

The search for the causitive agent for Paederis dermatitis, an inflammatory condition that results from contact with beetles of the Paederis family, resulted in the isolation of pederin (1).[1] Pederin subsequently inspired a substantial wave of investigation that led to the determination of its correct structure,[2] the observation of its potent cytotoxicity,[3] the postulation of protein synthesis inhibition as its likely mode of biological activity,[4] the identification of the 60S subunit of the ribosome as its potential biological target,[5] and the establishment of bacterial symbionts as its true biogenetic source.[6] Pederin’s interesting biological activity and challenging structural features have spawned significant synthetic efforts, resulting in several total, formal, and analog syntheses.[7] A noteworthy advance in pederin accessibility was recently disclosed by Rawal,[7f] who reported that pederin could be prepared through a 12 step (longest linear sequence) route. In accord with our interest in the pederin family of molecules,[8] we report our total synthesis of 1. In addition to the brief linear sequence, this approach highlights the utility of a late stage multicomponent construction of the N-acylaminal structure that allows for the efficient construction of analogs with structural variation in each of three distinct subunits.
We envisioned pederin to arise (Scheme 1) from the union of a pederic acid derivative (2), a tetrahydropyranyl nitrile (3), and MeOH through our recently reported[9] multicomponent amide synthesis. This late stage construction of the N-acyl aminal unit is well-suited for analog synthesis through variations on the pederic acid unit, the nitrile, and the alcohol. The nitrile can be prepared from lactol 4, which can be accessed from keto alcohol 5, a known compound[8c] that can be prepared in multigram quantities and with high enantiomeric purity in three steps from isobutyraldehyde.
Scheme 1.
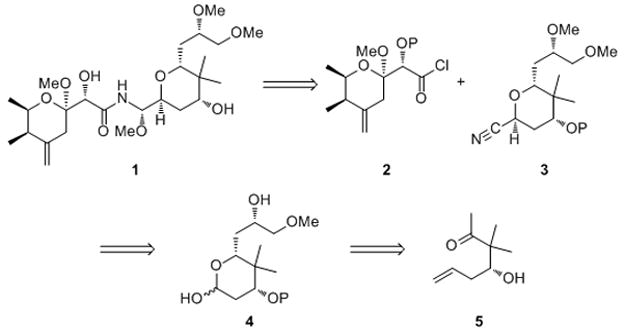
Retrosynthetic analysis of pederin.
Silylation of 5 proceeded through standard conditions to yield ether 6. We postulated that the remaining carbons of the right-hand fragment could be introduced through an aldol reaction between an enolate of 6 and methoxyacetaldehyde. High levels of 1,5-anti-diastereocontrol have been reported for aldol reactions between aldehydes and dialkylboron enolates of β-alkoxy ketones, but β-silyloxy enolates show poor levels of control.[10] Thus we employed Paterson’s pinene-derived boron enolate aldol strategy[11] to effect the conversion of 6 to 7 in 88% yield as a 15:1 mixture of diastereomers upon oxidative cleavage of the borinate intermediate. This degree of stereocontrol was gratifying in consideration of the modest levels of selectivity that are normally observed for aldol reactions with (Ipc)2B enolates of methyl ketones. Stereoselective reduction of 7 with Et2BOMe and NaBH4 yielded 8 efficiently. While the yields and stereocontrol for the aldol and reduction reactions were satisfactory, diol 8 could be accessed directly as a single stereoisomer in 80% yield by treating the borinate intermediate from the aldol reaction with LiBH4[12] prior to oxidative cleavage. This one pot sequence also facilitated product separation from the pinene-derived by-products. Ozonlytic cleavage of the terminal alkene led to the formation of lactol 9.
We proposed that the lactol could be ionized by BiBr3 selectively in the presence of the unprotected secondary alcohol and the resulting oxocarbenium ion would react with TMSCN[13] to afford the desired nitrile by analogy to a similar transformation in our synthesis of leucascandrolide A.[14] This strategy would allow for the introduction of the cyano group into the structure while avoiding steps to incorporate leaving or protecting groups. Exposing 9 to TMSCN and BiBr3 followed by an acidic work-up provided 10, though in only a 35% isolated yield. Further examination revealed that lactol silylation was competitive with ionization, and that the lactol silyl ether was unreactive toward ionization by BiBr3. This problem was solved by adding BF3•OEt2 to the mixture following the initial reaction with BiBr3 and TMSCN. The stronger Lewis acid promoted ionization of the lactol silyl ether to yield 10 in 63% isolated yield as a single stereoisomer. Minimal product formation was observed when BF3•OEt2 was used in the absence of BiBr3. Methylation of 10 was achieved with MeOTf and 2,6-di-tert-butylpyridine to provide 11, the nitrile component for the multicomponent acyl aminal formation, in 86% yield.
