

Abstract
Genome-wide pathway association studies provide novel insight into the biological mechanism underlying complex diseases. Current pathway association studies primarily focus on single important disease phenotype, which is sometimes insufficient to characterize the clinical manifestations of complex diseases. We present a multi-phenotypes pathway association study(MPPAS) approach using principle component analysis(PCA). In our approach, PCA is first applied to multiple correlated quantitative phenotypes for extracting a set of orthogonal phenotypic components. The extracted phenotypic components are then used for pathway association analysis instead of original quantitative phenotypes. Four statistics were proposed for PCA-based MPPAS in this study. Simulations using the real data from the HapMap project were conducted to evaluate the power and type I error rates of PCA-based MPPAS under various scenarios considering sample sizes, additive and interactive genetic effects. A real genome-wide association study data set of bone mineral density (BMD) at hip and spine were also analyzed by PCA-based MPPAS. Simulation studies illustrated the performance of PCA-based MPPAS for identifying the causal pathways underlying complex diseases. Genome-wide MPPAS of BMD detected associations between BMD and KENNY_CTNNB1_TARGETS_UP as well as LONGEVITYPATHWAY pathways in this study. We aim to provide a applicable MPPAS approach, which may help to gain deep understanding the potential biological mechanism of association results for complex diseases.
Introduction
Genome-wide association studies(GWAS) are successful for identifying common genetic variation underlying complex diseases in recent years [1]. In spite of the great power of GWAS, it may miss the causal genes with moderate genetic effects due to the stringent significant threshold of GWAS [2]. Moreover, the clinical manifestations of complex diseases usually arise from the interplay of multiple genetic and environmental risk factors through epigenetic and dynamic mechanism. Single gene can also participate in various biological processes. Identifying a small number of significant genes in GWAS may be insufficient to delineate the pathogenesis of complex diseases [3]. It is increasing recognized that a joint test of association between complex diseases and a group of functionally related genes, may provide more useful biological interpretations of association results [4], [5].
Motivated by the gene set enrichment analyses of microarray data [6], researchers proposed pathway association study approaches, which detected associations between complex diseases and a group of genes within a defined gene ontology or biological pathways [2]. Compared with SNP association studies, pathway association studies combine the association evidence of multiple functionally related genes, and potentially have greater power for revealing the biological mechanism underlying complex diseases [2]. For instance, a causal pathway with genes individually having weak genetic effects, but jointly contributing greatly to disease risks, is more likely to be detected at pathway level than at SNP level. Various pathway association study approaches were developed [7], [8], [9], [10], [11], [12], and successfully applied to genetic studies of complex diseases, such as osteoporosis and coronary heart disease [13], [14].
Current pathway association studies primarily focus on single important phenotype of complex diseases. A potential limitation of single phenotype pathway association studies is that single phenotype is sometimes insufficient to characterize complex diseases due to its complicated clinical manifestations. For example, obesity can be measured by body mass index, fat mass and proportions of fat mass in total body mass in practice. To address this issue, some researchers collected a set of disease-related phenotypes, and conducted pathway association tests of each phenotype ignoring the correlation among multiple disease phenotypes [15]. Given the difference of genetic structure underlying different disease phenotypes, it may be difficult to get replicated associations among different single phenotype pathway association studies. Additionally, multiple testing corrections were usually requested to ensure normal type I error rates in these studies. Because of the correlation among multiple disease phenotypes, multiple testing corrections (for example Bonferroni), may be too strict to miss the pathways with moderate association signals.
Recently, multivariable analyses approaches were applied to SNP association studies, which could simultaneously detect associations between SNPs and multiple disease phenotypes [16], [17]. The causal genes with moderate association signals in single phenotype SNP association studies are likely to present strong association signals in multiple phenotypes SNP association studies avoiding multiple testing corrections. It may be reasonable to consider that combining the genetic information of multiple disease phenotypes was potentially able to enhance the association signals of causal pathways, and therefore increased the power of pathway association studies of complex diseases. However, to the best of our knowledge, few multiple phenotypes pathway association study(MPPAS) approach is available now.
In this study, we present a flexible MPPAS approach using principle component analyses (PCA). In our approach, PCA is first applied to multiple correlated quantitative phenotypes for exacting a set of orthogonal phenotypic components. The extracted phenotypic components are then included into pathway association analyses instead of original disease phenotypes. Four statistics combining the association evidence of multiple genes within testing pathways, were proposed for assessing the overall association strength of the pathways with target traits. To illustrate the application of our method, extensive simulation studies using the real data from the HapMap project, were conducted to evaluate the power and type I error rates of PCA-based MPPAS under various scenarios, considering sample sizes, additive and interactive genetic effects. PCA-based MPPAS can be applied to GWAS data. A real GWAS data set of osteoporosis was analyzed by PCA-based MPPAS in this study.
