

Abstract
A method is described for performing global alignment of noncoding DNA sequences based on an evolutionary model parameterized by the frequency distribution of lengths of insertion/deletion events (indels) and their rate relative to nucleotide substitutions. A stochastic hill-climbing algorithm is used to search for the most probable alignment between a pair of sequences or three sequences of known phylogenetic relationship. The performance of the procedure, parameterized according to the empirical distribution of indel lengths in noncoding DNA of Drosophila species, is investigated by simulation. We show that there is excellent agreement between true and estimated alignments over a wide range of sequence divergences, and that the method outperforms other available alignment methods.
The genomes of higher eukaryotes contain large amounts of noncoding DNA in the form of intergenic DNA and introns. Unraveling the functional significance of noncoding DNA is a central problem in genome science, research into which has been stimulated by the rapid increase in the quantity of noncoding DNA sequences deposited in public data bases, and by the surprising finding that the genomes of multicellular eukaryotes contain substantially fewer genes than expected. Sequence alignment is a major issue for the evolutionary analysis of noncoding DNA.
Unless sequence divergence is high there is usually little difficulty in producing convincing alignments of protein-coding DNA sequences, because indels almost invariably occur in multiples of three base pairs (bp), and rarely cross codon boundaries. However, unless sequence divergence is low, indels cause severe problems for the alignment of noncoding DNA. The essence of the problem is that relative to the true alignment, putative alignments with too many gaps or gaps that are overfragmented tend to have too few nucleotide differences, whereas alignments with too few gaps tend to have too many differences. This is illustrated by the following example of a pair of sequences having two plausible alignments: Without other information it would be impossible to know which alignment is correct, although they have radically different proportions of nucleotide differences. Heuristic alignment methods produce alignments by minimizing a scoring function that depends on parameters, such as the extent by which gaps are penalized relative to nucleotide changes, whose values are chosen in a more or less arbitrary fashion. Inferences based on such alignments, such as estimates of the degree of sequence divergence or conservation, are therefore almost certainly biased (Thorne et al. 1991). Such heuristic methods have frequently been used for alignment of noncoding DNA from diverse species (e.g., Gu and Li 1995; Jareborg et al. 1999; Shabalina and Kondrashov 1999; Bergman and Kreitman 2001; Shabalina et al. 2001; Mouse Genome Sequencing Consortium 2002). A major problem with parameterizing scoring functions is that it is unclear how the relative penalties for substitution and indel events relate to parameters of DNA sequence evolution. Explicit model based approaches are therefore highly desirable.
Table 1.
| Alignment 1: | TTATA––––CAG | three nucleotide differences; |
| TTAGCTAAGCCG | ||
| Alignment 2: | TTA––TA––CAG | one nucleotide difference |
| TTAGCTAAGCCG |
By building on methods introduced by Bishop and Thompson (1986), Thorne, Kishino, and Felsenstein (1991; TKF hereafter) introduced a likelihood-based approach with an evolutionary model of indel evolution to estimate evolutionary parameters from a pair of related sequences. Initially, the model allowed only single-base pair indels (Thorne et al. 1991), and this was subsequently extended to allow a geometric distribution of indel lengths, under the rather unrealistic assumption that multibase insertions could only be deleted as a single unit and vice versa (Thorne et al. 1992). TKF's approach has been widely used as a basis for DNA alignment methods involving evolutionary models (e.g., Holmes and Bruno 2001; Metzler et al. 2001; Steel and Hein 2001). Here, an approach for the alignment of noncoding DNA is described that diverges in several ways from TKF's. First, the alignment is estimated assuming a model that allows an arbitrary distribution of indel lengths, whereas current implementations of the TKF model (e.g., Holmes and Bruno 2001) assume single-base pair indels only. Secondly, the distribution of indel lengths is derived empirically from additional data. As an example, the model is parameterized according to the frequency distribution of indel lengths in intronic DNA of the closely related species Drosophila simulans and D. sechellia, for which alignments are essentially unambiguous, which can then be used to estimate alignments from the more distantly related D. melanogaster and D. yakuba (Fig. 1). Thirdly, TKF use a dynamic programming algorithm that guarantees to find the (or one of the joint) most probable alignment(s), whereas we use a stochastic hill-climbing algorithm that searches for more probable alignment(s) and terminates if no improvement is found after a predetermined number of iterations. We have implemented our method for only the simplest model of nucleotide substitution, the Jukes-Cantor (1969) model, but we describe how the method can be extended to more realistic models. Our procedure is intended for global alignment of noncoding DNA sequences that are known to be homologous, as opposed to local alignment of complete genomes or of long contigs containing substantial nonhomologous stretches of DNA.
Figure 1.

The phylogeny of Drosophila species closely related to D. simulans (sim), including D. sechellia (sec), D. melanogaster (mel), and D. yakuba (yak).
The statistical properties of our method are examined using simulations. We employ simulations rather than real data because in the latter case the true alignment cannot be known, making quantification of accuracy impossible. We show that alignments generated by our method lead to estimates of sequence divergence that are better (less biased and more efficient) and to alignments that are closer to the true alignments than alignments generated by other available methods, for almost all combinations of parameter values we have considered.
