

Abstract
We report 80,388 ESTs from 23 Atlantic salmon (Salmo salar) cDNA libraries (61,819 ESTs), 6 rainbow trout (Oncorhynchus mykiss) cDNA libraries (14,544 ESTs), 2 chinook salmon (Oncorhynchus tshawytscha) cDNA libraries (1317 ESTs), 2 sockeye salmon (Oncorhynchus nerka) cDNA libraries (1243 ESTs), and 2 lake whitefish (Coregonus clupeaformis) cDNA libraries (1465 ESTs). The majority of these are 3′ sequences, allowing discrimination between paralogs arising from a recent genome duplication in the salmonid lineage. Sequence assembly reveals 28,710 different S. salar, 8981 O. mykiss, 1085 O. tshawytscha, 520 O. nerka, and 1176 C. clupeaformis putative transcripts. We annotate the submitted portion of our EST database by molecular function. Higher- and lower-molecular-weight fractions of libraries are shown to contain distinct gene sets, and higher rates of gene discovery are associated with higher-molecular weight libraries. Pyloric caecum library group annotations indicate this organ may function in redox control and as a barrier against systemic uptake of xenobiotics. A microarray is described, containing 7356 salmonid elements representing 3557 different cDNAs. Analyses of cross-species hybridizations to this cDNA microarray indicate that this resource may be used for studies involving all salmonids.
Gene and genome duplications are thought to be primary mechanisms of increasing the number of coding sequences subject to selection, leading to new proteins, morphogenic variations, and phenotypes (Ohno 1970; Holland et al. 1994; Sidow 1996). Members of the teleost family Salmonidae, including salmon, trout, char, grayling, and whitefish, all diverged from a common ancestor that is believed to have undergone a tetraploidization event 25 to 100 million years ago, after the teleost radiation (Allendorf and Thorgaard 1984). This relatively recent putative genome duplication in the salmonid lineage is supported by karyological and genome size data. Members of the family Clupeidae (e.g., herring, alewife), thought to maintain the ancestral diploid status, have 48 to 52 mostly acrocentric chromosomes per 2N cell and genome sizes of 0.8 to 1.4 pg/N, whereas salmonids have 52 to 102 chromosomes per 2N cell (over half metacentric or submetacentric) and genome sizes of 1.9 to 3.8 pg/N (Ohno et al. 1968; Phillips and Ráb 2001; Gregory 2002). Because extant salmonids exhibit quadrivalents in meiosis (primarily in males; Ohno et al. 1965; Allendorf and Thorgaard 1984) and disomic and tetrasomic inheritance at different loci (Allendorf and Danzmann 1997), they appear to be in the process of re-establishing diploidy. Remarkably, ∼50% of examined salmonid loci persist as functional duplicates (Bailey et al. 1978). Research on salmonid genomes will shed light on poorly understood evolutionary phenomena such as genome duplication and duplicate gene silencing.
In addition to their scientific importance as recent tetraploids, salmonids also serve as prominent models for studies involving environmental toxicology (Katchamart et al. 2002), carcinogenesis (Bailey et al. 1996), comparative immunology (Shum et al. 2001), and the molecular genetics and physiology of the stress response (Basu et al. 2002), olfaction (Zhang et al. 2001), vision (Faillace et al. 2002), osmoregulation (Tipsmarck et al. 2002), growth (Devlin et al. 2001), and gametogenesis (Madigou et al. 2002). Furthermore, Atlantic salmon (AS; Salmo salar) are of particular importance to the global aquaculture industry. GRASP (Genomics Research on Atlantic Salmon Project), an initiative funded by Genome Canada, is intended to improve understanding of physiological and evolutionary processes influencing the survival and phenotype of salmonids and other fish in natural and aquaculture environments. GRASP has developed genomics resources to help achieve these goals. There is a rich literature in salmonid genetics, physiology, and ecology to support these genomics research tools.
A previously reported S. salar EST project surveyed 1152 ESTs from six cDNA libraries, with 510BLAST-identified sequences representing 178 salmon genes (Davey et al. 2001). There are currently (August 2003) ∼60,000 S. salar nucleotide sequences in GenBank, of which >51,000 were submitted by GRASP. In addition to forming an EST database containing >80,000 sequences from five salmonid species, GRASP has built a microarray from 3557 unique salmonid cDNAs. Initial cross-species testing of this microarray has shown it to be effective in hybridizations with salmon, trout, and whitefish targets.
RESULTS AND DISCUSSION
EST Survey
This report describes ESTs obtained from high-complexity normalized and non-normalized, directionally cloned cDNA libraries, as well as subtracted cDNA libraries, from the following species: S. salar (23 libraries/library groups representing 16 adult tissues and whole juvenile), O. mykiss (six libraries/library groups from three adult tissues, and whole embryo and juvenile), O. tshawytscha (two libraries from adult mixed tissue), O. nerka (two libraries from adult brain and whole juvenile), and C. clupeaformis (two libraries from adult brain; see Table 2 below). The set of S. salar cDNA libraries represents most principal tissues in adult fish. EST clones are available from the corresponding author.
Table 2.
Salmonid cDNA Library Summary Statisticsa

The 95,320 clones from these cDNA libraries (71,144 S. salar, 19,093 O. mykiss, 1824 O. tshawytscha, 1051 O. nerka, and 2208 C. clupeaformis) were M13 forward-sequenced and quality checked. For all libraries except SSH (suppression subtractive hybridization), M13 forward sequences of properly oriented inserts should include 3′ UTR. Because of low conservation in 3′ UTRs and the pseudotetraploidy of salmonid genomes, we focused on 3′ sequencing to allow differentiation between paralogs arising from the recent salmonid genome duplication. 5′ (reverse) sequencing was attempted on 7487 of the 71,144 S. salar clones. The 80,388 high-quality ESTs (55,082 forward and 6737 reverse S. salar, 14,544 forward O. mykiss, 1317 forward O. tshawytscha, 1243 forward O. nerka, and 1465 forward C. clupeaformis) were assembled by using PHRAP under high stringency to identify EST clusters (contiguous sequences, or contigs) representing redundant transcripts (Tables 1, 2). The average trimmed PHRED20 length of these ESTs is 546 bases. The 61,819 S. salar ESTs were assembled into 11,560 contigs (with 17,150 singletons remaining), 14,544 O. mykiss ESTs formed 2370 contigs (6611 singletons), 1317 O. tshawytscha ESTs formed 136 contigs (949 singletons), 1243 O. nerka ESTs formed 291 contigs (229 singletons), and 1465 C. clupeaformis ESTs formed 138 contigs (1038 singletons; Table 1). There are 28,710 assembled S. salar sequences (putative transcripts), 8981 O. mykiss putative transcripts, 1085 O. tshawytscha putative transcripts, 520 O. nerka putative transcripts, and 1176 C. clupeaformis putative transcripts (Table 1). Results of alternate assemblies (CAP3 and stackPACK) of this EST collection are available at http://web.uvic.ca/cbr/grasp. The largest S. salar contig contains 252 ESTs (prolactin); the largest O. mykiss contig is size 93 (parvalbumen β); the largest O. tshawytscha contig is size 10 (cytochrome c oxidase subunit II); the largest O. nerka contig is size 21 (similar to ribosomal protein L41); and the largest C. clupeaformis contig is size 28 (ependymin; Table 1). BLAST alignments of ESTs against combined ribosomal and mitochondrial sequence databases (see Methods) identified 1052 S. salar, 396 O. mykiss, 103 O. tshawytscha, 40 O. nerka, and 157 C. clupeaformis reads.
Table 1.