The key fragment coupling reaction (Scheme 3) proceeded through the hydrozirconation of 11 followed by the addition of acid chloride 12, prepared through our variant[8d] of Nakata’s route,[15] to provide acylimine intermediate 13. The acylation needed to be conducted at low temperature because, in contrast to studies on simpler systems, tautomerization of 13 was observed at room temperature. Analogs of 12 that employed a silyl ether to protect the C7 hydroxyl group were unreactive acylating agents as a result of steric hindrance around the carboxyl group. The addition of MeOH and Mg(ClO4)2 to 13 led to the formation of acyl aminal 14 as a single stereoisomer in 43% yield. We attribute the high stereocontrol[16] to a reactive conformation in which Mg(ClO4)2 coordinates the tetrahydropyranyl oxygen and the acylimine nitrogen, forcing the MeOH to approach from the less congested face. The total synthesis of pederin was completed through an efficient one pot cleavage of both protecting groups (Bu4NF/THF, followed by LiOH/MeOH/H2O).[7f] All spectral data for the final compound matched reported[7] values. The Supporting Information contains tabulated comparisons of 1H NMR, 13C NMR, and optical rotation data.
Scheme 3.
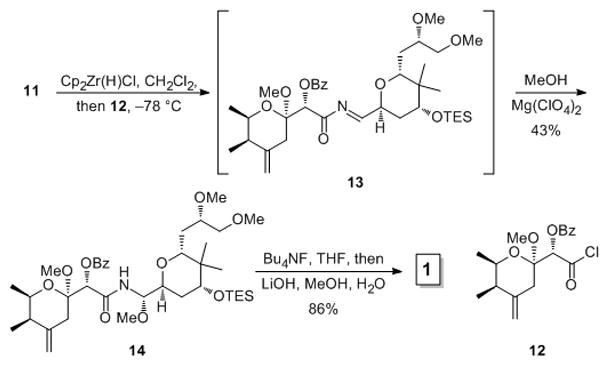
Completion of the total synthesis.
The brevity of the sequence and the opportunities that the late stage multicomponent reaction presents for diversification led us to explore the preparation of a number of pederin analogs. The syntheses of these compounds are shown in Scheme 4.[17] Analogs 15-18 were designed to study the importance of the alkoxy component at the C10 position on biological activity. The alkoxy groups of 15 and 16 are sterically similar, but the trifluoroethoxy group can ionize more readily, as evidenced by the isolation of enamide 19 as a significant byproduct in the preparation of 16. The alkene geometry in compound 19 was assigned based on a NOESY cross peak between the vinyl hydrogen of the enamide and a C12 hydrogen in the tetrahydropyran ring. This lability could be useful if biological activity requires that the N-acylaminal group serve as a latent acyliminium ion. This mechanism has been suggested as a possible source of activity for the mycalamide family of molecules.[18] Benzylic ethers 17 and 18 were designed to test the steric tolerance at this site while also being able to serve as precursors to N-acyl hemiaminal analogs. The C13 methyl ether analog was prepared by subjecting 20[19] to the multicomponent reaction and deprotection sequence to form 21. The corresponding hydroxyl site is methylated in the structurally related and highly cytotoxic mycalamide family of natural products,[20] and incorporation of the methyl ether could remove one step from the sequence. The exocyclic alkene at the C4 site is a source of chemical instability but was shown[21] to be unnecessary for biological activity in mycalamide analogs. Therefore we synthesized acid chloride 22[19] and employed it in the multicomponent reaction and deprotection sequence to yield 23. This structural alteration also shortened the synthesis of the left fragment by two steps. Hydrogenolytic benzyl ether cleavage and concomitant alkene reduction of 17 provided a product that was unstable toward chromatographic purification but the formation of N-acyl hemiaminal 24 was consistent with 1H NMR analysis, and the presence of [M+Na]+ and [M−H]− ions from LCMS experiments using cation and anion detection, respectively. Attempts to cleave the dimethoxybenzyl ether of 20 with DDQ[22] resulted in decomposition. The ability to prepare N-acyl hemiaminal analogs provides support for their potential role as biogenetic precursors of pederin that could be converted to the natural product through a methylase enzyme.[23] Amide 25, a minor by-product in the multicomponent reactions that arises from the reaction of acylimine 13 with excess Schwartz reagent, was converted to 26. While indirect evidence has suggested that the C10 methoxy group is important for the biological activity of these molecules,[24] this hypothesis has never been tested. Moreover this structure can be used to determine whether it could serve as a biogenetic precursor to pederin through amide oxidation.[25]
Scheme 4.