Statistical Framework
The simplest statistical framework within which we can estimate the alignment between a set of sequences is the Bayesian one (Gelman et al. 2003), in which the behaviors of all variables are modeled by probability distributions. Let a be a discrete variable describing the alignment, t be a (possibly vector) parameter of sequence evolution, and S be the observed sequence data. Joint inference about a and t is accomplished via the conditional probability
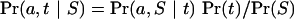 |
(1) |
and inference about a alone, treating t as a “nuisance” parameter, is made via the marginal probability
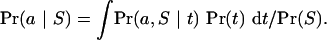 |
(2) |
Equations (1) and (2) show the central role played in inference by the probability Pr(a,S | t), which is supplied by the model described below. The unconditional Pr(S) is constant (because S is fixed) but in general is difficult to calculate; we therefore make inference using relative probabilities that are related to Pr(a,t | S) and Pr(a | S) by an unknown constant 1/Pr(S).
Probability Model of Sequence Evolution
Pr(a,S | t) is the product of two components that we derive separately. The first is the probability of the alignment or indel pattern, Pr(a | t), and the second is the probability of the observed nucleotide sequences given that indel pattern, Pr(S | a,t).
For two sequences there is a single parameter of sequence evolution t = (t12), and for three sequences a second parameter is added so that t is a vector, t = (t12, t(12)3). Here t12 is the total time of evolution between the two (most closely related) sequences, and t(12)3 is the time of evolution between the common ancestor of these two sequences and the more distantly related sequence, where time is measured in units where on average one nucleotide substitution occurs per site per unit time. We assume throughout that the branch lengths from sequences 1 and 2 to the join with sequence 3 are equal.
In the common ancestor the two sequences were identical to the ancestral sequence, and there were no indels in the alignment. We suppose that, since that time, insertions and deletions occurred as independent processes with a total rate θ per inter-base site relative to nucleotide substitutions. Insertions and deletions are treated together because (for the two-sequence case, and in our representation) they have identical effects on the alignment. Because we ignore multiple hits with respect to indels, the probability of an indel is θt12 per interbase site. The proportion of indels of length i is wi, with Σiwi=1 when we neglect the very rare possibility of a deletion that is longer than the entire sequence. A particular alignment a is characterized by gi gaps of length i and m interbase sites at which indels could have occurred but did not, which we call non-indels. We approximate the number of non-indels by the number of adjacent nucleotide pairs. This approximation will be good when the amount of alignment consisting of indels is small and when insertions and deletions occur at approximately equal rates.
Conditioning on the observed length of the sequences, the probability of a given alignment a with gi gaps of length i and m non-indels is then
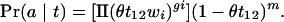 |
(3) |
The parameters θ and wi are treated as if they are known, although in practice they must be estimated from external data (see below).
To derive Pr(S | a,t) we use the Jukes-Cantor (1969) model of nucleotide substitution, which assumes that nucleotide substitutions occur as Poisson processes at equal rates, independent from each other and from indels. Then the probability of the observed sequences S given the alignment a and some value of t12 is
 |
(4) |
where
 |
(5) |
is the probability of a substitution difference between two homologous bases. In Equation (4), n is the number of nucleotide differences, l is the number of nucleotides that are not opposed by a gap, and u is the number of unopposed nucleotides, that is, those that are opposite a gap.
Deriving Pr(a,S | t) for three sequences is considerably more complicated than for two sequences because insertions and deletions do not have equivalent effects, and because substitutions on different branches of the tree do not have equivalent effects. An exact calculation would involve summing over all possible combinations of insertions, deletions, and substitutions that could give rise to the observed S. Here we take the simpler but approximate approach of calculating the probability of only the most parsimonious of those combinations. Let sequence 3 be known to be the outgroup to sequences 1 and 2. Parsimony is used to assign indel and nucleotide substitution events to either one of the ingroup branches, or to the outgroup branch. The total probability is simply the product of the probabilities for differences specific to the ingroups, and the probabilities for differences between (1,2) and 3. Because we are using parsimony to reconstruct the ancestral sequence, it seems inconsistent to account for multiple hits, and we replace Equation (5) with the linear relationship k12 = t12.
Bayesian inference requires an unconditional prior Pr(t). Here we consider only two very simple priors. The first is “uninformative” about the substitution probabilities, with k12 (and k(12)3 if three sequences are analyzed) uniformly distributed on the interval [0,0.75]. This implies independent exponential priors on t12 and t(12)3 with means 0.75. For many real data sets, however, there will be external information about the relative divergences. For such cases we consider a second prior that retains the uniform distribution for k12 and write t(12)3 = λ t12 with λ > 1 and treat λ as if it was known with complete certainty, so that t is effectively scalar. [We require λθ<1 for the multinomial gap distribution model; Equation (3).]
Alignment Algorithm
We were unable to develop a dynamic programming algorithm to find the most probable alignment(s) or to calculate likelihood functions that require summation over all alignments. We therefore developed a stochastic or Monte Carlo (MC) hill-climbing algorithm that searches for the alignment (or alignments) with (joint) highest probability Pr(a | S), conditional on some input sequences S. The algorithm essentially guarantees to converge to a local optimum, and can move between local optima to attempt to find a global optimum.
Our algorithm searches Pr(a | S) by visiting possible alignments one by one. In the case of the two-way alignments, we evaluate the integral in Equation (2) numerically for each alignment a that is visited. For three-way alignments, we made the following approximation, which greatly reduces execution time:
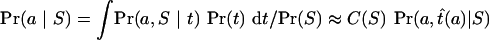 |
(6) |
where t̂(a) is the value of t that maximizes Pr(a,t | S) and C(S) is a constant that does not depend on a. This approximation works because we wish to integrate a function over t, with a held fixed, and the height of that function at its highest point, the “peak height” Pr(a,t̂(a)|S), turns out to be a good approximation to the area under the peak. For alignments with probability greater than 0.01 times the probability of the most probable (MP) alignment the correlation between Pr(a,t̂(a)|S) and Pr(a | S) exceeds 0.98, for cases we have examined. The unknown constant C(S) can be neglected because we only use relative probabilities for our inference.