Salmonid EST Project Summary Statisticsa
| Atlantic salmonb | Rainbow troutc | Chinook salmond | Sockeye salmone | Lake whitefishf | |
|---|---|---|---|---|---|
| Number of good sequencesg | 61,819h | 14,544 | 1317 | 1243 | 1465 |
| Average trimmed EST length (bp)i | 563 | 484 | 492 | 456 | 486 |
| Number of contigsj | 11,560 | 2370 | 136 | 291 | 138 |
| Number of singletons | 17,150 | 6611 | 949 | 229 | 1038 |
| Number of putative transcripts | 28,710 | 8981 | 1085 | 520 | 1176 |
| Max. assembled sequence size (no. of ESTs) | 252 | 93 | 10 | 21 | 28 |
| Average assembled sequence size (no. of ESTs) | 2.15 | 1.61 | 1.21 | 2.39 | 1.24 |
| Number of assembled ESTs withk | |||||
| Significant BLASTX hits | 10,511 | 3562 | 239 | 337 | 253 |
| Significant BLASTN hits | 13,459 | 4337 | 462 | 331 | 466 |
| No significant BLAST hits | 11,802 | 3667 | 566 | 118 | 663 |
| Percentage with no significant BLAST hitsk | 41.1 | 40.8 | 52.2 | 22.7 | 56.4 |
| Number of contigs containingi | |||||
| 2 ESTs | 5606 | 1360 | 90 | 108 | 97 |
| 3 ESTs | 2322 | 454 | 26 | 96 | 21 |
| 4-5 ESTs | 2030 | 350 | 12 | 48 | 9 |
| 6-10 ESTs | 1149 | 145 | 8 | 32 | 8 |
| 11-20 ESTs | 331 | 41 | 0 | 6 | 1 |
| 21-30 ESTs | 67 | 12 | 0 | 1 | 2 |
| 31-50 ESTs | 36 | 4 | 0 | 0 | 0 |
| >50 ESTs | 19 | 4 | 0 | 0 | 0 |
Assembled from the March 3, 2003, version of the GRASP EST database using PHRAP. Results of CAP3 and stackPACK assemblies of the March 3, 2003 GRASP EST database are available at http://web.uvic.ca/cbr/grasp
Salmo salar
Oncorhynchus mykiss
Oncorhynchus tshawytscha
Oncorhynchus nerka
Coregonus clupeaformis
A sequence is considered “good” if its trimmed PHRED20 length is at least 100 bases.
Includes 55.082 good forward (3′) and 6737 good reverse (5′) reads. Of 5606 good reverse reads from clones with good forward reads, 2268 overlap/cluster with the corresponding forward reads.
Vector, low-quality, and contaminating bacterial sequences are trimmed.
A contig (contiguous sequence) contains two or more ESTs.
Threshold for BLASTN and BLASTX significance: 10-5
Preliminary analysis of aligned S. salar and O. mykiss assembled ESTs identifies 1892 sequence pairs with >80% identity (see Methods). Of these, 1429 (∼76%) were contained within a distinct peak from 90%–97% identity (average ∼94%) at the nucleotide level. As it is difficult to distinguish orthologs from sequence pairs related by paralogy resulting from gene or genome duplications, a more focused study is underway.
REPuter (Kurtz et. al. 2001) identifies 11.9% of the total length of assembled sequences (TLAS) as known classes of repeats; 6.7% of the TLAS is composed of SINEs (predominately HpaI), whereas satellites, pseudogenes (including a large number of transposable element-associated sequences), and transposable elements account for 3.4%, 1.1%, and 0.7% of the TLAS, respectively.
Library Complexity and Gene Discovery
By using the March 3, 2003, versions of our EST database and GenBank databases, each library's ESTs were BLASTN- and BLASTX-aligned against a database composed of all nonredundant nucleic and amino acid sequences from that species in GenBank plus our collection of nonredundant ESTs. Percentage of singleton values for each library were calculated by using the August 25, 2003, version of the GRASP database (Table 2). Higher “percent new,” higher “percent no significant BLAST hit,” and higher “percent singleton” values indicate higher rates of gene discovery and higher complexity in a given library. For several of our libraries, higher- and lower-molecular-weight (MW) fractions were cloned separately. By using all three metrics, higher-MW fractions are of higher complexity (and higher rates of new gene identification) than their corresponding lower-MW fractions (Table 2). For example, the lower-MW S. salar head kidney library (average insert size of 1031 bp) has values of 15.0% new, 12.7% no BLAST hit, and 12.0% singletons, whereas the corresponding higher-MW library (average insert size of 2307 bp) values are 38.2%, 38.4%, and 35.3% respectively (Table 2). In addition, different suites of genes are identified in lower- and higher-MW fractions of a single cDNA library. This qualitative difference is evident in a list of the largest EST clusters in select non-normalized libraries/library groups (Table 3). Excluding ribosomal and mitochondrial clusters, the most abundant transcripts in the S. salar pyloric caecum lower-MW library are apolipoprotein A-I (2 forms of the gene in separate EST clusters), apolipoprotein E, 28 kD – 1e apolipoprotein, and galectin, whereas the largest EST contigs in the associated higher-MW library are selenoprotein Pa, MHC class I heavy chain, meprin A α, an unknown, and type II keratin E2, (Table 3). Likewise, in the head kidney, different sets of highly prevalent transcripts are seen in lower- and higher-MW library groups (Table 3). These results indicate that the preparation and characterization of higher-MW fractions of cDNA libraries improved the rate of gene discovery in the GRASP EST project.
Table 3.
Largest EST Clusters in Select Single-Tissue, Nonnormalized Atlantic Salmon Libraries/Library Groupsa
| Tissue (library)a | Total ESTs | ESTs in cluster | BLASTbE-value | Length (% identity)c | GenBank hit acc. no.b | Gene identification (species) of top BLAST hitb |
|---|---|---|---|---|---|---|
| Brain | 1161 | 23 | (X) 1.5E-60 | 113 (95.6%) | P28770 | Ependymin I (Oncorhynchus mykiss) |
| 19 | (X) 2.9E-128 | 221 (99.5%) | P28772 | Ependymin II (Salmo salar) | ||
| 17 | (X) 7.2E-10 | 119 (29.1%) | 062680 | Membrane attack complex inhibition factor (Sus scrofa) | ||
| 15 | (X) 5.6E-6 | 97 (32.9%) | P06906 | Myelin basic protein (Pan troglodytes) | ||
| 8 | n/ad | n/ad | n/ad | unknown | ||
| Esophagus | 749 | 88 | (X) 1.3E-24 | 111 (58.9%) | BAA86981 | Novel member of chitinase family (Homo sapiens)e |
| 32 | (X) 4.8E-59 | 315 (59.9%) | BAA86981 | Novel member of chitinase family (H. sapiens)e | ||
| 9 | (X) 4.7E-58 | 154 (75.3%) | AAD56283 | Pepsinogen A form IIa (Pseudopleuronectes americanus) | ||
| 8 | (X) 0 | 461 (99.5%) | AAG38613 | Elongation factor 1 α (S. salar) | ||
| 8 | (X) 9.3E-83 | 175 (97.7%) | CAA37852 | Creatine kinase (O. mykiss) | ||
| Gill | 2308 | 14 | (X) 1.6E-64 | 116 (98.2%) | AAG17525 | β-2 microglobulin (S. salar) |
| 13 | (X) 5.6E-132 | 244 (100%) | CAA49726 | MHC Class II β chain (S. salar) | ||
| 11 | (X) 6.6E-22 | 62 (62.9%) | AAG30024 | C-type lectin 2-1 (O. mykiss) | ||
| 10 | (N) 0 | 346 (99.1%) | BG936357 | unknown spleen clone SS1-0729 (S. salar) | ||
| 9 | (N) 0 | 876 (98.4%) | BG934637 | unknown kidney clone SK1-0954 (S. salar) | ||
| Head kidney (lower MW) | 784 | 57 | (X) 1.2E-79 | 148 (94.5%) | CAA65945 | β globinA (S. salar)f |
| 53 | (X) 1.1E-83 | 148 (100%) | CAA49580 | β globinB (S. salar)f | ||
| 41 | (X) 2.0E-82 | 148 (97.9%) | CAA65945 | β globinC (S. salar)f | ||