Results
Simulations
The power of PCA-based MPPAS using 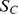 ,
, 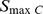 ,
, 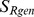 and
and 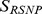 statistics, were evaluated by the simulation studies considering sample sizes, additive and interactive genetic effects. Figure 1A presents the power comparison results of
statistics, were evaluated by the simulation studies considering sample sizes, additive and interactive genetic effects. Figure 1A presents the power comparison results of 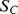 ,
, 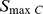 ,
, 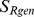 and
and 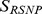 under various sample sizes. As expect, the power of PCA-based MPPAS trended to increase with increasing sample sizes in this study.
under various sample sizes. As expect, the power of PCA-based MPPAS trended to increase with increasing sample sizes in this study. 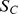 performed better than other statistics, and attained the highest power 92.07% with 2000 samples.
performed better than other statistics, and attained the highest power 92.07% with 2000 samples. 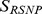 performed slightly worse than
performed slightly worse than 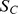 , but outperformed
, but outperformed 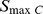 and
and 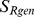 .
.
Figure 1. Power simulating results of PCA-based MPPAS using 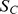 ,
, 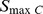 ,
, 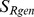 and
and 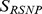 statistics under various sample sizes (A) and genetic effects(B&C).
statistics under various sample sizes (A) and genetic effects(B&C).

Figure 1B summarizes the power comparison results of 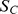 ,
, 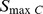 ,
, 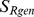 and
and 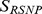 under various genetic effects. We observed significant impact of genetic effects on the performance of PCA-based MPPAS. The power of
under various genetic effects. We observed significant impact of genetic effects on the performance of PCA-based MPPAS. The power of 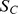 ,
, 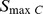 ,
, 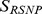 and
and 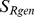 increased with increased genetic effects of causal pathways in this studies. Consistent with the simulation results of sample sizes,
increased with increased genetic effects of causal pathways in this studies. Consistent with the simulation results of sample sizes, 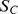 attained the highest power, following by
attained the highest power, following by 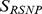 ,
, 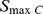 and
and 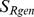 under various genetic effects investigated by this study. The simulation results of interactive genetic effects are presented in Figure 1C. We observed increased power of PCA-based MPPAS as the interactive genetic effects of causla pathways increasing.
under various genetic effects investigated by this study. The simulation results of interactive genetic effects are presented in Figure 1C. We observed increased power of PCA-based MPPAS as the interactive genetic effects of causla pathways increasing. 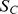 outperformed
outperformed 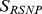 ,
, 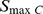 and
and 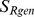 under various interactive genetic effects investigated by this study.
under various interactive genetic effects investigated by this study.
Figure 2 plot the type I error rates of PCA-based MPPAS using 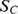 ,
, 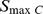 ,
, 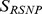 and
and 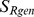 for testing association under various sample sizes. The type I error rates of
for testing association under various sample sizes. The type I error rates of 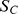 ,
, 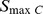 ,
, 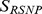 and
and 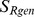 are not significant different from normal level (α = 0.05) under various simulating parameters investigated by this study.
are not significant different from normal level (α = 0.05) under various simulating parameters investigated by this study.
Figure 2. Type I error rate simulating results of PCA-based MPPAS using 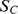 ,
, 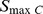 ,
, 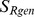 and
and 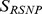 statistics under various sample sizes.
statistics under various sample sizes.

Genome-wide MPPAS of BMD
Figure 3 and figure 4 summarizes the genome-wide MPPAS results of BMD at spine and hip. With PCA-based MPPAS using 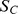 , we identified 2 pathways associated with BMD, including KENNY_CTNNB1_TARGETS_UP(p = 4.62×10−5) and LONGEVITYPATHWAY (p = 3.59×10−5). Detailed description of KENNY_CTNNB1_TARGETS_UP and LONGEVITYPATHWAY pathways can be found at GSEA Molecular Signatures Database (http://www.broadinstitute.org).
, we identified 2 pathways associated with BMD, including KENNY_CTNNB1_TARGETS_UP(p = 4.62×10−5) and LONGEVITYPATHWAY (p = 3.59×10−5). Detailed description of KENNY_CTNNB1_TARGETS_UP and LONGEVITYPATHWAY pathways can be found at GSEA Molecular Signatures Database (http://www.broadinstitute.org).
Figure 3. Q-Q plot of genome-wide MPPAS results of BMD at spine and hip.

Figure 4. Plot of genome-wide MPPAS results of BMD.

The significant pathways are highlighted in red. Significant pathways were defined by p values≤5.19×10−5 after Bonferroni correction(0.05/963).