The search algorithm explores the space of possible alignments by taking the current alignment a1 and generating a proposal alignment a2 by applying a randomly chosen transformation. The proposal alignment is accepted, and becomes the current alignment, with probability
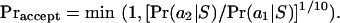 |
(7) |
If the proposal alignment is not accepted then the current alignment stays unchanged. Because better alignments are accepted with certainty and worse alignments are sometimes accepted, the algorithm tends to climb hills but can also move away from local optima to attempt to find a global optimum. The ratio of the probabilities of the two alignments is raised to the power 1/10 because the probability surface being explored is very rugged (or it is difficult to design proposal distributions that are good for all S). This 1/10 power increases the typical fraction of proposals accepted from ∼0.15 to ∼0.4. The algorithm records the alignment(s) amax that (jointly) maximizes Pr(a | S) over all alignments visited thus far.
For two sequences, one of the following random transformations is chosen at random each iteration:
Add a gap pair in random sites of opposing sequences.
Remove a random gap pair, or parts of a random gap pair, from opposing sequences.
Move a randomly selected gap within a sequence.
Split a randomly selected gap within a sequence.
Merge a pair of randomly selected adjacent gaps within a sequence.
These operations are applied to gaps that are close together, where possible by sampling the sizes of the inter-gap distances from a geometric distribution with a mean of four bases.
The three-way gap manipulation routines are substantially more complexthan the pairwise alignment routines. Gaps are inserted and deleted in pairs in order to maintain the length of the sequences. Gap moving, splitting, and joining are carried out on random gaps; if a gap is fully overlapping between a pair of sequences from the three, then the gap that is common to the pair may or may not be manipulated as a unit, according to a random draw.
In order to avoid becoming stuck sampling implausibly poor alignments, values of Pr(ai | S) less than 0.01Pr(amax | S) which occur for more than 100 consecutive iterations cause the algorithm to reset to the current alignment to be amax, and searching resumes from there. This means that the search algorithm cannot be guaranteed to be irreducible; that is, there may be alignments that can never be reached from other regions of the state space. We do not think that this concern is important in practice. Searching stops a predetermined number of iterations, typically 106, after the last increase in probability for the best alignment has been found.
Initial Alignment Algorithm
We used a heuristic method that is similar to the “divide-and-conquer” algorithm (e.g., Tönges et al. 1996; Stoye 1998), which searches for the stretch of fewest mismatches between a pair of sequences, divides the sequence pair at this stretch, then recursively aligns the sequences on either side of the match. In practice, the search algorithm is run for a predetermined number of iterations, starting from several initial alignments that are generated with different gap penalty parameters and a match length parameter. The search algorithm is then restarted for the final maximization run using the best alignment found in any of these initial runs.
Parameterization of Models of Indel Evolution
The model of noncoding DNA evolution is parameterized according to the empirical distribution of indel lengths and their overall rate relative to nucleotide substitutions from species for which essentially unambiguous alignments can be made. It is intended that the analysis can then be applied to more distantly related species. Consider a parameterization by intronic data of D. simulans and D. sechellia (Table 1). In a total of 6302 nucleotides, there are 193 substitutions, the total number of indels (Σg) is 44 with 198 bp of indel in total, and the total number of non-indel sites (m) is 6328. The average fraction of nucleotide differences is 0.0306, which is equal to t if multiple hits are ignored. The rate of indels per interbase site, relative to the rate of nucleotide substitutions, is θ = Σg /t(m + Σg) = 0.225. The frequency distribution of indel lengths is shown in Figure 2.
Table 1.
Numbers of Substitutions and Indels and Distribution of Indel Lengths in Introns and Noncoding DNA Between 23 D. simulans and D. sechellia Loci
|
Numbers of indels in length category (bp)
|
Lengths >6 bp
|
||||||||
|---|---|---|---|---|---|---|---|---|---|
| Locus | Length | Subs. | 1 | 2 | 3 | 4 | 5 | 6 | |
| Zw | 118 | 2 | 1 | 29 | |||||
| Adh | 120 | 7 | 1 | 1 | 1 | 7 | |||
| ci | 118 | 1 | |||||||
| Est-6 | 50 | 4 | 1 | ||||||
| hb | 280 | 2 | 1 | 1 | |||||
| per | 191 | 8 | 1 | ||||||
| Sxl | 224 | 5 | 1 | 1 | 14 | ||||
| w | 205 | 7 | 3 | ||||||
| zeste | 182 | 6 | 1 | ||||||
| CecC | 70 | 4 | |||||||
| ocn | 102 | 2 | 9 | ||||||
| fru | 352 | 17 | 1 | 1 | 1 | 37 | |||
| Adhr | 480 | 8 | 1 | 1 | |||||
| OdsH | 1413 | 29 | 3 | 1 | 1 | 2 | 12 | ||
| Sod | 730 | 33 | 1 | ||||||
| Amyrel | 57 | 15 | 1 | ||||||
| Mlc1 | 563 | 6 | 1 | ||||||
| Acp70A | 65 | 1 | 8 | ||||||
| Top2 | 519 | 15 | 2 | 1 | 8 | ||||
| Mst26Aa | 56 | 1 | |||||||
| Mst26Ab | 61 | 4 | |||||||
| janA | 163 | 8 | 2 | ||||||
| janB | 120 | 7 | 1 | ||||||
| yp2 | 63 | 1 | 1 | ||||||
| Totals | 6302 | 193 | 20 | 8 | 2 | 2 | 0 | 4 | 8 |
The length of each locus is the number of nucleotides excluding gaps, and “Subs.” is the number of nucleotide substitutions. Numbers of indels of lengths of up to 6 bp, and lengths of >6-bp indels are tabulated.