| 7 | (X) 1.1E-83 | 148 (100%) | CAA65945 | β globinD (S. salar)f | ||
| 7 | (X) 4.6E-66 | 119 (100%) | CAA65944 | α globin (S. salar) | ||
| Head kidney (higher MW) | 867 | 24 | (X) 9.4E-146 | 252 (100%) | 042161 | β actin (S. salar) |
| 5 | (X) 0 | 443 (96.2%) | A46533 | Immunoglobulin heavy chain constant region (S. salar)g | ||
| 4 | (X) 1.1E-141 | 287 (100%) | S21175 | dnaK-type molecular chaperone hsc71 (O. mykiss) | ||
| 4 | (X) 0 | 443 (100%) | A46533 | Immunoglobulin heavy chain constant region (S. salar)g | ||
| 4 | (N) 0 | 695 (99.7%) | AJ424426 | unknown kidney clone k09F03 (S. salar) | ||
| Mixed gut (stomach, mid, & hind) | 3753 | 47 | (X) 1.9E-85 | 239 (79.1%) | JH0472 | Apolipoprotein A-I (S. salar) |
| 20 | (X) 0 | 330 (97.8%) | AAK69705 | Procathepsin B (O. mykiss) | ||
| 18 | (X) 3.8E-34 | 76 (92.1%) | CAC45057 | Type II keratin E2 (O. mykiss) | ||
| 17 | (X) 0 | 461 (99.5%) | AAG38613 | Elongation factor 1 α (S. salar) | ||
| 16 | (X) 4.8E-89 | 325 (51.6%) | CAC87888 | Toad pancreatic chitinase (Bufo japonicus) | ||
| Ovary | 2664 | 13 | (X) 1.2E-48 | 139 (59.7%) | AAO43606 | Serum lectin isoform 2 (S. salar)h |
| 11 | (X) 4.3E-48 | 139 (59.1%) | AAO43606 | Serum lectin isoform 2 (S. salar)h | ||
| 6 | n/ad | n/ad | n/ad | unknown | ||
| 5 | n.ad | n/ad | n/ad | unknown | ||
| 5 | (X) 1.5E-16 | 127 (41.0%) | P56733 | Avidin-related protein 3 (Gallus gallus) | ||
| Pituitary Gland | 2883 | 123 | (N) 0 | 545 (98.5%) | X84787 | Prolactin (S. salar) |
| 96 | (N) 6.5E-166 | 1084 (94.9%) | X69809 | Proopiomelanocortine B (O. mykiss) | ||
| 48 | (N) 5.9E-73 | 173 (94.7%) | X69808 | Proopiomelanocortine A (O. mykiss) | ||
| 44 | (X) 1.0E-26 | 50 (100%) | AAA49558 | Growth hormone (S. salar) | ||
| 43 | (X) 3.1E-60 | 114 (100%) | AAA49407 | Gonadotropin-I α (Oncorhynchus keta) | ||
| Pyloric caeca (lower MW) | 329 | 8 | (X) 5.1E-123 | 262 (98.0%) | AAA88542 | Apolipoprotein A-I (S. salar)i |
| 7 | (X) 9.9E-67 | 268 (49.2%) | CAB65320 | Apolipoprotein E (O. mykiss) | ||
| 5 | (X) 9.7E-55 | 254 (46.0%) | BAB40965 | 28 kD-1e apolipoprotein (Anguilla japonica) | ||
| 5 | (X) 8.1E-27 | 129 (44.1%) | AAF61069 | Galectin (Paralichthys olivaceus) | ||
| 4 | (X) 3.9E-100 | 238 (92.4%) | JH0472 | Apolipoprotein A-I (S. salar)i | ||
| Pyloric caeca (higher MW) | 716 | 16 | (X) 1.2E-48 | 168 (58.2%) | AAG53688 | Selenoprotein Pa (Danio rerio) |
| 10 | (X) 4.5E-141 | 286 (86.4%) | AAG02508 | MHC Class I heavy chain (O. mykiss) | ||
| 8 | (X) 1.4E-32 | 122 (53.2%) | AAL85339 | Meprin A α; PABA peptide hydrolase (H. sapiens) | ||
| 7 | (N) 2.0E-12 | 115 (84.3%) | BG935597 | unknown liver clone SL1-0959 (S. salar) | ||
| 6 | (X) 3.8E-34 | 76 (92.1%) | CAC45057 | Type II keratin E2 (O. mykiss) |
Compiled using the August 16, 2003 version of the GRASP EST database and excluding ribosomal and mitochondrial EST clusters. For notes on libraries and library groups, see Table 2.
Most significant BLASTIN (N) or BLASTX (X) hit is reported. BLASTX hit reported if top BLASTN hit not associated with a named gene.
Extent of BLAST hit aligned region, and percent identity over the aligned region. Length (and percent identity) refers to amino acids if BLASTX reported, and nucleic acids if BLASTN reported.
Not applicable, as there are no significantly similar (E < 10-5) sequences in non-redundant GenBank nucleotide or amino acid sequence databases.
The C-terminal 111 amino acids of the aligned translations of these 2 EST contigs are 85.6% identical, indicating 2 distinct forms of the gene.
The aligned translations of β globins A, B, and C differ from the translation of β globin D at 8 of 148 (94.6% identity), 2 of 148 (98.6% identity), and 3 of 148 (98.0% identity) residues, respectively.
The aligned translations of these two EST clusters differ at 17 of 443 residues (96.2% identity).
The aligned translations of these two EST clusters differ at four of 139 residues (97.1% identity). They are ∼60% identical, at the amino acid level, to a third ovary lectin cluster containing four ESTs (not shown in table).
The aligned translations of these two EST clusters (238 amino acids) are 84.0% identical.
Insert orientation in various types of cDNA library was analyzed to determine its potential influence on gene discovery rates. All libraries in this database were classified by type (i.e. normalized, subtracted), and insert orientations in two libraries from each class were determined (see Supplemental table at http://web.uvic.ca/cbr/grasp for data, method, and discussion of bias). Incidences of reverse-oriented inserts were as follows: 4.5% in non-normalized, nonfractionated libraries (average of two analyzed libraries' values); 29% in non-normalized, higher-MW libraries; 20% in non-normalized, lower-MW libraries; 10% in normalized libraries; and 71.5% in subtracted (randomly cloned) libraries. The weighted average across all four directionally cloned library types (contributing 84.9% of the ESTs in the database) is 9.1% reverse orientation. M13 forward-read ESTs from reverse-oriented inserts give 5′ sequence.
The somewhat higher incidence of reverse-oriented inserts in higher-MW fraction libraries might contribute to the higher “% new” and “% singleton” values of these libraries over their lower-MW counterparts in our database (Table 2). However, insert orientation differences between library classes do not explain the dramatically higher “% no BLAST hit” values seen in higher-MW libraries (Table 2). Because most EST projects contributing to GenBank databases are biased toward 5′ sequencing, the higher “% no BLAST hit” values of our higher-MW libraries are likely conservative indices of the elevated rates of gene discovery associated with these libraries.
Assembled S. salar and O. mykiss ESTs were checked for open reading frames (ORFs) >200 bp (Fig. 1A). The chance of a random 66 codon (198 bp) ORF is (61/64)66 = 0.04206 (P < 0.05). Most of our ESTs are 3′ reads. The average observed 3′ UTR in this database is 264 bases (603′ ESTs considered; range, 59 to 592 bases), and average trimmed EST lengths are 484 to 563 bases (Table 1). Therefore, we believe that screening for 200-bp ORFs allows for adequate evaluation of the coding portion of the ESTs without excessive bias against genes with longer 3′ UTRs.
Figure 1.

Open reading frame (ORF) and BLAST results. Numbers of assembled Salmo salar (A) and Oncorhynchus mykiss (B) ESTs with and without 200 base ORFs are given. Within each of these categories, proportions of assembled ESTs with and without significant (E < 10–5) BLASTX hits against GenBank nonredundant protein database are shown, as are proportions of assembled ESTs with and without significant (E < 10–5) BLASTN hits against the nonredundant nucleotide database. The lengths and putative identifications (gene names of best BLASTX hits) of the longest ORFs in each species are given.