Discussion
Pathway association studies are based on the fact that different causal genes of a complex disease are likely to be functionally related, for instance belonging to same biological pathways [18]. Therefore, examining the overall association strength of a pathway may provide improved power for pathogenetic studies of complex diseases, especially for the pathways with each gene having small phenotypic effects, but all genes jointly contributing greatly to disease risks. However, current pathway association studies primarily focus on single important phenotype of complex diseases, which may miss the pathways with weak genetic effects. In this study, we presented a simple PCA-based MPPAS approaches, which can simultaneously test multiple correlated quantitative phenotypes. Simulations were conducted to evaluate the performance of PCA-based MPPAS using 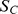 ,
, 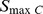 ,
, 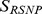 or
or 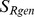 , and illustrated the application of PCA-based MPPAS for pathway association studies of complex diseases. We also observed significant impact of sample sizes and genetic effects on the performance of PCA-based MPPAS. PCA-based MPPAS using
, and illustrated the application of PCA-based MPPAS for pathway association studies of complex diseases. We also observed significant impact of sample sizes and genetic effects on the performance of PCA-based MPPAS. PCA-based MPPAS using 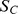 statistic appeared to outperform PCA-based MPPAS using
statistic appeared to outperform PCA-based MPPAS using 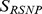 ,
, 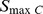 or
or 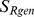 statistics in this study.
statistics in this study.
The PCA-based MPPAS have potentially two advantages over single phenotype pathway association studies. First, single phenotype is sometimes insufficient for characterizing complex diseases. In this situation, one strategy is to collect multiple disease phenotypes, and conduct single phenotype pathway association studies following by picking up the shared pathways with significant association signals among different studies. One issue of this approach is that the association finding of common causal pathways may be difficult to be replicated across various single phenotype pathway association studies, due to the difference of mechanism underlying different disease phenotypes. Second, multiple testing corrections are usually requested by this approach. Because of the stringent significant threshold after multiple testing corrections, the causal pathways with moderate but meaningful associations may be missed by single phenotype association studies. In contrast, MPPAS incorporate the genetic information of multiple correlated disease phenotypes into single test statistic. The causal pathways with moderate association signals in single phenotype pathway association studies, are likely to present strong association signals in MPPAS avoiding multiple testing corrections.
PCA-based MPPAS can be applied to GWAS data. A real GWAS data of BMD was used to assess the performance of PCA-based MPPAS in this study. We observed significant associations with BMD for KENNY_CTNNB1_TARGETS_UP and LONGEVITYPATHWAY pathways. Previous studies may provide some hints for understanding the associations detected by this study. For instance, previous studies found that the GHR, GH1, ATK1, IGF1 and IGF1R genes of LONGEVITYPATHWAY (containing 14 genes) contributed to the variation of BMD [19], [20], [21], [22]. KENNY_CTNNB1_TARGETS_UP consists of a set of genes being the target of Wnt pathway, which plays an important in the regulation of bone mass accrual [23], [24]. Further studies may be needed to validate the associations detected by this study.
A potential extension of our approach is that haplotype association studies may also be applied to PCA-based MPPAS instead of SNP association studies used by this study. It is known that haplotype association studies preserving the polymorphism and linkage disequilibrium information of multiple adjacent SNPs, was more powerful for detecting rare genetic variants than SNP association studies in some cases [25], [26]. For instance, the causal genes with multiple SNPs jointly having significant phenotypic effects, but individual SNP making a small contribution, is likely to be missed by SNP association studies. PCA-based MPPAS using haplotype as basic unit for association testing, may provide additional information for reveal the biological mechanism of complex diseases. Further studies may be worth to investigate the performance of MPPAS using haplotype as association testing unit.
Population stratification is a problem in population–based SNP association studies. SNP association studies conducted in an admixed population with subpopulations having different allele frequency distribution, may result in spurious association results [27]. Because most of current pathway association studies are based on the results of SNP association studies, the performance of pathway association studies may also suffer from the impact of population stratification. The best solution is to collect genetic unrelated subjects as study samples. Additionally, some statistical methods can also be applied to SNP association studies for correcting population stratification, such as Structure and Eigensoft [28], [29]. Linkage disequilibrium(LD) is another concern with pathway association studies, which may result in extensive spurious associations [7]. In this study, the significance levels of testing statistics of PCA-based MPPAS were evaluated by Monte Carlo permutations, which used the same individuals and maintained the same LD structure between original datasets and subsequent randomized datasets. PCA-based MPPAS do not depend on specific statistical assumption, for example the normality assumption of target traits. This approach minimizes the impact of LD on the performance of PCA-based MPPAS. The computational cost of PCA-based MPPAS is also acceptable in practice. For instance, our genome-wide PCA-based MPPAS of BMD needed about 12 days(1000 subjects and 50,000 replicates).
