a set of SNP markers present in expressed genes of the apple. Genomics 92: 353–358 [DOI] [PubMed] [Google Scholar]
- Chang S, Puryear J, Cairney J. (1993) A simple and efficient method for isolating RNA from pine trees. Plant Mol Biol Rep 11: 113–116 [Google Scholar]
- Cipollini ML. (2000) Secondary metabolites of vertebrate-dispersed fruits: evidence for adaptive functions. Rev Chil Hist Nat 73: 421–440 [Google Scholar]
- Espley RV, Brendolise C, Chagné D, Kutty-Amma S, Green S, Volz R, Putterill J, Schouten HJ, Gardiner SE, Hellens RP, et al. (2009) Multiple repeats of a promoter segment causes transcription factor autoregulation in red apples. Plant Cell 21: 168–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espley RV, Hellens RP, Putterill J, Stevenson DE, Kutty-Amma S, Allan AC. (2007) Red colouration in apple fruit is due to the activity of the MYB transcription factor, MdMYB10. Plant J 49: 414–427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Force A, Lynch M, Pickett FB, Amores A, Yan YL, Postlethwait J. (1999) Preservation of duplicate genes by complementary, degenerative mutations. Genetics 151: 1531–1545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gleave AP. (1992) A versatile binary vector system with a T-DNA organisational structure conducive to efficient integration of cloned DNA into the plant genome. Plant Mol Biol 20: 1203–1207 [DOI] [PubMed] [Google Scholar]
- Grattapaglia D, Sederoff R. (1994) Genetic linkage maps of Eucalyptus grandis and Eucalyptus urophylla using a pseudo-testcross: mapping strategy and RAPD markers. Genetics 137: 1121–1137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grotewold E, Athma P, Peterson T. (1991) Alternatively spliced products of the maize P gene encode proteins with homology to the DNA-binding domain of myb-like transcription factors. Proc Natl Acad Sci USA 88: 4587–4591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon S, Lethiec F, Duroux P, Gascuel O. (2005) PHYML Online: a Web server for fast maximum likelihood-based phylogenetic inference. Nucleic Acids Res 33: W557–W559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellens RP, Allan AC, Friel EN, Bolitho K, Grafton K, Templeton MD, Karunairetnam S, Gleave AP, Laing WA. (2005) Transient expression vectors for functional genomics, quantification of promoter activity and RNA silencing in plants. Plant Methods 1: 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertog MGL, Feskens EJM, Hollman PCH, Katan MB, Kromhout D. (1993) Dietary antioxidant flavonoids and risk of coronary heart disease: the Zutphen Elderly Study. Lancet 342: 1007–1011 [DOI] [PubMed] [Google Scholar]
- Higo K, Ugawa Y, Iwamoto M, Korenaga T. (1999) Plant cis-acting regulatory DNA elements (PLACE) database: 1999. Nucleic Acids Res 27: 297–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollister JD, Gaut BS. (2009) Epigenetic silencing of transposable elements: a trade-off between reduced transposition and deleterious effects on neighboring gene expression. Genome Res 19: 1419–1428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaeger SR, Harker FR. (2005) Consumer evaluation of novel kiwifruit: willingness-to-pay. J Sci Food Agric 85: 2519–2526 [Google Scholar]
- Jordano P, García C, Godoy JA, García-Castaño JL. (2007) Differential contribution of frugivores to complex seed dispersal patterns. Proc Natl Acad Sci USA 104: 3278–3282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurka J, Kapitonov VV, Pavlicek A, Klonowski P, Kohany O, Walichiewicz J. (2005) Repbase Update, a database of eukaryotic repetitive elements. Cytogenet Genome Res 110: 462–467 [DOI] [PubMed] [Google Scholar]