Of the 28,710 assembled S. salar sequences, 22,622 (79%) have ORFs >200 bp (Fig. 1A). Of these, 10,123 (45%) have significant (E < 10–5) BLASTX hits, and 9822 (43%) have significant (E < 10–5) BLASTN hits (Fig. 1A). Novel salmonid genes may be included in the 12,499 assembled ESTs containing 200-bp ORFs but without BLASTX matches (Fig. 1A). Of the 6088 assembled S. salar ESTs without 200-bp ORFs, 388 (6%) have significant BLASTX hits (likely representing cDNAs coding for short proteins) and 1664 (27%) have significant BLASTN hits (likely representing cDNAs for short proteins as well as previously identified salmonid intronic and untranslated sequences; Fig. 1A). The 4424 assembled S. salar ESTs having neither 200-bp ORFs nor BLASTN hits (Fig. 1A) probably include novel salmonid cDNAs with long 3′ UTRs. The ORF and BLAST results for O. mykiss assembled sequences are very similar to those for S. salar (Fig. 1B).
Using Functional Annotation to Infer Putative Organ Functions
The gene ontology (GO) statistics presented in this report reflect the state of our database on March 3, 2003. For the collective S. salar libraries, and for each S. salar library group, assembled ESTs were assigned putative molecular functions based on BLASTX similarity to functionally annotated human protein sequences (Gene Ontology Consortium 2001).
To illustrate ways in which this salmonid EST database may be mined for information on putative organ functions, we focused on a selection of S. salar organ-specific library groups: gill, mixed gut (stomach + mid-gut + hind-gut, not including pyloric caecum), ovary, pyloric caecum, and pituitary gland (Table 4). Overall, 26% of S. salar assembled ESTs matched sequences in the GO database (Table 4). For organ-specific libraries or library groups, the percentage of assembled ESTs hitting GO sequences ranged from 25% (ovary) to 40% (pyloric caecum; Table 4). Z-statistics were used to determine if, for a given GO classification, the proportion of assembled ESTs in an organ-specific S. salar library group differed significantly from the proportion of assembled ESTs from remaining S. salar library groups (see Methods, Table 4, and Supplemental data at http://web.uvic.ca/cbr/ grasp). Because putative organ functions are sought, only those GO categories with disproportionately high numbers of assembled ESTs will be discussed. The gill library has disproportionately high numbers of assembled ESTs in the “iron binding,” “oxidoreductase, acting on heme group of donors,” and “transporter” GO categories (Table 4). Disproportionately high numbers of ovary assembled ESTs are seen in GO categories related to heavy metal (copper, iron, and zinc) binding and enzyme inhibition (Table 4). The pituitary gland library has disproportionately high numbers of assembled ESTs in “iron binding,” “hormone binding,” and “oxidoreductase, acting on heme group of donors” categories (Table 4).
Table 4.
Gene Ontologya (Molecular Function) of Assembled ESTs From Organ-Specific Libraries/Library Groupsb

This approach was used to acquire putative functional information on a poorly characterized organ, the pyloric caecum. The teleost pyloric caecum, a large elaborate set of finger-like extensions off the gut, is known to play an important role in nutrient uptake (Buddington and Diamond 1986). There is scant literature on the fish pyloric caecum. Douglas et al. (1999b) classified 147 winter flounder pyloric caecum ESTs by putative function. Winter flounder pyloric caecum libraries were used to isolate cDNA clones encoding trypsinogen (Douglas and Gallant 1998) and aminopeptidase N (Douglas et al. 1999a). Here we report 6559 S. salar pyloric caecum ESTs, facilitating a deeper understanding of gene expression in this organ.
Both mixed gut and pyloric caecum library groups have disproportionately high numbers of assembled ESTs in the “enzyme,” “oxidoreducatase, acting on heme group of donors,” and “transporter” GO categories (Table 4). These may point to general functions along the digestive tract. “Iron binding” and “hydrolase” GO categories have disproportionately high numbers of assembled ESTs in mixed gut but not pyloric caecum (Table 4). There are disproportionately high numbers of assembled ESTs in the “cytochrome P450,” “selenium binding,” “oxidoreductase, acting on NADH or NADPH,” “oxidoreductase, acting on peroxide as acceptor,” and “transferring sulfur-containing groups” categories in pyloric caecum but not mixed gut library groups (Table 4), indicating putative specialized roles for the pyloric caecum. Selenium is a component of selenoprotein P and glutathione peroxidases, antioxidant enzymes that protect cells from oxidative injury (Deplancke and Gaskins 2002; Burk et al. 2003; Schomburg et al. 2003). Selenoprotein P is one of the largest EST contigs in the pyloric caecum library group (Table 5), and this library group contains at least eight different assembled ESTs identified by BLAST as glutathione peroxidases. At least 10 different pyloric caecum assembled ESTs are identified as cytochromes P450, a class of heme-containing monooxygenases involved in metabolism of foreign compounds such as environmental pollutants and agrochemicals (Danielson 2002). Collectively, these results indicate that the salmon pyloric caecum functions in redox control and as a barrier against the systemic uptake of xenobiotics.
Table 5.
Largest EST Clustersa in the Pyloric Caecum Library Group,b and Locations of Other Members of Clusters Across S. salar cDNA Library Groupsb

Additional hypothetical functions of the pyloric caecum may be proposed by examining the largest EST clusters (representing highly expressed genes) in the pyloric caecum library group, and locating other members of these clusters across all S. salar library groups (Table 5). Several defense-relevant EST clusters, including CC chemokine macrophage inflammatory protein (MIP)-3a, galectin, and GDP-d-mannose-4,6-dehydratase (GMD), derive most of their ESTs from pyloric caecum libraries (Table 5). Galectins serve as master regulators of immune cell homeostasis during innate immune responses (Rabinovich et al. 2002). GMD is required for the synthesis of fucosylated oligosaccharides, selectin ligands involved in leukocyte extravasation (Ohyama et al. 1998; Eshel et al. 2001). These data indicate an innate defense function of the salmonid pyloric caecum. That previously unknown EST types, frequencies, and distributions have been observed among pyloric caecum and other organs by this analysis indicates general utility for this approach in revealing unknown functions of many other organ and cellular systems.
Application of a Salmonid cDNA Microarray to Different Species
A preliminary cDNA microarray (available from corresponding author), composed of 6440AS and 916 rainbow trout (RT) cDNA elements or spots (Table 6), was hybridized with labeled targets from three members of the order Salmoniformes (AS, RT, and lake whitefish [LW]) and one member of the order Osmeriformes (rainbow smelt; Fig. 2A) to explore the validity of using this microarray with other fish species. Hybridization performance of each species' labeled target to the salmonid elements was judged from the numbers of AS and RT elements passing a hybridization signal threshold, and mean total raw signals from AS and RT elements (see Methods; Table 6). No transformations or normalizations were performed on the data. Data and statistics for all slides are given in Table 6.
Table 6.
Analysis of Cross-Species Hybridization to 7356 Element Salmonid cDNA Microarraya

Figure 2.

Evolutionary relationships, genome sizes, and microarray hybridization characteristics of three salmonids relative to smelt. (A) Phylogenetic tree, based on morphological characters, showing evolutionary relationships among teleosts relevant to this study, and other fish orders with genome projects (Nelson 1994). (B) Phylogenetic tree, based on morphological characters, showing evolutionary relationships of select salmonids (Smith and Stearley 1989; Kido et al. 1991). Arrows indicate putative genome duplication events (Wolfe 2001). (C, D) Mean total signals on Atlantic salmon (AS) or rainbow trout (RT) chip elements/spots (Table 6) are converted to “smelt units” by dividing by 0.661E7 for AS chip elements, or 0.824E6 for RT chip elements. Genome sizes for AS (Salmo salar), RT (Oncorhynchus mykiss), and smelt (Osmerus eperlanus, close relative of Osmerus mordax used in this study) were measured by DNA flow cytometry (Vinogradov 1998). Genome size of lake whitefish (LW, Coregonus clupeaformis) was measured by Feulgen densitometry (Booke 1968). Error bars (C) show mean total signal SEM values (Table 6) converted to “smelt units” as above. n indicates number of microarrays hybridized with labeled target from each species.