In summary, we present a flexible PCA-based MPPAS approach avoid multiple testing corrections. Simulations and real GWAS data analyses results illustrated the application of PCA-based MPPAS for identifying causal pathways underlying complex diseases. PCA-based MPPAS may help to overcome the limitations of single phenotype pathway association studies, and gain deep understanding the molecular mechanism of association results for complex diseases.
Materials and Methods
Ethics Statement
All studies were approved by the Institutional Review Boards of Xi'an Jiaotong University. Informed-consent documents were read and signed by all study participants.
General Model
Suppose a sample of n unrelated subjects and k quantitative phenotypes, which was determined by a biological pathway with m genotyped SNPs. Let 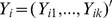 denote the k×1 phenotype vector, and
denote the k×1 phenotype vector, and 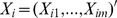 denote the m×1 genotype vector of subject i (i = 1,…,n). In this study, we coded
denote the m×1 genotype vector of subject i (i = 1,…,n). In this study, we coded 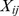 (j = 1,…,m) to be 0, 1 or 2, representing the copy number of minor allele of subject i at the jth SNP.
(j = 1,…,m) to be 0, 1 or 2, representing the copy number of minor allele of subject i at the jth SNP. 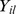 (l = 1,…,k) can be formulated as
(l = 1,…,k) can be formulated as
| (1) |
where 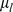 denotes the mean of the lth quantitative phenotypes.
denotes the mean of the lth quantitative phenotypes. 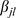 denotes the additive genetic effect of SNP j for the lth quantitative phenotypes.
denotes the additive genetic effect of SNP j for the lth quantitative phenotypes. 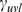 denotes the interactive genetic effect between SNP u and SNP v for the lth quantitative phenotypes.
denotes the interactive genetic effect between SNP u and SNP v for the lth quantitative phenotypes. 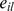 denotes the residual environmental effect of subject i for the lth quantitative phenotype.
denotes the residual environmental effect of subject i for the lth quantitative phenotype.
Extracting Phenotypic Components by PCA
Because different phenotypes may be measured using different units in practice, we first standardize the original quantitative phenotypes. Let 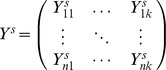 denotes the matrix of k standardized quantitative phenotypes for n subjects. The matrix element
denotes the matrix of k standardized quantitative phenotypes for n subjects. The matrix element 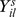 can be computed by
can be computed by
| (2) |
where 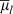 and
and 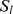 denote the mean and standard deviation of the lth quantitative phenotype, respectively.
denote the mean and standard deviation of the lth quantitative phenotype, respectively.
PCA(implemented by R software, http://www.r-project.org/) is then applied to 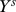 for extracting k orthogonal phenotypic components. Following standard PCA precedure, let
for extracting k orthogonal phenotypic components. Following standard PCA precedure, let 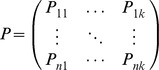 denotes the matrix of k extracted phenotypic components for n subjects. The matrix element
denotes the matrix of k extracted phenotypic components for n subjects. The matrix element 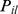 is calculated by
is calculated by
| (3) |
where 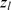 is calculated by PCA and denotes the eigenvector of the lth phenotypic components.
is calculated by PCA and denotes the eigenvector of the lth phenotypic components. 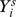 denotes the standardized phenotypic vector of subject i.
denotes the standardized phenotypic vector of subject i.
Pathway Association Testing Statistics
The phenotypic components extracted by PCA are included into pathway association analysis instead of original quantitative phenotypes. Suppose a pathway with r genes and m genotyped SNPs. For a given gene within the pathway, we first detect associations between each SNP of the gene and each phenotypic component. For each gene, the largest statistic of all SNPs mapped to the gene is assigned to the gene as the statistic of the gene [7]. Let 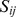 (i = 1,…,k and j = 1,…r) denotes the largest statstic of gene j for the ith phenotypic component. Let
(i = 1,…,k and j = 1,…r) denotes the largest statstic of gene j for the ith phenotypic component. Let 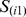 ≥
≥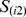 ≥…≥
≥…≥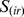 denote the ordered statstics of the pathway for the ith phenotypic component. Based on the idea that a pathway with more genes associated with target traits, is more likely to be disease-causing pathway, we present four statistics to evaluate the overall association strength of a pathway with target traits. The first one takes a linear combination of statistics of all genes within the pathway, defined by
denote the ordered statstics of the pathway for the ith phenotypic component. Based on the idea that a pathway with more genes associated with target traits, is more likely to be disease-causing pathway, we present four statistics to evaluate the overall association strength of a pathway with target traits. The first one takes a linear combination of statistics of all genes within the pathway, defined by
| (4) |
where 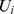 is computed by PCA and denotes the proportion of phenotypic variance explained by the ith phenotypic component. The phenotypic information harboring by different components are different, and can be measured by the explained proportions of phenotypic variation in PCA.