Figure 2.

The empirical distribution of indel lengths in noncoding DNA between D. simulans and D. sechellia, and the indel length frequency distribution model assumed for the MC analysis.
Because we treat the frequencies of different indel lengths as known, a distribution with non-zero indel length frequencies is desirable, so some smoothing of the empirical distribution was performed. The rates for 1-bp and 2-bp indels are relatively accurately known (Table 1), so we directly use their observed frequencies in the model, that is, 0.455 and 0.182, respectively. For indels in the range 3–40 bp, the frequencies, wx, for the model were obtained by minimizing the sum over 3≤x≤40 of the squared differences between the observed frequency distribution and wx = β/αx. Here β is a normalizing constant, α is the parameter, and x is indel length. The estimate for α was 1.167, and this gave the distribution shown in Figure 2. Indels of length >40 are assigned the probability of an indel of length 40 in the analysis.
Performance Evaluation
We tested our algorithm by examining the fraction of correctly aligned sites and the statistical properties of an estimator of divergence time. We calculated the fraction of correctly aligned sites by counting the number of base pairs or bases-to-gaps which were correctly aligned in a comparison to the true alignment. As an estimator of divergence time, we considered the standard estimator found by setting k equal to the observed fraction of nucleotide differences in Equation (5) and solving for t.
For each simulated data set, we observed the mean and variance of the fraction of correctly aligned bases or the estimator of t calculated from both the true alignment and the alignments estimated using a range of methods including MCALIGN. Because none of the estimators examined are perfectly unbiased, we express the variance as the estimated (root) mean square error (m.s.e.). For t, this is m.s.e. = Σ (test – ttrue)2/N when there were N simulations. The ratio of the mean square errors is the relative efficiency of different estimators of t. Insofar as calculating maximum likelihood estimate (MLE) of t using the known alignment is most efficient, there is an upper limit on the possible efficiency of an estimator when the alignment is unknown, and efficiency is an indication of the scope for further improvement of such estimators.
RESULTS
Performance of MCALIGN
We assessed the performance of the alignment method in simulations in which the simulation model either conformed or did not conform to the model of indel evolution assumed by the analysis.
Tables 2 and 3 show the results of simulations for two sequences, with 200 replicates for each combination of values of t12 and θ, for the case of the same simulation and analysis model. The means of the estimator of t are very close to the true values, showing that the estimator is close to unbiased over the range of parameter values simulated (Table 2; see also Fig. 3) As expected, the (root) m.s.e. is larger when the alignment must be estimated. The efficiency of our estimator of t, which is the ratio of the two m.s.e. values, is over 95% for t12 = 0.05 but drops to less than 80% for t12 = 0.30. Therefore, there is room for improvement, either by more accurate estimation of the alignment or by integrating over alignments as in the sum method of TKF. The estimated efficiencies shown in Table 2 fluctuate because they are ratios of variances, which have large associated sampling error; we expect that the true efficiency is a monotonically decreasing function of t12. The fraction of correctly aligned bases also declines as sequence divergence increases (Table 3).
Table 2.
Performance of MCALIGN and Other Alignment Methods Compared by Simulation
|
Simulated
|
Alignment known
|
Alignment estimated
|
MCALIGN efficiency
|
||||||
|---|---|---|---|---|---|---|---|---|---|
| t | θ | DIALIGN | LAGAN | AVID | CLUSTAL | HANDEL | MCALIGN | ||
| 0.05 | 0.225 | 0.0488 (0.0159) | 0.0529 (0.0192) | 0.0549 (0.0194) | 0.0499 (0.0161) | 0.0525 (0.0185) | 0.0485 (0.0165) | 0.0487 (0.0162) | 0.97 |
| 0.10 | 0.225 | 0.0995 (0.0232) | 0.1121 (0.0327) | 0.1125 (0.0330) | 0.1024 (0.0255) | 0.1115 (0.0364) | 0.1000 (0.0328) | 0.0988 (0.0241) | 0.93 |
| 0.15 | 0.225 | 0.1519 (0.0311) | 0.1818 (0.0561) | 0.1749 (0.0511) | 0.1585 (0.0352) | 0.1807 (0.0656) | 0.1562 (0.0480) | 0.1510 (0.0324) | 0.92 |
| 0.20 | 0.225 | 0.2043 (0.0366) | 0.2491 (0.0769) | 0.2443 (0.0719) | 0.2132 (0.0435) | 0.2518 (0.0946) | 0.2107 (0.0652) | 0.2022 (0.0388) | 0.89 |
| 0.25 | 0.225 | 0.2502 (0.0426) | 0.3216 (0.1055) | 0.3039 (0.0824) | 0.2629 (0.0467) | 0.3122 (0.1011) | 0.2537 (0.0631) | 0.2519 (0.0438) | 0.94 |
| 0.30 | 0.225 | 0.2968 (0.0470) | 0.3876 (0.1181) | 0.3670 (0.1029) | 0.3103 (0.0469) | 0.3799 (0.1281) | 0.2923 (0.0703) | 0.2974 (0.0531) | 0.75 |
| 0.15 | 0.1 | 0.1531 (0.0317) | 0.1612 (0.0359) | 0.1628 (0.0391) | 0.1539 (0.0321) | 0.1592 (0.0366) | 0.1538 (0.0383) | 0.1524 (0.0315) | 1.01 |
| 0.15 | 0.3 | 0.1504 (0.0276) | 0.1958 (0.0656) | 0.1904 (0.0625) | 0.1652 (0.0351) | 0.1909 (0.0666) | 0.1573 (0.0382) | 0.1504 (0.0276) | 1.00 |
| 0.15 | 0.4 | 0.1498 (0.0303) | 0.2140 (0.0899) | 0.2085 (0.0812) | 0.1731 (0.0469) | 0.2185 (0.1030) | 0.1621 (0.0489) | 0.1480 (0.0325) | 0.87 |
Estimates of sequence divergence, t, from 200 replicates per t value, with sequences of length 200 base pairs. Estimated root mean square error (e.r.m.s.e.) is shown in parentheses.
