

Abstract
A histone deacetylase (HDAC)-based yeast assay employing a URA3 reporter gene was applied as a primary screen to evaluate a marine-derived actinomycete extract library and identify human class III HDAC (SIRT) inhibitors. Based on the bioassay-guided purification, a new compound designated as streptosetin A (1) was obtained from one of the active strains identified through the yeast assay. The gross structure of the new compound was elucidated from the 1D and 2D NMR data. The absolute stereostructure of 1 was determined based on X-ray crystal structure analysis and simulation of ECD spectra using time-dependent density functional theory (TD-DFT) calculations. This compound showed weak inhibitory activity against yeast Sir2p, and human SIRT1 and SIRT2.
Class III HDACs (SIRT1-7) are NAD-dependent epigenetic enzymes unlike classical zinc-dependent class I, II and IV HDACs, and are known to be insensitive to zinc-dependent HDAC inhibitors such as trichostatin A.1 Among SIRT isoforms, SIRT1 and SIRT2 have received increased attention as targets for anticancer molecules because the former is overexpressed in cancer cells2 and the latter is involved in the cell cycle.3 More importantly, SIRT1 and SIRT2 interact with the same non-histone substrates as those regulated by classical HDACs, which include the tumor suppressor protein p53,4 and α-tubulin.5 In addition, the synthetic SIRT1/SIRT2 inhibitor sirtinol demonstrated apoptotic and autophagic cell death in an MCF-7 breast cancer cell line.6 Given the evidence listed above and the FDA approvals of two classical HDAC inhibitors, SAHA (vorinostat, 2006) and FR901228 (istodax, 2009), for the treatment of cutaneous T-cell lymphoma (CTCL), as well as LBH589 (panobinostat) currently being evaluated in phase I/II/III clinical trials,7 class III HDAC (SIRT) inhibitors are anticipated to be anticancer drug candidates. Crucial SIRT inhibitors have not been discovered from natural sources yet, except for a few mild SIRT1/SIRT2 inhibitors including tanikolide dimer,8 (+)-guttiferone G9 and amurensin G.10 On the other hand, a number of synthetic SIRT inhibitors including a potent SIRT1 selective inhibitor EX-527,11 have been developed in the past decade.
The SIRT family is evolutionally conserved from bacteria to mammals.12 Among the human SIRT enzymes, SIRT1 is the closest homolog of the yeast silent information regulator 2 protein (Sir2p).13 Furthermore, the seven human isoforms possess close sequence identity, where their catalytic and NAD+ binding domains are also conserved.1 Consequently, Sir2p inhibitors are likely to inhibit human SIRT enzymes. In fact, sirtinol14 and splitomicin,15 the most common molecular probes for SIRT1 and SIRT2, have been independently identified as Sir2p inhibitors from a synthetic chemical library using a genetically modified yeast strain as a screening tool. As described in the article previously reported by the Schreiber group,14 this yeast screening employs a URA316 reporter gene embedded in the telomere region of the yeast chromosome, which is activated by Sir2p inhibitors. Selective Sir2p inhibitory activity is visually observed by adding 5-fluoroorotic acid (5-FOA) in the culture medium. The reporter gene activated by a Sir2p inhibitor converts 5-FOA into the cytotoxic compound 5-fluorouracil (5-FU), which leads to the death of yeast cells, whereas yeast cells survive the Sir2p inhibitor in the absence of 5-FOA. We have begun a program to further apply such a yeast screen to evaluate natural products libraries and isolate SIRT inhibitors. Reported below are outcomes based on the evaluation of extract libraries created from marine-derived actinomycetes. The yeast screening has led to the discovery of a new compound designated as streptosetin A (1).
Results and Discussion
Prior to screening our chemical library, we first verified the sensitivity of the two yeast strains DMY284317 and UCC100115 with a URA3 reporter gene in the telomere region using the known Sir2p inhibitor splitomicin. The DMY2843 strain showed more pronounced activity at 20 μM than those observed in the UCC1001 strain (Figure S3). Thus, the yeast strain, DMY2843, was chosen for further screening in this study. A mini-library composed of 506 extracts created from the same number of marine-derived actinomycetes was tested against the DMY2843 strain at 20 μg/mL as the final concentration. This screening identified a total of 53 active strains, which were divided into two groups: (1) selective activity (19 strains, 3.8%), and (2) non-selective activity (34 strains, 6.7%). In the former group, yeast growth was inhibited only in the presence of 5-FOA based on Sir2p inhibition, while yeast cell death was observed both in the presence and absence of 5-FOA in the latter group. The non-selective activity originates either from potent Sir2p inhibitory activity or from different modes of action. Among the selective active strains, Streptomyces sp. CP13-10 was selected for further study. A peak library created from the extract (30 mg) of this strain was subjected to the same yeast screening to pinpoint compounds responsible for the selective activity. The screening result indicated that the compound eluting at 15 min showed Sir2p inhibitory activity. To characterize the active compound, this strain was cultured in liquid medium (6 L) containing starch (1%), yeast extract (0.4%), peptone (0.2%), CaCO3 (0.1%), Fe2SO4 · 7H2O (0.004%) in artificial seawater (6 L) for 7 days at 30 °C, shaking at 200 rpm. The bacterial pellet and culture broth were independently extracted with MeOH. The combined MeOH extract was then partitioned between EtOAc and H2O. The organic layer was dried under reduced pressure to afford an extract. The active compound designated as streptosetin A (1) was purified from the extract using reversed phase HPLC.