is computed by PCA and denotes the proportion of phenotypic variance explained by the ith phenotypic component. The phenotypic information harboring by different components are different, and can be measured by the explained proportions of phenotypic variation in PCA. 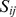 are weighted by the explained proportion of phenotypic variance, which gives higher weight to the phenotypic components explained larger part of variance of original k quantitative phenotypes.
are weighted by the explained proportion of phenotypic variance, which gives higher weight to the phenotypic components explained larger part of variance of original k quantitative phenotypes.
Because of combining the association evidence of all genes within the pathway, 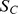 may be susceptive to the impact of pathway sizes. Consider an extreme case that we have a vary large pathway with only one significant gene. In this situation, the true association signal of causal gene may be masked by the nosie of other genes within the pathway. Therefore, we proposed the second statistic
may be susceptive to the impact of pathway sizes. Consider an extreme case that we have a vary large pathway with only one significant gene. In this situation, the true association signal of causal gene may be masked by the nosie of other genes within the pathway. Therefore, we proposed the second statistic 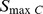 , which taked the maximum value of averaged statistics within the pathway.
, which taked the maximum value of averaged statistics within the pathway. 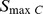 is defined by
is defined by
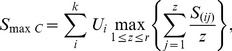 |
(5) |
where 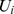 is defined in equation 4.
is defined in equation 4.
Recently, SNP raio tests were proposed for pathway association studies [10]. This approach compared the ratio of significant and no-significant SNPs within a pathway to the distribution of ratios derived from GWAS results of randomized phenotypes. The pathways with larger part of genes or SNPs associated with disease phenotypes is more likely to contribute to disease risks. In this study, we extended the ratio tests to PCA-based MPPAS, and considered two ratio testing approaches, SNP ratio tests and gene ratio tests. Let 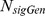 and
and 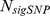 denote the numbers of significant genes and SNPs within testing pathways. The pathway ratio testing statistics can be expressed as
denote the numbers of significant genes and SNPs within testing pathways. The pathway ratio testing statistics can be expressed as
| (6) |
where r and m denotes respectively the numbers of genes and SNPs within the pathway.
For statistical tests, a permutation precedure was implemented to evaluate the significance levels of 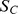 ,
, 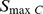 ,
, 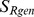 and
and 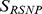 in this study. During each permutation, the sample labels were randomly assigned to individuals following by computation of
in this study. During each permutation, the sample labels were randomly assigned to individuals following by computation of 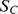 ,
, 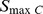 ,
, 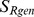 and
and 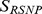 , respectively. 2,000 Monte Carlo permutations were conducted to obtain the empirical distributions of
, respectively. 2,000 Monte Carlo permutations were conducted to obtain the empirical distributions of 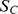 ,
, 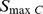 ,
, 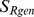 and
and 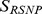 . The significance levels of
. The significance levels of 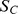 ,
, 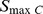 ,
, 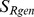 and
and 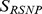 were finally evaluated according to the obtained empirical distributions.
were finally evaluated according to the obtained empirical distributions.
Simulations
Genotype simulation
HAPGEN program was used here for genotype simulations [30], [31]. Based on known haplotype data, HAPGEN can simulate whole-genome genotype data by implementing a hidden Markov model [30], [31]. Specific for this study, the genome-wide haplotype data, minor allele frequencies (MAF) and D′ of Caucasian were downloaded from the HapMap website(http://hapmap.ncbi.nlm.nih.gov/downloads/index.html.en). HAPGEN was then used to simulate genome-wide genotype data of Caucasian with default running parameters recommended by HAPGEN developers.
333 pathways or gene ontology with sizes ranging from 20 to 40, were collected from four public pathway databases, including BioCarta(http://www.biocarta.com), KEGG(http://www.genome.jp/kegg/), Ambion GeneAssist Pathway Atlas(http://www5.appliedbiosystems.com/tools/pathway/), and GSEA Molecular Signatures Database(http://www.broadinstitute.org). The obtained pathway-gene annotation file was used to link pathway and gene information in following pathway simulation studies.