Table 3.
Proportions of Correctly Matched Bases From MCALIGN and Other Alignment Methods Compared by Simulation
|
Simulated
|
Proportion of matched bases
|
||||||
|---|---|---|---|---|---|---|---|
| t | θ | DIALIGN | LAGAN | AVID | CLUSTAL | HANDEL | MCALIGN |
| 0.05 | 0.225 | 0.986 (0.0014) | 0.987 (0.0014) | 0.990 (0.0011) | 0.983 (0.0022) | 0.969 (0.0015) | 0.991 (0.00085) |
| 0.10 | 0.225 | 0.963 (0.0023) | 0.966 (0.0023) | 0.977 (0.0016) | 0.960 (0.0035) | 0.944 (0.0022) | 0.980 (0.0015) |
| 0.15 | 0.225 | 0.925 (0.0036) | 0.932 (0.0037) | 0.949 (0.0029) | 0.913 (0.0058) | 0.874 (0.0033) | 0.954 (0.0026) |
| 0.20 | 0.225 | 0.892 (0.0044) | 0.901 (0.0046) | 0.928 (0.0035) | 0.871 (0.0078) | 0.820 (0.0043) | 0.935 (0.0033) |
| 0.25 | 0.225 | 0.839 (0.0050) | 0.848 (0.0053) | 0.887 (0.0042) | 0.812 (0.0079) | 0.776 (0.0039) | 0.894 (0.0046) |
| 0.30 | 0.225 | 0.776 (0.0058) | 0.792 (0.0066) | 0.838 (0.0055) | 0.751 (0.0096) | 0.702 (0.0047) | 0.844 (0.0057) |
| 0.15 | 0.1 | 0.973 (0.0019) | 0.978 (0.0020) | 0.984 (0.0015) | 0.973 (0.0029) | 0.948 (0.0034) | 0.986 (0.0012) |
| 0.15 | 0.3 | 0.903 (0.0041) | 0.909 (0.0043) | 0.933 (0.0032) | 0.894 (0.0062) | 0.854 (0.0036) | 0.944 (0.0027) |
| 0.15 | 0.4 | 0.863 (0.0041) | 0.902 (0.0037) | 0.902 (0.0037) | 0.822 (0.0085) | 0.800 (0.0035) | 0.916 (0.0031) |
Data are from 200 replicates per parameter value combination, with sequences of length 200 base pairs. Standard error of the mean is shown in parentheses.
Figure 3.

Estimated numbers of nucleotide substitutions plotted against observed numbers. Each point is the outcome from an alignment involving one simulation of sequences of length 200 bp.
The performance of the procedure was also investigated for cases of the alignment of three sequences, using the same model of indel evolution in the simulation and analysis. In the simulation results presented in Table 4, alignment probability was evaluated for the case of known branch lengths if ingroup and outgroup branch lengths are equal. The results suggest that the performance is nearly as good as for aligning a pair of sequences (Table 2). As might be expected there is a slight drop-off in performance for higher values of t simulated. If branch lengths are unknown, and t12 and t(12)3 need to be estimated separately, the performance of the procedure is somewhat poorer (data not shown). In Table 4, the estimated and true t12 and t(12)3 tend to be higher and lower than the actual values simulated, respectively; we suspect that this bias is caused by using parsimony to assign the ancestral state in situations where all three nucleotides vary.
Table 4.
MC Analysis With Simulations of Three-Way Alignments in Which Equal Branch Lengths Are Assumed in MC Analysis
| Actual t1,2 and t(1,2)3 | Observed t1,2 (ermse) | Estimated t1,2 (ermse) | Observed t(1,2)3 (ermse) | Estimated t(1,2)3 (ermse) |
|---|---|---|---|---|
| 0.05 | 0.0500 (0.0138) | 0.0473 (0.0140) | 0.0531 (0.0178) | 0.0507 (0.0166) |
| 0.1 | 0.1017 (0.0282) | 0.0976 (0.0277) | 0.1006 (0.0211) | 0.0955 (0.0208) |
| 0.15 | 0.1521 (0.0280) | 0.1462 (0.0289) | 0.1403 (0.0290) | 0.1390 (0.0313) |
| 0.2 | 0.2136 (0.0340) | 0.2055 (0.0349) | 0.1797 (0.0381) | 0.1742 (0.0460) |
| 0.25 | 0.2757 (0.0519) | 0.2672 (0.0540) | 0.2258 (0.0371) | 0.2109 (0.0538) |
| 0.3 | 0.3068 (0.0412) | 0.2984 (0.0510) | 0.2520 (0.0546) | 0.2600 (0.0599) |
The internal and external branch lengths are equal (i.e., t1,2 and t(1,2)3), and alignment probability is evaluated under this model.