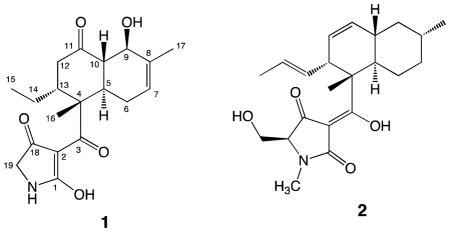
The molecular formula of streptosetin A (1) was established as C19H25NO5 from the HRESIMS data. The initial 13C NMR spectrum showed only 17 carbon signals (Table 1), which were classified into the following five groups from DEPT and HSQC experiments: (1) three carbonyl groups (C-3, C-11, C-18), (2) a trisubstituted double bond carbons (C-7, C-8), (3) four sp3 methines (C-5, C-9, C-10, C-13) including one oxygen bearing methine (δH-9 70.1, δH-9 4.44), (4) four sp3 methylenes (C-6, C-12, C-14, C-19) including one nitrogen bearing methylene (δH-19 51.2, δH-19 3.79), and (5) three methyl groups (C-15, C-16, C-17) including one vinyl methyl (δH-17 19.4, δH-17 1.71). The COSY analysis provided two spin systems (C-7-C-6-C-5-C-10-C-9 and C-12-C-15) as indicated in boldfaced lines in Figure 2. An analysis of the HMBC data led to the two substructures (Figure 2). The former spin system was incorporated into a cyclohexene with an oxygen atom at the allylic position based on the HMBC correlations from H-7 to C-9/C-17, from H-9 to C-7/C-8, and from H-17 to C-7/C-8/C-9. This cyclohexene and the latter spin system were further connected via C-4 and C-11 to form a decalin system, substructure A, based on the HMBC correlations from H-5 to C-4, from H-9 to C-11, from H-10 to C-11, and from H-12 to C-4/C-11. Finally, substructure A incorporated a methyl group (C-16) and a ketone (C-3) at C-4 due to HMBC correlations from H-5 to C-3/C-16, and from H-16 to C-3/C-4/C-5/C-13. In addition, it was expected that C-9 possessed an alcohol group as no other carbons bearing an oxygen atom were observed in the 13C NMR spectrum. A second substructure B with a nitrogen atom was defined by the HMBC correlation from the nitro-methylene H2-19 to C-18. At this point the unaccounted atoms consisted of C2H2O, which were invisible in the both the 1H and 13C NMR spectra. Subsequently, as shown in Table 1 these carbons were visualized by a 13C NMR spectrum acquired at 75 MHz and were eventually assigned to C-1 (δC 180.1) and C-2 (δC 100.6), as discussed further below. The two hydrogens were assigned as exchange protons since they were not detected in the 1H NMR spectrum measured in CD3OD. Thus, substructure C with C2O was proposed.
Table 1.
NMR Data of 1 in CD3OD (500 MHz for 1H, 125 MHz for 13C)
| position | δC, type | δH, mult (J in Hz) | COSY | HMBC | NOESY |
|---|---|---|---|---|---|
| 1 | 180.1,a C | ||||
| 2 | 100.6,a C | ||||
| 3 | 202.4, C | ||||
| 4 | 51.6, C | ||||
| 5 | 40.9, CH | 2.61, ddd (12.7, 10.7, 3.8) | 6ax, 10 | 3, 4, 6, 7, 9, 10, 16 | 6eq, 9, 14a |
| 6ax | 29.0, CH2 | 2.01, m | 5, 6eq, 9, 17 | 6eq, 7, 10 | |
| 6eq | 2.20, br. d (16.7) | 6ax, 7 | 5, 7, 8, 10 | 5, 6ax, 7 | |
| 7 | 124.8, CH | 5.48, dq (6.1, 2.3) | 6eq, 17 | 5, 6, 9, 17 | 6ax, 6eq, 17 |
| 8 | 135.6, C | ||||
| 9 | 70.1, CH | 4.44, br. d (8.6) | 6ax, 10, 17 | 5, 7, 8, 10, 11 | 5, 10, 17 |
| 10 | 55.2, CH | 2.48, dd (12.7, 8.6) | 5, 9 | 5, 6, 9, 11 | 6ax, 9, 12ax, 16 |
| 11 | 213.9, C | ||||
| 12ax | 40.2, CH2 | 2.88, dd (13.7, 4.8) | 12eq, 13 | 4, 11, 13, 14 | 10, 12eq, 13, 16 |
| 12eq | 2.37, dd (13.7, 3.1) | 12ax | 4, 10, 11, 13, 14 | 12ax, 13, 15 | |
| 13 | 44.3, CH | 3.07, br. d (9.8) | 12ax, 14a | 12ax, 12eq, 14b, 15, 16 | |
| 14a | 23.5, CH2 | 0.98, ddq (14.0, 11.6, 7.1) | 13, 14b, 15 | 12, 13, 15 | 5, 14b, 15 |
| 14b | 1.15, m | 14a, 15 | 4, 12, 13, 15 | 13, 14a, 15 | |
| 15 | 12.3, CH3 | 0.79, t (7.2) | 14a, 14b | 13, 14 | 12eq, 13, 14a, 14b |
| 16 | 16.0, CH3 | 1.79, s | 3, 4, 5, 13 | 6ax, 10, 12ax, 13 | |
| 17 | 19.4, CH3 | 1.71, br. dd (2.3, 1.2) | 7, 6ax, 9 | 7, 8, 9 | 7, 9 |
| 18 | 192.4, C | ||||
| 19 | 51.2, CH2 | 3.79 (2H), m | 1, 18 |
Detected at 75 MHz
Figure 2.