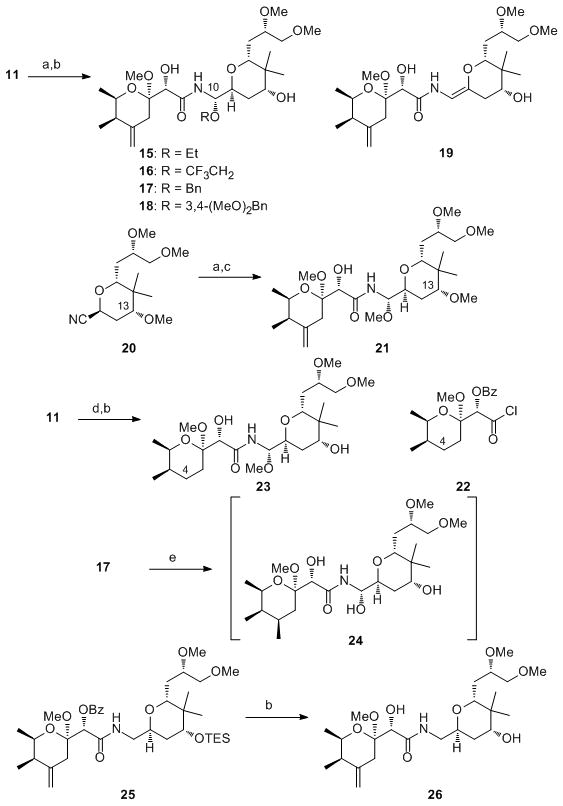
Analog synthesis. Reagents and conditions: a) Cp2Zr(H)Cl, CH2Cl2, then 12, −78 °C, then Mg(ClO4)2, ROH, −78 °C to −40 °C, 40% for EtOH (dr = 15:1), 21% for CF3CH2OH, 29% for DMBOH (dr = 4:1), 30% from 20; b) Bu4NF, THF, 0 °C, then LiOH, MeOH, H2O, 78% for 15, 64% for 16 (+ 29% 19), 21% for 17 (two steps), 75% for 18, 82% for 23, 80% for 26; c) LiOH, MeOH, 80%; d) Cp2Zr(H)Cl, CH2Cl2, then 22, −78 °C, then Mg(ClO4)2, then MeOH, 35%; e) H2, Pd/C, EtOH. DMBOH = 3,4-dimethoxybenzyl alcohol.
We have reported a ten step (longest linear sequence) synthesis of pederin. This route is the shortest sequence to pederin that has been reported, and features a late stage multicomponent reaction to unite the subunits and form the challenging acylaminal group. Application of the method to analog synthesis yielded compounds that will be used to test hypotheses regarding the biological activity and biosynthesis of this intriguing natural product. While the multicomponent reactions did not proceed in excellent yields, the use of this process as a strategy level step provided useful amounts of products, a significant increase in molecular complexity, and unprecedented access to analogs. The results from biological assays will be reported in a full account of this work.
Scheme 2.