To evaluate the effect of element (cDNA spotted onto the microarray slide) and target (labeled cDNA hybridized to the slide) species affiliations on hybridization characteristics, data and statistics for AS and RT microarray elements were compiled separately. On AS probes, AS target gave the highest signal (mean of three slides: 2.01E7, SEM 4.99E5), followed by RT (mean of three slides: 1.88E7, SEM 8.16E4), LW (mean of three slides: 1.54E7, SEM 3.31E5), and rainbow smelt (mean of three slides: 6.61E6, SEM 5.37E5; Table 6). On RT probes, RT target gave the highest signal (mean of three slides: 2.53E6, SEM 4.19E4), followed by AS (mean of three slides: 1.93E6, SEM 5.25E4), LW (mean of three slides: 1.88E6, SEM 1.64E4), and rainbow smelt (mean of three slides: 8.24E5, SEM 7.56E4; Table 6).
The ranking of hybridization performances conformed to expectations, given the evolutionary relationships of the species tested (Fig. 2A,B). AS and RT, members of the subfamily Salmoninae, diverged in the Miocene 8 to 20 million years ago (Stearley 1992; Devlin 1993; Coe et al. 1995). Phylogenies based on morphological (Nelson 1994) and molecular (Phillips and Oakley 1997) data show that the genera Salmo and Oncorhynchus are more closely related to one another than either group is related to Coregonus, the genus of LW. On both AS and RT chip elements, hybridization performance of LW target ranks third behind AS and RT targets (Table 6, Fig. 2C,D). Because the mean numbers of AS and RT elements passing threshold are comparable for AS, RT, and LW targets (Table 6), the lower signal from LW-hybridized slides likely reflects lower percentage of identity between salmonine probe and coregonine target sequences. These hybridization results match predicted distances of divergence for the salmonid species tested (Fig. 2B). Our preliminary analysis of AS and RT putatively orthologous EST contigs (primarily 3′) shows ∼94% identity and is in agreement with the success of these species' targets on one anothers' probes (Table 6, Fig. 2C,D). Our EST database does not yet contain adequate numbers of LW EST contigs to permit large-scale alignment of putative orthologous sequences. However, the high performances of LW targets on AS and RT probes (Table 6, Fig. 2C,D) are suggestive of high similarity between LW and salmonine orthologous cDNAs. Hybridization performances of rainbow smelt targets were less than half those of salmonid (AS, RT, or LW) targets (Table 6, Fig. 2C,D), likely due to lower similarity (reflecting longer time since divergence) between cDNAs from members of the order Salmoniformes and orthologous sequences from members of the order Osmeriformes (Fig. 2A).
Identification of Candidate Duplicated Genes
Osmerids are diploid and salmonids are degenerate tetraploids (Ohno et al. 1968; Fig. 2C,D), placing the putative, salmonid-specific genome duplication event after the divergence of Osmeridae and Salmonidae (Fig. 2A). Because at least 50% of recent gene duplicates are thought to persist in salmonids (Bailey et al. 1978), it is expected that gene family expansion (the presence of multiple expressed paralogs) would be widespread in this group. Preliminary comparisons of robust EST clusters (in single AS libraries/library groups) that have identical top BLASTX hits reveal the presence of multiple distinct forms (not splice variants) of several genes (i.e., novel member of chitinase family, β-globin, and serum lectin, Table 3; 28 kD – 1e apolipoprotein, Table 5). Further work (i.e., molecular phylogenetics, fluorescence in situ hybridization) will be required to distinguish paralogs arising during the recent salmon-specific genome duplication from those with origins in other gene/genome duplication events. The GRASP EST database, and an improved salmonid presence in GenBank databases, will facilitate identification of additional members in gene families, contributing to a better understanding of the evolution of related genes within and between genomes.
METHODS
Aquaculture and Sampling
S. salar (McConnell strain) juveniles were obtained from Heritage Aquaculture (British Columbia, Canada), and cultured throughout their life history. Subadult S. salar were sampled from various tissues at 2.75 years of age (Fisheries and Oceans Canada, West Vancouver, British Columbia) and used for generating all adult cDNA libraries and labeled targets for microarray hybridizations. For juvenile cDNA libraries, S. salar (McConnell strain) and O. mykiss (Tzenzaicut Lake strain) were obtained from SeaSpring Hatchery (Duncan, British Columbia) and Vancouver Island Trout Hatchery (Duncan, British Columbia), respectively. For labeled targets used in microarray hybridizations, embryonic stages of O. mykiss were derived from a domesticated strain (Spring Valley Trout Farm, Langley, British Columbia) and cultured to ∼80g before sampling. O. mykiss gonadal tissues (≥1.5 years; Spring Valley Strain), used to generate subtractive cDNA libraries, were obtained from Mountain Trout Sales (Sooke, British Columbia). O. tshawytscha tissues were obtained from 4-year-old females (Robertson Creek, British Columbia); O. nerka tissues were obtained from whole juvenile fish (Dr. L.J. Albright, Simon Fraser University); C. clupeaformis brain and liver were obtained from 3-year-old animals (Laboratoire Bernatchez, Université Laval, Quebec), and Osmerus mordax livers were obtained from adult smelt (NRC Institute for Marine Biosciences).
Fish were raised in fiberglass tanks with natural lighting and at densities <10kg/m3 with water input rate >1 L min–1 kg–1. S. salar and O. tshawytscha were reared in fresh 10°C well water until smolt stage (1.5 years) and then transferred to sea water until sexual maturation. O. mykiss were cultured only in fresh 10°C well water. Most fish were fed to satiation three times per day with commercial salmon diets (Pacific Apollo 1000, Moore Clarke, Vancouver, British Columbia) comprised of 40% protein and 25% lipid.
Fish were killed by a blow to the head, followed by rapid dissection. Tissues were flash-frozen in liquid nitrogen and stored at –80°C until RNA extraction. For gut tissues, discrete sections were excised and the lumen gently rinsed free of food and feces with a stream of ice-cold phosphate-buffered saline (10 mM PO4, 138 mM NaCl, and 27 mM KCl at pH 7.4).
cDNA Libraries
Flash-frozen tissues were ground by using baked (5 h, 220°C) mortars and pestles under liquid N2, and poly(A)+ RNA was purified by using MicroPoly(A)Pure kits (Ambion) or Oligotex Direct mRNA Micro Kits (Qiagen). With the exception of the O. nerka libraries, the normalized S. salar mixed tissue library, and the suppression subtractive hybridization (SSH) libraries, cDNA libraries were directionally constructed (5′ EcoRI, 3′ XhoI), using the pBluescript II XR cDNA Library Construction Kit, following manufacturer's instructions (Stratagene). Size fractionation, performed on XhoI-digested cDNAs immediately prior to ligation into vector, was by 1% agarose gel extraction (Qiagen). O. nerka libraries were size-fractionated by using CHROMA Spin-400 columns (Clontech), and directionally constructed (5′ SfiIA, 3′ SfiIB) in pDNR-LIB using the Creator SMART cDNA Library Construction Kit (Clontech). Select cDNA libraries were normalized to Cot = 5 by using the Soares method (Soares et al. 1994; Bonaldo et al. 1996). The normalized (Cot = 10) S. salar mixed tissue (spleen, head kidney, brain) library was directionally constructed in pCMV Sport6.1 (ResGen). SSH libraries were constructed by using the PCR-Select cDNA Subtraction Kit (Clontech) following manufacturer's instructions, and were TA cloned into pCR 4-TOPO (Invitrogen). Insert sizes of cDNA libraries were determined by visual comparison of clone restriction fragments with the DNA size markers λHindIII (GIBCO-BRL) and 1-kb ladder (GIBCO-BRL).
Sequencing, Sequence Analysis, and Contig Assembly
Libraries were manually arrayed in 96-well microtiter plates or were robotically arrayed in 384-well plates. Glycerol stocks of overnight cultures were prepared in 96-well or 384-well format. Plasmid DNAs were extracted and BigDye Terminator (ABI) cycle sequenced on ABI 3700 and 377 sequencers by using conventional procedures and the following primers: 5′-T18-3′, M13 forward (5′-GTAAAACGACGGCCAGT-3′), and M13 reverse (5′-AACAGCTATGACCATG-3′ or 5′-AACAGCTATGACCAT-3′). Base-calling from chromatogram traces was performed by using PHRED (Ewing and Green 1998; Ewing et al. 1998). Vector, poly-A tails, and low-quality regions were trimmed from EST sequences; sequences that had <100 good-quality bases after trimming were discarded.