In order to evaluate the robustness of the procedure, cases were also investigated in which the model assumed by the MC analysis differed from the simulation model. We explored the effect of varying the simulated value of θ (the rate of indels relative to nucleotide substitutions), while assuming a constant value for θ in the MC analysis. For simulated values of θ lower than the value assumed by the MC analysis, the MC procedure tends to add gaps inappropriately and thereby masks off a proportion of the single-base pair differences, hence the estimates for t tend to be very slightly lower than the observed values (Table 5). Conversely, if θ is higher than the value assumed by the MC analysis, there are too few gaps in the MC alignments and the estimates of t tend to be higher than the observed. The bias in this direction can be quite substantial.
Table 5.
MC Analysis for Cases of Discrepancy Between the Relative Frequency of Indels in the Simulation Model and MC Analysis Model
| θ simulated | Actual t | Observed t (ermse) | Estimated t (ermse) |
|---|---|---|---|
| 0.112 | 0.1 | 0.107 (0.0263) | 0.105 (0.0262) |
| 0.450 | 0.103 (0.0254) | 0.107 (0.0318) | |
| 0.112 | 0.2 | 0.206 (0.0306) | 0.204 (0.0336) |
| 0.450 | 0.205 (0.0346) | 0.245 (0.0716) | |
| 0.112 | 0.3 | 0.298 (0.0489) | 0.288 (0.0462) |
| 0.450 | 0.295 (0.0490) | 0.379 (0.1080) |
Pairs of sequences were simulated with the value of θ shown (the rate of indels relative to nucleotide substitutions). The value of θ assumed in the MC alignment analysis was 0.225. There were 50 replicates per value simulated.
Comparison With Other Methods
We compared MCALIGN with implementations of several heuristic alignment methods: The first three, DIALIGN (Morgenstern 1999), AVID (Bray et al. 2003), and LAGAN (Brudno et al. 2003), are all intended for alignment of genomic contigs including noncoding DNA. The fourth, CLUSTAL (Thompson et al. 1994), is not in fact designed for aligning noncoding DNA, but is widely used for this purpose. For all of these methods we used the default parameter values provided by the implementation, a procedure which would be most likely to be followed by a user faced with aligning real noncoding DNA sequence data. We were able to improve the performance of CLUSTAL on our simulated data sets by seeking a scoring function that led to reasonably unbiased estimates of t, but to do this required knowing the true t in advance. It is likely that a similar procedure would also improve the performance of other methods, although the method of AVID does not have any configurable parameters.
We also compared MCALIGN with the HANDEL package of Holmes and Bruno (2001), which implements the TKF (1991) model for arbitrary numbers of sequences. Because the TKF model allows only single-base pair indels, we parameterized HANDEL with an equivalent number of base pairs of indels per base substitution, that is
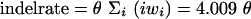 |
for the parameterization under which we simulated our data. We first estimated t from the unaligned sequences using HANDEL's TKFDISTANCE program, and then searched for an alignment conditioning on this time using HANDEL's TKFALIGN program, allowing 10,000 iterations with greedy progressive refinement every 100 iterations. In all cases the estimates of t obtained from this most probable alignment were as or less biased than the estimates of t obtained directly from the unaligned sequences.
As shown in Tables 2 and 3, all of the methods examined here performed acceptably for small divergences, t ≤ 0.1. However, at larger divergences and/or higher rates of indels, the programs DIALIGN, LAGAN, and CLUSTAL all performed relatively poorly over the range of values of t and θ simulated, in the sense that estimates of t are biased and the e.r.m.s.e. are large.
Alignments produced by AVID exhibit a small but consistent upward bias in the corresponding estimates of t, and for both AVID and HANDEL the efficiency relative to MCALIGN is less than one (i.e., they have greater r.m.s.e.) for most parameter value combinations. AVID outperforms MCALIGN for only one case, t = 0.30 θ = 0.225, of all the parameter combinations we considered. In the case of HANDEL we attribute its lower efficiency to its tendency to produce alignments with indels that are more fragmented than the true alignment, because of HANDEL's assumption of a single-base pair indel model. Similar results are obtained for alignments of sequences of 1000 bp (data not shown). The rankings of the different methods are similar if performance is measured as the fraction of correctly aligned bases (Table 3), although HANDEL performs the poorest, presumably due to overfragmentation of indels.
Execution Time and Analysis of Real Data
The execution time of MCALIGN was evaluated on a 2.8-GHz Intel processor. In aligning simulated data, execution time increases nonlinearly with sequence length, and increases as a function of sequence divergence (Fig. 4). These execution times are very long in comparison to DIALIGN, LAGAN, and AVID, which have execution times for 1000-bp sequences of the order of 0.1 sec. However, alignment of sequences totaling several Mb is feasible with MCALIGN if segments of ∼1 kb are aligned (see below). MCALIGN execution times for alignment of mouse and Drosophila sequences are similar to that for simulated data.
Figure 4.

Execution time of MCALIGN plotted against sequence length for sequence divergences of 0.1, 0.2, and 0.3. Execution times were estimated from the average of five simulations.