Phenotype simulations
Genetic epistatic model was applied here for quantitative phenotype simulations. Suppose a complex disease underlying by a biological pathway, was mesured by three correlated quantitaitve phenotypes, Q1, Q2 and Q3. During each phenotype simulation, we first randomly selected a pathway as the causal pathway. Three SNPs (SNP1, SNP2 and SNP3) were then randomly selected from different genes of the causal pathway as the causal loci of Q1. The same precedure was also conducted for Q2 and Q3, respectively. Let 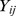 denotes the jth quantitative phenotype value of subject i, defined by
denotes the jth quantitative phenotype value of subject i, defined by
| (7) |
where 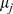 denotes the mean of the jth quantitative phenotype.
denotes the mean of the jth quantitative phenotype. 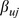 denotes the additive genetic effect of SNP u for the jth quantitative phenotype.
denotes the additive genetic effect of SNP u for the jth quantitative phenotype. 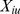 (
(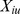 = 0, 1 or 2) denotes the copy number of minor allele of subject i at SNP u.
= 0, 1 or 2) denotes the copy number of minor allele of subject i at SNP u. 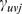 denotes the interactive genetic effect between SNP u and SNP v for the jth quantitative phenotype. Without loss of generality, we assume that there was an interactive genetic effect between SNP1 and SNP3 for Q1 in this study.
denotes the interactive genetic effect between SNP u and SNP v for the jth quantitative phenotype. Without loss of generality, we assume that there was an interactive genetic effect between SNP1 and SNP3 for Q1 in this study. 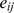 denotes the residual environmental effect of subject i for the jth quantitative phenotype, and follow a zero-mean normal distribution with variance
denotes the residual environmental effect of subject i for the jth quantitative phenotype, and follow a zero-mean normal distribution with variance 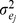 .
.
Data analysis
The simulated genotype and phenotype data were simultaneously analyzed by PCA-based MPPAS using 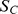 ,
, 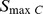 ,
, 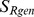 and
and 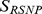 , respectively. Sample sizes, additive and interactive genetic effects were controlled to simulate various scenarios of pathway association studies in practice. Detailed parameter designs are presented in Table 1. 1,000 replicates were conducted for each parameter setting. Power and type I error rates were calculated respectively as the proportions of positive association results (p values≤0.05) obtained from the pathways simulated with and without genetic effects in 1,000 replicates. All our data simulations and analyses were implemented with statistical package R [32], except for SNP association tests implemented by PLINK [33].
, respectively. Sample sizes, additive and interactive genetic effects were controlled to simulate various scenarios of pathway association studies in practice. Detailed parameter designs are presented in Table 1. 1,000 replicates were conducted for each parameter setting. Power and type I error rates were calculated respectively as the proportions of positive association results (p values≤0.05) obtained from the pathways simulated with and without genetic effects in 1,000 replicates. All our data simulations and analyses were implemented with statistical package R [32], except for SNP association tests implemented by PLINK [33].
Table 1. Parameter configurations in the simulation studies.
| Sample size | Genetic effecta | ||||
| SNP1 | SNP2 | SNP3 | SNP1×SNP3 | ||
| Simulation 1 | 600 | 3.00% | 1.50% | 1.00% | 1.50% |
| 800 | 3.00% | 1.50% | 1.00% | 1.50% | |
| 1000 | 3.00% | 1.50% | 1.00% | 1.50% | |
| 2000 | 3.00% | 1.50% | 1.00% | 1.50% | |
| Simulation2 | 1000 | 1.00% | 0.50% | 0.25% | 0.50% |
| 1000 | 2.00% | 1.00% | 0.50% | 1.00% | |
| 1000 | 3.00% | 1.50% | 1.00% | 1.50% | |
| 1000 | 3.50% | 2.00% | 1.50% | 2.00% | |
| Simulation3 | 1000 | 2.00% | 1.00% | 0.50% | 1.00% |
| 1000 | 2.00% | 1.00% | 0.50% | 1.50% | |
| 1000 | 2.00% | 1.00% | 0.50% | 2.00% | |
| 1000 | 2.00% | 1.00% | 0.50% | 3.00% | |
denote the phenotypic variance explained by the additive genetic effects of SNP1, SNP2 and SNP3 as well as an interactive effect between SNP1 and SNP3, respectively.
333 pathways with sizes varying from 20 to 40, were collected from public pathway databases and used for pathway simulations.