Vectors were screened by using cross_match (part of the PHRAP package, version 0.990329), with minscore = 18. This is more sensitive than Consed, allowing detection of adaptor sequences in subtractive libraries. All vector was trimmed from the ends of the sequence. If there was remaining vector in the middle, it was removed and the shorter of the two remaining fragments trimmed with it.
To trim poly-A tails, sequences were scanned from their ends forward to the beginning of the last run of consecutive As. If the tail of the sequence up to that point was at least 60% A, then it was considered part of the tail. This test was repeated from that point forward until it failed. The portion of the sequence that passed was considered poly-A tail. If this test found nothing, then the last 100 bases of the sequence were scanned for a run of at least 15 consecutive As. If found, then the trailing sequence was assumed to be bad or vector, and all sequence up to and including the run of As was trimmed. To scan for poly-T tails, the same tests were performed on reverse-complemented sequences. Sequences were not considered poly-A or poly-T tails if they were <10 bases in length.
PHRAP (http://www.genome.washington.edu/UWGC), under stringent clustering parameters (minimum score, 100; repeat stringency, 0.99), was used to assemble ESTs into contigs. Contig consensus sequences and singleton sequences were aligned with nonredundant GenBank nucleotide and amino acid sequence databases by using BLASTN and BLASTX, respectively (Altschul et al. 1990, 1997). Results of EST clustering using CAP3 (40-bp overlap, 95% identity, other parameters default) and stackPACK (using RepeatMasked sequences without quality scores) are available at the GRASP Web site (http://web.uvic.ca/cbr/grasp). To determine the approximate amount of ribosomal and mitochondrial sequence in the GRASP EST database, each species' ESTs were aligned against a BLAST database containing the same species' GenBank sequences annotated as ribosomal plus the GenBank mitochondrial sequences from that species or its closest relative. BLAST hits with E values <10–5 qualified ESTs as ribosomal or mitochondrial.
Assembled EST contigs were scanned for repeats by using REPuter (Kurtz et al. 2001). Candidate repeats (length >50 bases, fewer than eight mismatches, and E < 10–4) were assembled into contigs by using PHRAP, and compared with GenBank nr and nt databases by using BLASTX and BLASTN, respectively. Threshold for a significant BLAST hit was set at 10–15. BLAST results were deposited in a database, and a Web interface for querying was implemented (http://woodstock.ceh.uvic.ca/nkuipers/public_html/).
O. mykiss orthologs to S. salar contigs were detected by semiglobal (end-gaps-free) pairwise alignment of forward and reverse-complement contigs. Alignments with overlaps of <100 nucleotides were discarded. O. mykiss contigs were considered orthologous to an S. salar contig if either the forward- or reverse-complement alignment showed at least 80% identity.
Functional Characterization of EST Contigs
By using the March 3, 2003, version of the GRASP EST database, assembled S. salar ESTs from select organ-specific libraries or library groups (pyloric caecum, gill, mixed gut, ovary, and pituitary gland), and all S. salar libraries collectively, were compared via BLASTX with annotated protein sequences from the GO database (November 2002 version; Table 4). Sequences with significant matches (E-value < 10–5) were classified according to the GO classification(s) of their strongest hit. For several GO functional categories of genes, Z-statistics were used to determine if there were significant differences between the proportions of assembled ESTs in an organ-specific library/library group (i.e., gill) and the proportions of assembled ESTs in remaining (i.e., non-gill) libraries. Z-statistics, used for the comparison of two sample proportions (Anderson and Finn 1997), were calculated by using the following equation:
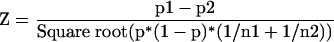 |
where p1 is the proportion of assembled ESTs in the organ of interest (2/444 for gill library, antioxidant GO category; Table 4), p2 is the proportion of assembled ESTs in nonorgan libraries (9/6869 for nongill, antioxidant; see Supplemental data at http://web.uvic.ca/cbr/grasp), p is the overall proportion (10/6937 for antioxidant; Table 4), n1 is the number of organ-specific assembled ESTs (444 for gill; Table 4), and n2 is the number of nonorgan assembled ESTs (6869 for nongill; see Supplemental data at http://web.uvic.ca/cbr/grasp). Z has a standard normal distribution, so P-values are computed as 1 – (CDF (abs(Z)) × 2), where CDF is the cumulative distribution function of the standard normal distribution and abs is absolute value. This P-value gives a two-tailed test for the probability that the proportions of organ and nonorgan EST contigs in a given molecular function category are equal.
Microarray Fabrication and Quality Control
The 3557 clones from 18 high-complexity salmonid cDNA libraries/library groups (Table 2) were selected with an emphasis on immune relevant genes. Clones were robotically rearrayed from daughter glycerol stock 384-well plates into 96-well plates prefilled with 7% glycerol in LB + ampicillin, incubated overnight at 37°C, and checked for uniform optical density. Plasmid inserts were PCR-amplified in a Tetrad PTC-200 thermocycler (MJ Research) by using 1 μL overnight culture, 0.2 μM M13/pUC forward primer (5′-CCCAGTCACGACGTTGTAAAACG-3′), 0.2 μM M13/pUC reverse primer (5′-AGCGGATAACAATTTCACACAGG-3′), 2 mM MgCl2, 10 mM Tris-HCl, 50 mM KCl, 250μM dNTPs, 1U AmpliTaq (PerkinElmer), and nuclease-free H2O (GIBCO) to 100 μL. PCR conditions were as follows: 2 min at 95°C denaturation; 35 cycles of 30 sec at 95°C, 45 sec at 60°C, and 3 min at 72°C; and 7 min at 72°C. Five microliters of each PCR product were run on a 1% agarose gel to assess yield and quality. Out of 3557 clones, there were 3312 strong single bands (93%), 170 absent (5%), and 75 multiple bands (2%). PCR products were robotically cleaned (Qiagen) and consolidated into 384-well plates, lyophilized by speed-Vac, and resuspended in 15 μL 3× SSC.
All cDNAs were printed as double, side-by-side spots on Telechem Superamine slides (Arrayit) with the Biorobotics Microgrid II microarray printer (Apogent Discoveries). Microspot 10K quill pins (Biorobotics) in a 48-pin tool were used to deposit ∼0.5 nL (0.2 ng cDNA) per spot onto the slide. The resulting microarrays have a 4 × 12 subgrid layout with 132 spots per subgrid, each spot having approximate diameter and pitch of 100 μm and 250μm, respectively. A 280-bp GFP (green fluorescent protein) cDNA was amplified from a GFP clone (Clontech) by using the primers (5′-GAAACATTCTTGGACACAAATTGG-3′) and (5′-GCAGCTGTTACAAACTCAAGAAGG-3′), and printed in subgrid corners to assist in placing on the grid. The slides were crosslinked in a UV Stratalinker 2400(Stratagene) at 120mJ. Spot morphology was assessed by visual inspection, SYBR Green 1 (Molecular Probes) staining, or hybridization with labeled non-specific probe. To check clone tracking, 42 high-quality sequences were obtained from randomly selected wells of the cleaned, consolidated 384-well plates used for microarray printing. All 42 had BLAST identifiers matching gene identifications predicted from the rearray spreadsheet, indicating highly accurate clone tracking throughout the process of microarray fabrication.