- Katoh K, Misawa K, Kuma K, Miyata T. (2002) MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res 30: 3059–3066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi S, Goto-Yamamoto N, Hirochika H. (2004) Retrotransposon-induced mutations in grape skin color. Science 304: 982. [DOI] [PubMed] [Google Scholar]
- Kumar A, Bennetzen JL. (1999) Plant retrotransposons. Annu Rev Genet 33: 479–532 [DOI] [PubMed] [Google Scholar]
- Kumar S, Bink MCAM, Volz RK, Bus VGM, Chagné D. (2012) Towards genomic selection in apple (Malus × domestica Borkh.) breeding programmes: prospects, challenges and strategies. Tree Genet Genomes 8: 1–14 [Google Scholar]
- Le SQ, Gascuel O. (2008) An improved general amino acid replacement matrix. Mol Biol Evol 25: 1307–1320 [DOI] [PubMed] [Google Scholar]
- Liew M, Pryor R, Palais R, Meadows C, Erali M, Lyon E, Wittwer C. (2004) Genotyping of single-nucleotide polymorphisms by high-resolution melting of small amplicons. Clin Chem 50: 1156–1164 [DOI] [PubMed] [Google Scholar]
- Lin-Wang K, Bolitho K, Grafton K, Kortstee A, Karunairetnam S, McGhie TK, Espley RV, Hellens RP, Allan AC. (2010) An R2R3 MYB transcription factor associated with regulation of the anthocyanin biosynthetic pathway in Rosaceae. BMC Plant Biol 10: 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippman Z, Gendrel AV, Black M, Vaughn MW, Dedhia N, McCombie WR, Lavine K, Mittal V, May B, Kasschau KD, et al. (2004) Role of transposable elements in heterochromatin and epigenetic control. Nature 430: 471–476 [DOI] [PubMed] [Google Scholar]
- Maliepaard C, Alston FH, van Arkel G, Brown LM, Chevreau E, Dunemann F, Evans KM, Gardiner S, Guilford P, van Heusden AW, et al. (1998) Aligning male and female linkage maps of apple (Malus pumila Mill.) using multi-allelic markers. Theor Appl Genet 97: 60–73 [Google Scholar]
- Mazza G, Velioglu YS. (1992) Anthocyanins and other phenolic-compounds in fruits of red-flesh apples. Food Chem 43: 113–117 [Google Scholar]
- Newcomb RD, Crowhurst RN, Gleave AP, Rikkerink EHA, Allan AC, Beuning LL, Bowen JH, Gera E, Jamieson KR, Janssen BJ, et al. (2006) Analyses of expressed sequence tags from apple. Plant Physiol 141: 147–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira V. (2004) Insertion bias and purifying selection of retrotransposons in the Arabidopsis thaliana genome. Genome Biol 5: R79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potter D, Eriksson T, Evans RC, Oh S, Smedmark JEE, Morgan DR, Kerr M, Robertson KR, Arsenault M, Dickinson TA, et al. (2007) Phylogeny and classification of Rosaceae. Plant Syst Evol 266: 5–43 [Google Scholar]
- Regan BC, Julliot C, Simmen B, Vienot F, Charles-Dominique P, Mollon JD. (2001) Fruits, foliage and the evolution of primate colour vision. Philos Trans R Soc Lond Ser B Biol Sci 356: 229–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renaud S, de Lorgeril M. (1992) Wine, alcohol, platelets, and the French paradox for coronary heart disease. Lancet 339: 1523–1526 [DOI] [PubMed] [Google Scholar]
- Sanzol J. (2010) Dating and functional characterization of duplicated genes in the apple (Malus domestica Borkh.) by analyzing EST data. BMC Plant Biol 10: 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaffer RJ, Friel EN, Souleyre EJF, Bolitho K, Thodey K, Ledger S, Bowen JH, Ma J-H, Nain B, Cohen D, et al. (2007) A genomics approach reveals that aroma production in apple is controlled by ethylene predominantly at the final step in each biosynthetic pathway. Plant Physiol 144: 1899–1912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwinn K, Venail J, Shang YJ, Mackay S, Alm V, Butelli E, Oyama R, Bailey P, Davies K, Martin C. (2006) A small family of MYB-regulatory genes controls floral pigmentation intensity and patterning in the genus Antirrhinum. Plant Cell 18: 831–851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekido K, Hayashi Y, Yamada K, Shiratake K, Matsumoto S, Maejima T, Komatsu H. (2010) Efficient breeding system for red-fleshed apple based on linkage with S3-RNase allele in ‘Pink Pearl’. HortScience 45: 534–537 [Google Scholar]