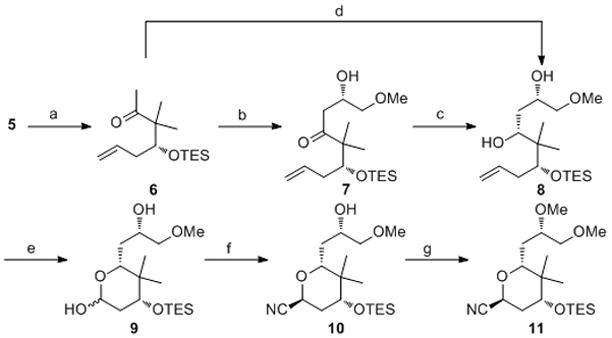
Synthesis of the nitrile component. Reagents and conditions: a) TESCl, imidazole, CH2Cl2, 100%; b) (+)-DIP-Cl, Et3N, MeOCH2CHO, Et2O, −78 °C, 88%, dr = 15:1; c) NaBH4, Et2BOMe, MeOH, THF, −40 °C, 95%; d) (+)-DIP-Cl, Et3N, Et2O, −78 °C, then LiBH4, −40 °C, 80%; e) O3, CH2Cl2, −78 °C, then Ph3P, 95%; f) TMSCN, BiBr3, CH3CN, 0 °C, then BF3•OEt2, −40 °C, 63%; g) MeOTf, 2,6-tBu2Py, CH2Cl2, 86%. DIP-Cl = B-chlorodiisopinocam-phenylborane, TMSCN = trimethylsilyl cyanide, MeOTf = methyl trifluoromethanesulfonate, 2,6-tBu2Py = 2,6-di-tert-butylpyridine.
Footnotes
We thank the National Science Foundation for generous support of this work through grant CHE-0848299 and the the NIH for the 700 MHz NMR at the University of Pittsburgh (S10RR023404).
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
References
- 1.Pavan M, Bo G. Phys Comp Oec. 1953;3:307. [Google Scholar]
- 2.a) Cardani C, Ghiringhelli D, Mondelli R, Quilico A. Tetrahedron Lett. 1965;8:2537. [Google Scholar]; b) Furusaki A, Watanabé T, Matsumoto T, Yanagiya M. Tetrahedron Lett. 1968;9:6301. [Google Scholar]
- 3.a) Soldati M. Experimentia. 1966;22:176. doi: 10.1007/BF01897720. [DOI] [PubMed] [Google Scholar]; b) Richter A, Kocienski P, Raubo P, Davies DE. Anticancer Drug Des. 1997;12:217. [PubMed] [Google Scholar]
- 4.Carrasco L, Fernandez-Puentes C, Vasquez D. Mol Cell Biochem. 1976;10:97. doi: 10.1007/BF01742203. [DOI] [PubMed] [Google Scholar]
- 5.Nishimura S, Matsunaga S, Yoshida M, Hirota H, Yokoyama S, Fusetani N. Bioorg Med Chem Lett. 2005;13:449. doi: 10.1016/j.bmc.2004.10.012. [DOI] [PubMed] [Google Scholar]
- 6.a) Piel J. Proc Natl Acad Sci USA. 2002;99:14002. doi: 10.1073/pnas.222481399. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Piel J, Butzke D, Fusetani N, Hui DQ, Platzer M, Wen GP, Matsunaga S. J Nat Prod. 2005;68:472. doi: 10.1021/np049612d. [DOI] [PubMed] [Google Scholar]
- 7.a) Matsuda F, Tomiyoshi N, Yanagiya M, Matsumoto T. Tetrahedron. 1988;44:7063. [Google Scholar]; b) Hoffmann RW, Schlapbach A. Tetrahedron. 1992;48:1959. [Google Scholar]; Kocienski P, Narquizian R, Raubo P, Smith C, Farrugia LJ, Muir K, Boyle FT. J Chem Soc-Perkin Trans. 2000;1:2357. [Google Scholar]; d) Takemura T, Nishii Y, Takahashi S, Kobayashi J, Nakata T. Tetrahedron. 2002;58:6359. [Google Scholar]; e) Jiang X, Williams N, De Brabander JK. Org Lett. 2007;9:227. doi: 10.1021/ol062656o. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Jewett JC, Rawal VH. Angew Chem. 2007;119:6622. doi: 10.1002/anie.200701677. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2007;46:6502. [Google Scholar]; g) Liu D, Xue J, Xie Z, Wei L, Zhang X, Li Y. Synlett. 2008:1526. [Google Scholar]
- 8.a) Rech JC, Floreancig PE. Org Lett. 2003;5:1495. doi: 10.1021/ol034273l. [DOI] [PubMed] [Google Scholar]; b) Green ME, Rech JC, Floreancig PE. Org Lett. 2005;7:4117. doi: 10.1021/ol051396s. [DOI] [PubMed] [Google Scholar]; c) Rech JC, Floreancig PE. Org Lett. 2005;7:5175. doi: 10.1021/ol0520267. [DOI] [PubMed] [Google Scholar]; d) Green ME, Rech JC, Floreancig PE. Angew Chem. 2008;120:7427. doi: 10.1002/anie.200802548. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2008;47:7317. [Google Scholar]