Microarray Hybridization and Analysis
This microarray experiment was designed to comply with MIAME guidelines (Brazma et al. 2001). All scanned microarray TIF images, an ImaGene grid, the gene identification file, and ImaGene quantified data files are available at http://web.uvic.ca/cbr/grasp. To minimize technical variability, all targets were synthesized in one round, and all hybridizations were conducted simultaneously on slides from a single batch (CL010, Table 6). Total RNA, prepared from flash-frozen adult liver tissues using TRIzol reagent and methods (Invitrogen), was quantified and quality-checked by spectrophotometer and agarose gel. Hybridizations were performed by using the Genisphere Array50 kit and instructions. Briefly, 15 μg total RNA were reverse-transcribed by using a special oligo d(T) primer with a 5′ unique sequence overhang for the Cy3 labeling reactions. Microarrays were prepared for hybridization by washing two times at 5 min in 0.1% SDS, washing five times at 1 min in MilliQ H2O, immersing 3 min in 95°C MilliQ H2O, and drying by centrifugation (5 min at 2000 rpm in 50-mL conical tube). The cDNA was hybridized to the salmon cDNA microarray in a formamide-based buffer (25% formamide, 4× SSC, 0.5% SDS, 2× Denhardt's solution) for 16 h at 48°C. The arrays were washed one time for 10 min at 48°C (2× SSC, 0.1% SDS), two times for 5 min in 2× SSC, 0.1% SDS at room temperature (RT), two times for 5 min in 1× SSC at RT, and two times for 5 min in 0.1× SSC at RT, and dried by centrifugation. The Cy3 3-dimensional fluorescent molecules (3DNA capture reagent, Genisphere) were hybridized to the bound cDNA on the microarray; the Cy3 3DNA capture reagent bound to its complementary cDNA capture sequence on the Cy3 oligo d(T) primer. The second hybridization was done for 3 h at 48°C, and washed and dried as before.
The fluorescent images of hybridized arrays were acquired by using ScanArray Express (PerkinElmer). The Cy3 cyanine fluor was excited at 543 nm, and the same laser power (90%) and photomultiplier tube (PMT) setting (75) were used for all slides in the study. Fluorescent intensity data was extracted by using Imagene 5.5 software (Biodiscovery). To avoid transformations associated with background correction (i.e., setting negative background corrected median signal values to zero), raw median signal values were analyzed. No normalization was applied to the data. From the raw Imagene fluorescence intensity report files, the gene lists were sorted, and median signal values from 1356 control elements (204 buffer alone, 912 bare glass, and 240 GFP cDNA) were analyzed. For each slide, threshold was calculated as the mean intensity for these 1356 controls plus 2 SD. For data analyses, the 6440 S. salar (AS) chip elements and 916 O. mykiss (RT) chip elements were considered separately. The mean numbers of AS and RT elements passing threshold, mean total slide signal (salmonid elements only) and SEM, mean total slide background (local background fluorescence intensities associated with salmonid elements) and SEM, and average signal and background per salmonid element were calculated by slide and by species. To assess array-wide performance, signal-to-background ratio was calculated as raw total signal divided by raw total background.
Acknowledgments
This research was supported by Genome Canada, Genome BC, and the Province of BC and, additionally, by the Natural Sciences and Engineering Research Council of Canada (B.K., W.D.). We would like to thank Carlo Biagi, Steve Dann, and Shelby Temple for their assistance in obtaining tissues for cDNA library construction; Bento Soares and Brian Berger for providing methods and advice on normalizing cDNA libraries, and all those at the BCCA Genome Sciences Centre who contributed to this work.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.1687304. Article published online before print in February 2004.
Footnotes
[Supplemental material is available online at http://web.uvic.ca/cbr/grasp. The sequence data from this study have been submitted to GenBank dbEST under accession nos.: Salmo salar, BU965588–BU965906, CA036414–CA039704, CA039711–CA064598, CA767613–CA770910, and CB498694–CB518126; Oncorhynchus mykiss, CB485850–CB498693; Oncorhynchus tshawytscha, CB484816–CB485849; Oncorhynchus nerka, CD510521–CD511184; and Coregonus clupeaformis, CB483540–CB484653. The following individuals kindly provided reagents, samples, or unpublished information as indicated in the paper: C. Biagi, S. Dann, S. Temple, and R. Roper stimulated the S. salar head kidney cells used to create one cDNA library group.]
References
- Allendorf, F.W. and Danzmann, R.G. 1997. Secondary tetrasomic segregation of MDH-B and preferential pairing of homologues in rainbow trout. Genetics 145: 1083–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allendorf, F.W. and Thorgaard, G.H. 1984. Tetraploidy and the evolution of salmonid fishes. In Evolutionary genetics of fishes (ed. B.J. Turner), pp. 1–53. Plenum Press, New York.
- Altschul, S.F., Gish, W., Miller, W., Myers, E.W., and Lipman, D.J. 1990. Basic local alignment search tool. J. Mol. Biol. 215: 403–410. [DOI] [PubMed] [Google Scholar]
- Altschul, S.F., Madden, T.L., Schäffer, A.A., Zhang, J., Zhang, Z., Miller, W., and Lipman, D.J. 1997. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 25: 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson, T.W. and Finn, J.D. 1996. The new statistical analysis of data. Springer-Verlag, New York.
- Bailey, G.S., Poulter, R.T., and Stockwell, P.A. 1978. Gene duplication in tetraploid fish: Model for gene silencing at unlinked duplicated loci. Proc. Natl. Acad. Sci. 75: 5575–5579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey, G.S., Williams, D.E., and Hendricks, J.D. 1996. Fish models for environmental carcinogenesis: The rainbow trout. Environ. Health Perspect. 104: 5–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu, N., Todgham, A.E., Ackerman, P.A., Bibeau, M.R., Nakano, K., Schulte, P.M., and Iwama, G.K. 2002. Heat shock protein genes and their functional significance in fish. Gene 295: 173–183. [DOI] [PubMed] [Google Scholar]
- Bonaldo, M.F., Lennon, G., and Soares, M.B. 1996. Normalization and subtraction: Two approaches to facilitate gene discovery. Genome Res. 6: 791–806. [DOI] [PubMed] [Google Scholar]
- Booke, H.E. 1968. Cytotaxonomic studies of the coregonine fishes of the Great Lakes, USA: DNA and karyotype analysis. J. Fish. Res. Board Can. 25: 1667–1687. [Google Scholar]
- Brazma, A., Hingamp, P., Quackenbush, J., Sherlock, G., Spellman, P., Stoeckert, C., Aach, J., Ansorge, W., Ball, C.A., Causton, H.C., et al. 2001. Minimum information about a microarray experiment (MIAME): Toward standards for microarray data. Nat. Genet. 29: 365–371. [DOI] [PubMed] [Google Scholar]
- Buddington, R.K. and Diamond, J.M. 1986. Aristotle revisited: The function of pyloric caeca in fish. Proc. Natl. Acad. Sci. 83: 8012–8014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burk, R.F., Hill, K.E., and Motley, A.K. 2003. Selenoprotein metabolism and function: Evidence for more than one function for selenoprotein P. J. Nutr. 133: 1517S–1520S. [DOI] [PubMed] [Google Scholar]
- Coe, I.R., von Schalburg, K.R., and Sherwood, N.M. 1995. Characterization of the Pacific salmon gonadotropin-releasing hormone gene, copy number, and transcription start site. Mol. Cell. Endocrinol. 115: 113–122. [DOI] [PubMed] [Google Scholar]
- Danielson, P.B. 2002. The cytochrome p450 superfamily: Biochemistry, evolution and drug metabolism in humans. Curr. Drug Metab. 3: 561–597. [DOI] [PubMed] [Google Scholar]
- Davey, G.C., Caplice, N.C., Martin, S.A., and Powell, R. 2001. A survey of genes in the Atlantic salmon (Salmo salar) as identified by expressed sequence tags. Gene 263: 121–130. [DOI] [PubMed] [Google Scholar]
- Deplancke, B. and Gaskins, H.R. 2002. Redox control of the transsulfuration and glutathione biosynthesis pathways. Curr. Opin. Clin. Nutr. Metab. Care 5: 85–92. [DOI] [PubMed] [Google Scholar]
- Devlin, R.H. 1993. Sequence of sockeye salmon type 1 and type 2 growth hormone genes and the relationship of rainbow trout with Atlantic and Pacific salmon. Can. J. Fish. Aquat. Sci. 50: 1738–1748. [Google Scholar]
- Devlin, R.H., Biagi, C.A., Yesaki, T.Y., Smailus, D.E., and Byatt, J.C. 2001. Growth of domesticated transgenic fish. Nature 409: 781–782. [DOI] [PubMed] [Google Scholar]
- Douglas, S.E. and Gallant, J.W. 1998. Isolation of cDNAs for trypsinogen from the winter flounder. Pleuronectes americanus. J. Mar. Biotechnol. 6: 214–219. [PubMed] [Google Scholar]
- Douglas, S.E., Bullerwell, C.E., and Gallant, J.W. 1999a. Molecular investigation of aminopeptidase N expression in the winter flounder. Pleuronectes americanus. J. Appl. Ichthyol. 15: 80–86. [Google Scholar]
- Douglas, S.E., Gallant, J.W., Bullerwell, C.E., Wolff, C., Munholland, J., and Reith, M.E. 1999b. Winter flounder expressed sequence tags: Establishment of an EST database and identification of novel fish genes. Mar. Biotechnol. 1: 458–464. [DOI] [PubMed] [Google Scholar]
- Eshel, R., Besser, M., Zanin, A., Sagi-Assif, O., and Witz, I.P. 2001. The FX enzyme is a functional component of lymphocyte activation. Cell. Immunol. 213: 141–148. [DOI] [PubMed] [Google Scholar]
- Ewing, B. and Green, P. 1998. Base-calling of automated sequencer traces using PHRED, II: Error probabilities. Genome Res. 8: 186–194. [PubMed] [Google Scholar]
- Ewing, B., Hillier, L., Wendl, M.C., and Green, P. 1998. Base-calling of automated sequencer traces using PHRED, I: Accuracy assessment. Genome Res. 8: 175–185. [DOI] [PubMed] [Google Scholar]
- Faillace, M.P., Julian, D., and Korenbrot, J.I. 2002. Mitotic activation of proliferative cells in the inner nuclear layer of the mature fish retina: Regulatory signals and molecular markers. J. Comp. Neurol. 451: 127–141. [DOI] [PubMed] [Google Scholar]
- Gregory, T.R. 2002. Animal genome size database. http://www.genomesize.com.