- Silfverberg-Dilworth E, Matasci CL, Van de Weg WE, Van Kaauwen MPW, Walser M, Kodde LP, Soglio V, Gianfranceschi L, Durel CE, Costa F, et al. (2006) Microsatellite markers spanning the apple (Malus × domestica Borkh.) genome. Tree Genet Genomes 2: 202–224 [Google Scholar]
- Smit AFA, Hubley R, Green P. (1996) RepeatMasker Open-3.0. http://www.repeatmasker.org (April 18, 2012)
- Stintzing FC, Carle R. (2004) Functional properties of anthocyanins and betalains in plants, food, and in human nutrition. Trends Food Sci Technol 15: 19–38 [Google Scholar]
- Stracke R, Werber M, Weisshaar B. (2001) The R2R3-MYB gene family in Arabidopsis thaliana. Curr Opin Plant Biol 4: 447–456 [DOI] [PubMed] [Google Scholar]
- Takos AM, Jaffé FW, Jacob SR, Bogs J, Robinson SP, Walker AR. (2006) Light-induced expression of a MYB gene regulates anthocyanin biosynthesis in red apples. Plant Physiol 142: 1216–1232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traveset A, Willson MF. (1998) Ecology of the fruit-colour polymorphism in Rubus spectabilis. Evol Ecol 12: 331–345 [Google Scholar]
- Umemura H, Shiratake K, Matsumoto S, Maejima T, Komatsu H. (2011) Practical breeding of red-fleshed apple: cultivar combination for efficient red-fleshed progeny production. HortScience 46: 1098–1101 [Google Scholar]
- van Ooijen JW, Voorrips RE. (2001) JoinMap 3.0. Plant Research International, Wageningen, The Netherlands
- Velasco R, Zharkikh A, Affourtit J, Dhingra A, Cestaro A, Kalyanaraman A, Fontana P, Bhatnagar SK, Troggio M, Pruss D, et al. (2010) The genome of the domesticated apple (Malus × domestica Borkh.). Nat Genet 42: 833–839 [DOI] [PubMed] [Google Scholar]
- Volz RK, Chagné D, Whitworth CJ, Espley RV, Allan AC, Carlisle CM, Oraguzie NC, Gardiner SE. (2006) Breeding for red flesh in apple. In CF Mercer, ed, 13th Australasian Plant Breeding Conference, Christchurch, New Zealand, pp 149–149
- Volz RK, Oraguzie NC, Whitworth CJ, How N, Chagné D, Carlisle CM, Gardiner SE. (2009) Red flesh breeding in apple: progress and challenges. Acta Hortic 814: 337–342 [Google Scholar]
- Wang H, Cao GH, Prior RL. (1997) Oxygen radical absorbing capacity of anthocyanins. J Agric Food Chem 45: 304–309 [Google Scholar]
- Wang J, Li R, Li Y, Fang X, Feng B, Li J. (2008) Genome resequencing and identification of variations by Illumina Genome Analyzer Reads. Protocol Exchange doi:10.1038/nprot.2008.238 [Google Scholar]
- Wendel JF. (2000) Genome evolution in polyploids. Plant Mol Biol 42: 225–249 [PubMed] [Google Scholar]
- Wolfe K, Wu XZ, Liu RH. (2003) Antioxidant activity of apple peels. J Agric Food Chem 51: 609–614 [DOI] [PubMed] [Google Scholar]
- Zhang P, Chopra S, Peterson T. (2000) A segmental gene duplication generated differentially expressed myb-homologous genes in maize. Plant Cell 12: 2311–2322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Evans KM, Peace C. (2011) Utility testing of an apple skin color MdMYB1 marker in two progenies. Mol Breed 27: 525–532 [Google Scholar]
- Zimmermann IM, Heim MA, Weisshaar B, Uhrig JF. (2004) Comprehensive identification of Arabidopsis thaliana MYB transcription factors interacting with R/B-like BHLH proteins. Plant J 40: 22–34 [DOI] [PubMed] [Google Scholar]