- 9.a) Wan S, Green ME, Park JH, Floreancig PE. Org Lett. 2007;9:5385. doi: 10.1021/ol702184n. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) DeBenedetto MV, Green ME, Wan S, Park JH. Org Lett. 2009;11:835. doi: 10.1021/ol802764j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.a) Paterson I, Gibson KR, Oballa RM. Tetrahedron Lett. 1996;37:8585. [Google Scholar]; b) Evans DA, Coleman PJ, Côte B. J Org Chem. 1997;62:788. [Google Scholar]; c) Evans DA, Côte B, Coleman PJ, Connell BT. J Am Chem Soc. 2003;125:10893. doi: 10.1021/ja027640h. [DOI] [PubMed] [Google Scholar]
- 11.Paterson I, Goodman JM, Lister MA, Schumann RC, McClure CK, Norcross RD. Tetrahedron. 1990;46:4663. [Google Scholar]
- 12.Paterson I, Perkins MV. Tetrahedron Lett. 1992;33:801. [Google Scholar]
- 13.Komatsu N, Uda M, Suzuki H, Takahashi T, Domae T, Wada M. Tetrahedron Lett. 1997;38:7215. [Google Scholar]
- 14.Jung HH, Seiders JR, II, Floreancig PE. Angew Chem. 2007;119:8616. doi: 10.1002/anie.200702999. [DOI] [PubMed] [Google Scholar]; Angew Chem, Int Ed. 2007;46:8464. [Google Scholar]
- 15.Trotter NS, Takahashi S, Nakata T. Org Lett. 1999;1:957. doi: 10.1021/ol991200m. [DOI] [PubMed] [Google Scholar]
- 16.For a related, though less selective reaction, see: Huang X, Shao N, Palani A, Aslanian R, Buevich A. Org Lett. 2007;9:2597. doi: 10.1021/ol071068n.
- 17.The stereochemical outcomes of the multicomponent reactions were assigned based on the highly predictable trends of the N–H and C10 hydrogens in the 1H NMR spectra of the desired and undesired stereoisomers. Please see the Supporting Information for tabulated data.
- 18.Hong CY, Kishi Y. J Org Chem. 1990;55:4242. [Google Scholar]
- 19.Please see the Supporting Information for details on the syntheses of 18 and 20.
- 20.Perry NB, Blunt JW, Munro MHG, Thomson AM. J Org Chem. 1990;55:223. [Google Scholar]
- 21.Fukui H, Tsuchiya Y, Fujita K, Nakagawa T, Koshina H, Nakata T. Bioorg Med Chem Lett. 1997;7:2081. [Google Scholar]
- 22.a) Smith AB, III, Safonov IG, Corbett RM. J Am Chem Soc. 2002;124:11102. doi: 10.1021/ja020635t. [DOI] [PubMed] [Google Scholar]; b) Rishel MJ, Hecht SM. Org Lett. 2001;3:2867. doi: 10.1021/ol010139u. [DOI] [PubMed] [Google Scholar]
- 23.Zimmermann K, Engeser M, Blunt JW, Munro MHG, Piel J. J Am Chem Soc. 2009;131:2780. doi: 10.1021/ja808889k. [DOI] [PubMed] [Google Scholar]
- 24.a) Matsunaga S, Fusetani N, Nakao Y. Tetrahedron. 1992;48:8369. [Google Scholar]; b) Simpson JS, Garson MJ, Blunt JW, Munro MHG, Hooper JNA. J Nat Prod. 2000;63:704. doi: 10.1021/np990431z. [DOI] [PubMed] [Google Scholar]
- 25.Fisch KM, Gurgui C, Heycke N, van der Sar SA, Anderson SA, Webb VL, Taudien S, Platzer M, Rubio BK, Robinson SJ, Crews P, Piel J. Nature Chem Biol. 2009;5:494. doi: 10.1038/nchembio.176. [DOI] [PubMed] [Google Scholar]