We have used MCALIGN to make alignments of a total of ∼80 kb of intronic and intergenic DNA sequences from D. melanogaster, D. simulans, and D. yakuba of up to ∼1.5 kb in length from ∼80 loci (Halligan et al. 2004). The ends of each intronic or 5′ or 3′ intergenic sequence were anchored by the coding sequence, and thus correct identification of homologous noncoding DNA segments was straightforward, and convincing alignments were obtained. These alignments can be downloaded from P. Keightley's Web site (http://homepages.ed.ac.uk/eang33/). We have also used MCALIGN for the larger-scale alignment of ∼6 Mb of intergenic and intronic DNA sampled from 300 loci from the mouse and rat genomes (Keightley and Gaffney 2003). We ensured the orthology between the mouse and rat noncoding DNA segments by sampling well annotated loci and aligning intergenic DNA adjacent to the corresponding coding sequences. It was feasible to align noncoding DNA segments of up to 6 kb in length; these were aligned by MCALIGN in 1000-bp segments. In some cases, alignment failed due to the presence of long insertions or due to errors in the mouse or rat sequence assemblies, which led to a complete breakdown in homology at some distance from the coding sequence. If the contigs contained microsatellite loci, these were successfully aligned, but were occasionally flanked by obviously nonhomologous runs of nucleotides; we assume that this is an artifact of the shotgun sequence assembly.
DISCUSSION
In many cases one would not wish to estimate an alignment for its own sake, but rather would wish to estimate some quantity based on the alignment, such as the number of nucleotide substitutions. In such situations the alignment is missing data and a proper treatment would involve integrating over all possible alignments (Thorne et al. 1991, 1992) or sampling over alignments (see, e.g., Holmes and Bruno 2001). At the present time such an approach has not been implemented for a biologically realistic model of indel lengths. We offer our approach as a pragmatic solution to the problem of estimating parameters in complexmodels of sequence evolution, in the face of uncertainty in the alignment. If one is going to estimate parameters based on a single alignment, it is clearly better to use an alignment estimated under the most realistic model available. We are therefore advocating an approach similar to that taken by Fu (1994) and Thomas and Hill (2000), who estimate a single genealogy or single sibship pattern respectively, and then go on to estimate the parameters of interest as if that estimate was correct. Our use of the single most probable alignment seems to give reasonable results, probably because the information supplied about the parameters of indel evolution sharpens the posterior density so that most of the probability piles up on the MP alignment. In this context we note that our simulation results using the HANDEL package of Holmes and Bruno (2001) suggest that less biased estimates of t are obtained by conditioning on the most probable alignment rather than by finding the true MLE for t by summing over all possible alignments. This unexpected behavior seems to be caused by simulating data under one model (multiple-base pair indels) and then analyzing under a different (single-base pair indels) model, because the expected behavior is observed when data are simulated under the model assumed by HANDEL.
We investigated simulations parameterized according to a model of the frequency distribution of indel and nucleotide substitution in Drosophila intronic DNA. We have also produced a parameterization appropriate to rodent intronic DNA, by using the empirical distribution of indel lengths and their relative frequency in orthologous introns of closely related mouse species (Mus domesticus, M. spretus, and M. caroli). In rodent introns our estimate for θ is 0.146, which is substantially lower than θ for Drosophila introns (0.225). There is also a higher relative frequency of 1-bp indels (0.57 vs. 0.45), and the frequency of longer indels drops off more quickly in rodents (α = 1.45 vs. 1.17). The rodent intron alignment parameters are available at the program download site (see below). In order to parameterize models appropriate to other taxa, it is necessary to obtain noncoding DNA sequences from closely related species or polymorphism data.
The simulations indicate that estimates of t are almost perfectly unbiased up to divergences of ∼30%. We do not report results for divergences above 30% because in many cases the MP alignment would contain one or more “opposing gaps” which are absent from the true alignment. These gaps on opposite strands mask off areas of low homology, and can imply that much of the sequence is not, in fact, aligned; such alignments may also have a higher probability than the true alignment. With the Drosophila indel model, opposing gaps begin to appear if t is above 0.3 (particularly under the three-way alignment, where there is no correction for multiple hits), and such sequences often appear to have “unalignable” segments.
The simulations also demonstrate that, from the point of view of estimating divergence between sequences or the fraction of correctly aligned bases, MCALIGN is generally superior to the other alignment methods we have tested. A major reason for this is that MCALIGN was supplied with parameters of the model of molecular evolution under which the data were simulated. An alternative program that is also able to use such information directly, HANDEL, performed less well because it assumes a model of single-base pair indels. A similar approach is taken by other Hidden Markov Model based approaches, such as HMMER (Eddy 2003; see also Durbin et al. 1998). This program allows one to train the algorithm on known alignments, but a crucial difference from our approach is that an HMM trained on alignments for one value of t will perform badly when used to align sequences with a different value of t. On the other hand, our approach performs well over a range of t using a single parameterization.
In the future, our approach could be extended to more complexmodels of nucleotide substitution, for example, by counting transition and transversion substitutions separately in Equations (4) and (5), and to more than three sequences. Investigators using the current implementation of our approach on data that obviously depart from the Jukes-Cantor (1969) model should proceed with appropriate caution and simulation work.
The MC alignment program is available from P. Keightley's Web site.
Acknowledgments
We thank Adam Eyre-Walker, Jotun Hein, Bill Hill, Stuart Baird, Kevin Dawson, Jeff Thorne, Peter Donnelly, and five reviewers for helpful advice and constructive comments. T.J. was supported by Wellcome Trust International Prize Travelling Research Fellowship #061530.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.1571904.