- Holland, P.W., Garcia-Fernandez, J., Williams, N.A., and Sidow, A. 1994. Gene duplications and the origins of vertebrate development. Development (Suppl.) 1994: 125–133. [PubMed]
- Katchamart, S., Miranda, C.L., Henderson, M.C., Pereira, C.B., and Buhler, D.R. 2002. Effect of xenoestrogen exposure on the expression of cytochrome P450 isoforms in rainbow trout liver. Environ. Toxicol. Chem. 11: 2445–2451. [PubMed] [Google Scholar]
- Kido, Y., Aono, M., Yamaki, T., Matsumoto, K., Murata, S., Saneyoshi, M., and Okada, N. 1991. Shaping and reshaping of salmonid genomes by amplification of tRNA-derived retroposons during evolution. Proc. Natl. Acad. Sci. 88: 2326–2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtz, S., Choudhuri, J.V., Ohlebusch, E., Schleiermacher, C., Stoye, J., and Giegerich, R. 2001. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 29: 4633–4642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madigou, T., Uzbekova, S., Lareyre, J.J., and Kah, O. 2002. Two messenger RNA isoforms of the gonadotrophin-releasing hormone receptor, generated by alternative splicing and/or promoter usage, are differentially expressed in rainbow trout gonads during gametogenesis. Mol. Reprod. Dev. 63: 151–160. [DOI] [PubMed] [Google Scholar]
- Nelson, J.S. 1994. Fishes of the world, 3rd ed. John Wiley & Sons, New York.
- Ohno, S. 1970. Evolution by gene duplication. Springer-Verlang, Heidelberg, Germany.
- Ohno, S., Stenius, C., Faisst, E., and Zenzes, M.T. 1965. Post-zygotic chromosomal rearrangements in rainbow trout (Salmo irideus GIBBONS) Cytogenetics 4: 117–129. [DOI] [PubMed] [Google Scholar]
- Ohno, S., Wolf, U., and Atkin, N.B. 1968. Evolution from fish to mammals by gene duplication. Hereditas 59: 169–187. [DOI] [PubMed] [Google Scholar]
- Ohyama, C., Smith, P.L., Angata, K., Fukuda, M.N., Lowe, J.B., and Fukuda, M. 1998. Molecular cloning and expression of GDP-D-mannose-4,6-dehydratase, a key enzyme for fucose metabolism defective in Lec13 cells. J. Biol. Chem. 273: 14582–14587. [DOI] [PubMed] [Google Scholar]
- Phillips, R.B. and Oakley, T.H. 1997. Phylogenetic relationships among the Salmoninae based on nuclear and mitochondrial DNA sequences. In Molecular systematics of fishes (eds. T.D. Kocher and C.A. Stepien), pp. 145–162. Academic Press, San Diego, CA.
- Phillips, R. and Ráb, P. 2001. Chromosome evolution in the Salmonidae (Pisces): An update. Biol. Rev. 76: 1–25. [DOI] [PubMed] [Google Scholar]
- Rabinovich, G.A., Rubinstein, N., and Toscano, M.A. 2002. Role of galectins in inflammatory and immunomodulatory processes. Biochim. Biophys. Acta 1572: 274–284. [DOI] [PubMed] [Google Scholar]
- Schomburg, L., Schweizer, U., Holtmann, B., Flohe, L., Sendtner, M., and Kohrle, J. 2003. Gene disruption discloses role of selenoprotein P in selenium delivery to target tissues. Biochem. J. 370: 397–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shum, B.P., Guethlein, L., Flodin, L.R., Adkison, M.A., Hedrick, R.P., Nehring, R.B., Stet, R.J.M., Secombes, C., and Parham, P. 2001. Modes of salmonid MHC class I and II evolution differ from the primate paradigm. J. Immunol. 166: 3297–3308. [DOI] [PubMed] [Google Scholar]
- Sidow, A. 1996. Gen(om)e duplications in the evolution of early vertebrates. Curr. Opin. Genet. Dev. 6: 715–722. [DOI] [PubMed] [Google Scholar]
- Smith, G.R. and Stearley, R.F. 1989. The classification and scientific names of rainbow and cutthroat trouts. Fisheries 14: 4–10. [Google Scholar]
- Soares, M.B., Bonaldo, M.F., Jelene, P., Su, L., Lawton, L., and Efstratiadis, A. 1994. Construction and characterization of a normalized cDNA library. Proc. Natl. Acad. Sci. 91: 9228–9232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stearley, R.F. 1992. Historical ecology of the Salmoninae, with special reference to Oncorhynchus. In Systematic historical ecology and North American freshwater fishes (ed. R.L. Mayden), pp. 622–658. Stanford University Press, Stanford, CA.
- Tipsmarck, C.K., Madsen, S.S., Seidelin, M., Christensen, A.S., Cutler, C.P., and Cramb, G. 2002. Dynamics of Na+,K+,2Cl– cotransporter and Na+,K+-ATPase expression in the branchial epithelium of brown trout (Salmo trutta) and Atlantic salmon (Salmo salar). J. Exp. Zool. 293: 106–118. [DOI] [PubMed] [Google Scholar]
- Vinogradov, A.E. 1998. Genome size and GC-percent in vertebrates as determined by flow cytometry: The triangular relationship. Cytometry 31: 100–109. [DOI] [PubMed] [Google Scholar]
- Wolfe, K.H. 2001. Yesterday's polyploids and the mystery of diploidization. Nat. Rev. Genet. 2: 333–341. [DOI] [PubMed] [Google Scholar]
- Zhang, C., Brown, S.B., and Hara, T.J. 2001. Biochemical and physiological evidence that bile acids produced and released by lake char (Salvelinus namaycush) function as chemical signals. J. Comp. Physiol. B 171: 161–171. [DOI] [PubMed] [Google Scholar]
WEB SITE REFERENCES
- http://www.geneontology.org; the Gene Ontology Consortium (2001).
- http://web.uvic.ca/cbr/grasp; University of Victoria Centre for Biomedical Research.
- http://woodstock.ceh.uvic.ca/nkuipers/public_html/; Web interface for querying a database containing BLAST-identified candidate repeats in the GRASP EST database.
- http://www.genome.washington.edu/UWGC; University of Washington Genome Centre (PHRED version 0.990722.j; PHRAP version 0.990329).