Application to real GWAS Data of BMD
PCA-based MPPAS using 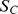 was applied to a real GWAS data consisting of 1,000 unrelated US whites. The sample characteristics and experimental design have been detailed in previous study [34]. Briefly, Affymetrix 500 k SNP arrays were used to genotype a total of 500,568 SNPs. After quality control, 312,172 SNPs covering 14,585 genes were retained for MPPAS of BMD in this study. Areal BMD of spine and hip were measured by dual-energy X-ray absorptiometry (DXA) with Hologic QDR 4500W densitometers (Hologic, Inc., Bedford, MA, USA). Age and sex were used to adjust the raw spine and hip BMD values as covariates for subsequent analyses. The adjusted BMD data were normally distributed. 963 pathways or gene ontology with sizes varying from 5 to 168, were derived from public pathway databases, including BioCarta, KEGG, Ambion GeneAssist Pathway Atlas, and GSEA Molecular Signatures Database. PCA-based MPPAS using
was applied to a real GWAS data consisting of 1,000 unrelated US whites. The sample characteristics and experimental design have been detailed in previous study [34]. Briefly, Affymetrix 500 k SNP arrays were used to genotype a total of 500,568 SNPs. After quality control, 312,172 SNPs covering 14,585 genes were retained for MPPAS of BMD in this study. Areal BMD of spine and hip were measured by dual-energy X-ray absorptiometry (DXA) with Hologic QDR 4500W densitometers (Hologic, Inc., Bedford, MA, USA). Age and sex were used to adjust the raw spine and hip BMD values as covariates for subsequent analyses. The adjusted BMD data were normally distributed. 963 pathways or gene ontology with sizes varying from 5 to 168, were derived from public pathway databases, including BioCarta, KEGG, Ambion GeneAssist Pathway Atlas, and GSEA Molecular Signatures Database. PCA-based MPPAS using 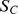 was used to detect association between each pathway and BMD. 50,000 replicates were conducted to evaluate the empirical p values of
was used to detect association between each pathway and BMD. 50,000 replicates were conducted to evaluate the empirical p values of 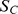 for each gene set investigated in this study. Significant pathway were defined by p values≤5.19×10−5 after Bonferroni correction(0.05/963).
for each gene set investigated in this study. Significant pathway were defined by p values≤5.19×10−5 after Bonferroni correction(0.05/963).
Funding Statement
The study was supported by National Natural Scientific Foundation of China (81102086), the Doctoral fund of Ministry of Education of China (20110201120057) and the Fundamental Research Funds for the Central Universities of China.The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Visscher PM, Brown MA, McCarthy MI, Yang J (2012) Five years of GWAS discovery. Am J Hum Genet 90: 7–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wang K, Li M, Hakonarson H (2010) Analysing biological pathways in genome-wide association studies. Nat Rev Genet 11: 843–854. [DOI] [PubMed] [Google Scholar]
- 3. Barabasi AL (2007) Network medicine–from obesity to the “diseasome”. N Engl J Med 357: 404–407. [DOI] [PubMed] [Google Scholar]
- 4. Askland K, Read C, Moore J (2009) Pathways-based analyses of whole-genome association study data in bipolar disorder reveal genes mediating ion channel activity and synaptic neurotransmission. Hum Genet 125: 63–79. [DOI] [PubMed] [Google Scholar]
- 5. Lesnick TG, Papapetropoulos S, Mash DC, Ffrench-Mullen J, Shehadeh L, et al. (2007) A genomic pathway approach to a complex disease: axon guidance and Parkinson disease. PLoS Genet 3: e98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, et al. (2005) Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102: 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wang K, Li M, Bucan M (2007) Pathway-based approaches for analysis of genomewide association studies. Am J Hum Genet 81: 1278–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Luo L, Peng G, Zhu Y, Dong H, Amos CI, et al. (2010) Genome-wide gene and pathway analysis. Eur J Hum Genet 18: 1045–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhang K, Cui S, Chang S, Zhang L, Wang J (2010) i-GSEA4GWAS: a web server for identification of pathways/gene sets associated with traits by applying an improved gene set enrichment analysis to genome-wide association study. Nucleic Acids Res 38: W90–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. O'Dushlaine C, Kenny E, Heron EA, Segurado R, Gill M, et al. (2009) The SNP ratio test: pathway analysis of genome-wide association datasets. Bioinformatics 25: 2762–2763. [DOI] [PubMed] [Google Scholar]
- 11. Yu K, Li Q, Bergen AW, Pfeiffer RM, Rosenberg PS, et al. (2009) Pathway analysis by adaptive combination of P-values. Genet Epidemiol 33: 700–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chen L, Zhang L, Zhao Y, Xu L, Shang Y, et al. (2009) Prioritizing risk pathways: a novel association approach to searching for disease pathways fusing SNPs and pathways. Bioinformatics 25: 237–242. [DOI] [PubMed] [Google Scholar]