References
- Bergman, C.M., and Kreitman, M. 2001. Analysis of conserved noncoding DNA in Drosophila reveals similar constraints in intergenic and intronic sequences. Genome Res. 11: 1335–1345. [DOI] [PubMed] [Google Scholar]
- Bishop, M.J. and Thompson, E.A. 1986. Maximum likelihood alignment of DNA sequences. J. Mol. Biol. 190: 159–165. [DOI] [PubMed] [Google Scholar]
- Bray, N., Dubchak, I., and Pachter, L. 2003. AVID: A global alignment program Genome Res. 13: 97–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brudno, M., Do, C.B., Cooper, G.M., Kim, M.F., Davydov, E., Green, E.D., Sidow, A., and Batzoglou, S. 2003. LAGAN and Multi-LAGAN: Efficient tools for large-scale multiple alignment of genomic DNA. Genome Res. 13: 721–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durbin, R., Eddy, S., Krogh, A., and Mitchison, G. 1998. Biological sequence analysis: Probabilistic models of proteins and nucleic acids. Cambridge University Press, Cambridge, UK.
- Eddy, S. 2003. HMMER: Profile HMMs for protein sequence analysis. http://hmmer.wustl.edu.
- Fu, Y.-X. 1994. A phylogenetic estimator of effective population size or mutation rate. Genetics 136: 685–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelman, A. Carlin, J.B., Stern, H.S., and Rubin, D.B. 2003. Bayesian Data Analysis, chapters 1 and 12. Chapman and Hall/CRC Press, New York.
- Gu, X. and Li, W.-H. 1995. The size distribution of insertions and deletions in human and rodent pseudogenes suggests the logarithmic gap penalty for sequence alignment. J. Mol. Evol. 40: 464–473. [DOI] [PubMed] [Google Scholar]
- Halligan, D.L., Eyre-Walker, A., Andolfatto, P., and Keightley, P.D. 2004. Patterns of evolutionary constraints in intronic and intergenic DNA of Drosophila. Genome Res. 14: 273–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes, I. and Bruno, W.J. 2001. Evolutionary HMMs: A Bayesian approach to multiple alignment. Bioinformatics 17: 803–810. [DOI] [PubMed] [Google Scholar]
- Jareborg, N., Birney, E., and Durbin, R. 1999. Comparative analysis of noncoding regions of 77 orthologous mouse and human gene pairs. Genome Res. 9: 815–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jukes, T.H. and Cantor, C.R. 1969. Evolution of protein molecules. In Mammalian protein metabolism (ed. H.N. Munro), pp. 21—123. Academic Press, New York.
- Keightley, P.D. and Gaffney, D.J. 2003. Functional constraints and frequency of deleterious mutations in noncoding DNA of rodents. Proc. Natl. Acad. Sci. 100: 13402–13406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzler, D., Fleissner, R., Wakolbinger, A., and von Haeseler, A. 2001. Assessing variability by joint sampling of alignments and mutation rates. J. Mol. Evol. 53: 660–669. [DOI] [PubMed] [Google Scholar]
- Morgenstern, B. 1999. DIALIGN 2: Improvement of the segment-to-segment approach to multiple sequence alignment. Bioinformatics 15: 211–218. [DOI] [PubMed] [Google Scholar]
- Mouse Genome Sequencing Consortium. 2002. Initial sequencing and comparative analysis of the mouse genome. Nature 420: 520–562. [DOI] [PubMed] [Google Scholar]
- Shabalina, S.A. and Kondrashov, A.S. 1999. Pattern of selective constraint in C. elegans and C. briggsae genomes. Genet. Res. 74: 23–30. [DOI] [PubMed] [Google Scholar]
- Shabalina, S.A., Ogurtsov, A.Y., Kondrashov, V.A., and Kondrashov, A.S. 2001. Selective constraint in intergenic regions of human and mouse genomes. Trends Genet. 17: 373–376. [DOI] [PubMed] [Google Scholar]
- Steel, M. and Hein, J. 2001. Applying the Thorne-Kishino-Felsenstein model to sequence evolution on a star-shaped tree. Appl. Math. Lett. 14: 679–684. [Google Scholar]
- Stoye, J. 1998. Multiple sequence alignment with the divide-and-conquer method Gene 211: GC45–GC56. [DOI] [PubMed] [Google Scholar]
- Thomas, S.C. and Hill, W.G. 2000. Estimating quantitative genetic parameters using sibships reconstructed from marker data. Genetics 155: 1961–1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson, J.D., Higgins, D.G., and Gibson, T.J. 1994. CLUSTAL W—Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrixchoice. Nucleic Acids Res. 22: 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorne, J.L., Kishino, H., and Felsenstein, J. 1991. An evolutionary model for maximum likelihood alignment of DNA sequences. J. Mol. Evol. 33: 114–124. [DOI] [PubMed] [Google Scholar]
- Thorne, J.L., Kishino, H., and Felsenstein, J. 1992. Inching toward reality–An improved likelihood model of sequence evolution. J. Mol. Evol. 34: 3–16. [DOI] [PubMed] [Google Scholar]
- Tönges, U., Perrey, S.W., Stoye, J., and Dress, A.W.M. 1996. A general method for fast multiple sequence alignment. Gene 172: GC33–GC41. [DOI] [PubMed] [Google Scholar]
WEB SITE REFERENCES
- http://homepages.ed.ac.uk/eang33/; Executables, source code, and user instructions for MCALIGN. Alignments of Drosophila noncoding DNA sequences.