- 13. Zhang L, Guo YF, Liu YZ, Liu YJ, Xiong DH, et al. (2010) Pathway-based genome-wide association analysis identified the importance of regulation-of-autophagy pathway for ultradistal radius BMD. J Bone Miner Res 25: 1572–1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. de Las Fuentes L, Yang W, Davila-Roman VG, Charles Gu C (2012) Pathway-based genome-wide association analysis of coronary heart disease identifies biologically important gene sets. Eur J Hum Genet [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Liu YJ, Guo YF, Zhang LS, Pei YF, Yu N, et al. (2010) Biological pathway-based genome-wide association analysis identified the vasoactive intestinal peptide (VIP) pathway important for obesity. Obesity (Silver Spring) 18: 2339–2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mei H, Chen W, Dellinger A, He J, Wang M, et al. (2010) Principal-component-based multivariate regression for genetic association studies of metabolic syndrome components. BMC Genet 11: 100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yang Q, Wu H, Guo CY, Fox CS (2010) Analyze multivariate phenotypes in genetic association studies by combining univariate association tests. Genet Epidemiol 34: 444–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Holmans P, Green EK, Pahwa JS, Ferreira MA, Purcell SM, et al. (2009) Gene ontology analysis of GWA study data sets provides insights into the biology of bipolar disorder. Am J Hum Genet 85: 13–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dennison EM, Syddall HE, Rodriguez S, Voropanov A, Day IN, et al. (2004) Polymorphism in the growth hormone gene, weight in infancy, and adult bone mass. J Clin Endocrinol Metab 89: 4898–4903. [DOI] [PubMed] [Google Scholar]
- 20. Ulici V, Hoenselaar KD, Agoston H, McErlain DD, Umoh J, et al. (2009) The role of Akt1 in terminal stages of endochondral bone formation: angiogenesis and ossification. Bone 45: 1133–1145. [DOI] [PubMed] [Google Scholar]
- 21. Dennison EM, Syddall HE, Jameson KA, Sayer AA, Gaunt TR, et al. (2009) A study of relationships between single nucleotide polymorphisms from the growth hormone-insulin-like growth factor axis and bone mass: the Hertfordshire cohort study. J Rheumatol 36: 1520–1526. [DOI] [PubMed] [Google Scholar]
- 22. Lakatos PL, Bajnok E, Tornai I, Folhoffer A, Horvath A, et al. (2004) Insulin-like growth factor I gene microsatellite repeat, collagen type Ialpha1 gene Sp1 polymorphism, and bone disease in primary biliary cirrhosis. Eur J Gastroenterol Hepatol 16: 753–759. [DOI] [PubMed] [Google Scholar]
- 23. Rawadi G, Roman-Roman S (2005) Wnt signalling pathway: a new target for the treatment of osteoporosis. Expert Opin Ther Targets 9: 1063–1077. [DOI] [PubMed] [Google Scholar]
- 24. Sims AM, Shephard N, Carter K, Doan T, Dowling A, et al. (2008) Genetic analyses in a sample of individuals with high or low BMD shows association with multiple Wnt pathway genes. J Bone Miner Res 23: 499–506. [DOI] [PubMed] [Google Scholar]
- 25. Stephens JC, Schneider JA, Tanguay DA, Choi J, Acharya T, et al. (2001) Haplotype variation and linkage disequilibrium in 313 human genes. Science 293: 489–493. [DOI] [PubMed] [Google Scholar]
- 26. Schaid DJ, Rowland CM, Tines DE, Jacobson RM, Poland GA (2002) Score tests for association between traits and haplotypes when linkage phase is ambiguous. Am J Hum Genet 70: 425–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Deng HW (2001) Population admixture may appear to mask, change or reverse genetic effects of genes underlying complex traits. Genetics 159: 1319–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pritchard JK, Stephens M, Rosenberg NA, Donnelly P (2000) Association mapping in structured populations. AmJHumGenet 67: 170–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, et al. (2006) Principal components analysis corrects for stratification in genome-wide association studies. NatGenet 38: 904–909. [DOI] [PubMed] [Google Scholar]
- 30. Spencer CC, Su Z, Donnelly P, Marchini J (2009) Designing genome-wide association studies: sample size, power, imputation, and the choice of genotyping chip. PLoS Genet 5: e1000477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Marchini J, Howie B, Myers S, McVean G, Donnelly P (2007) A new multipoint method for genome-wide association studies by imputation of genotypes. Nat Genet 39: 906–913. [DOI] [PubMed] [Google Scholar]
- 32.R-Development-Core-Team (2007) R: A language and environment for statistical computing. Vienna, Austria.
- 33. Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, et al. (2007) PLINK: a tool set for whole-genome association and population-based linkage analyses. AmJHumGenet 81: 559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Liu XG, Tan LJ, Lei SF, Liu YJ, Shen H, et al. (2009) Genome-wide association and replication studies identified TRHR as an important gene for lean body mass. Am J Hum Genet 84: 418–423. [DOI] [PMC free article] [PubMed] [Google Scholar]