Substructures and significant 2D NMR correlations for 1. Arrows indicate HMBC correlations. Bold lines show COSY spin systems.
The relative configuration and conformation of substructure A were further determined by detailed analysis of vicinal coupling constants and NOESY correlations (Figure 3). The large coupling constant between H-5 and H-10 (3J5,10 = 12.7 Hz) suggested a trans-fused decalin. NOE correlations between H-10 and H-12ax, H-10 and H3-16, H-12ax and H3-16 and H-13 and H3-16 denoted a chair conformation for the cyclohexane ring with H-10, H-12ax and 16-CH3 in axial positions and H-13 in an equatorial arrangement. NOEs between H-12eq and H3-15 and H-5 and H2-14a implied that H-5 and 13-C2H5 are arranged trans to 16-CH3 with both axial arrangement. The cyclohexene ring was deduced to exist in a twist-chair conformation due to relatively large coupling constants between H-5 and H-6ax (3J5,6ax = 10.7 Hz) and H-9 and H-10 (3J9,10 = 8.6 Hz) and NOEs between H-5 and H-9 and H-6ax and H-10. These observations suggested that 9-OH is pseudoequatorial, and oriented cis to H-6ax, H-10 and 16-CH3, and trans to H-5 and 13-C2H5.
Figure 3.

Key NOESY correlations for 1.
Our attention shifted next to elucidating the total structure of streptosetin A (1). The two carbons in substructure C were concluded to be sp2 quaternary carbons (a tetrasubstituted double bond) as no reasonable working structures with sp and sp3 quaternary carbons were generated. Finally, two working structures 1a and 1b with a conjugated diketo-enol motif were proposed (Figure 4). These working structures possessed a dynamic tautomerism system through the carbons, C-1, C-2, C-3 and C-18. Thus, the carbon signals C-1 and C-2 were not detected in the 13C NMR spectrum due to signal broadening. Based on biogenetic assumptions, working structure 1a appeared to be more reasonable because it was categorized as a relatively common tetramic acid derivative. It was extremely difficult to observe all the carbon signals in the tetramic acid moiety at 125.7 MHz at room temperature (25°C) due to tautomerism. This appears to be a common issue with tetramic acid derivatives,18 which could be solved by measuring the 13C NMR at low temperature as reported in the articles regarding this class of secondary metabolites.19,20 However, attempts to observe the carbon signals for C-1 and C-2 at −25 °C were unsuccessful. Interestingly, these two carbons were detected as broad signals at δC 100.6 (C-2) and 180.1 (C-1) at lower frequency (75.4 MHz). The latter carbon signal (C-1) was confirmed by the HMBC spectrum, in which a cross-peak was observed between H-19 and C-1.
Figure 4.

Working structures for 1.
An X-ray single crystal structure analysis was carried out for the final structure determination because NMR techniques could not distinguish between the working structures. The X-ray crystal structure21 was identical with 1a, which justified the configuration and conformational assignments for the decalin system obtained from the NMR data (Figure 5). In the X-ray structure, intramolecular hydrogen bonding was observed between the hydroxy group at C-1 and the carbonyl group at C-3. In addition, the absolute configurations of the stereogenic centers were determined to be 4R,5R,9R,10S,13R based on the comparison of intensities of Friedel pairs of reflections [Flack x parameter = 0.06(6),22 Hooft y parameter = 0.060(9)23; values of zero indicate the correct enantiomorph]. To confirm the absolute stereostructure of 1 suggested from analysis of the X-ray structure, the modified Mosher’s method24 was initially chosen as this molecule possessed an equatorial secondary alcohol at C-9. However this reaction yielded a complex mixture of products, in which the desired Mosher’s ester was not detected. This could be attributed to the β-hydroxyketone moiety in the decalin system, which could assist the dehydration between C-9 and C-10 and/or retro-aldol type reaction rather than esterification. Absolute configurations of the asymmetric centers in 1 were finally confirmed using ab initio electronic circular dichroism (ECD) spectral calculations.25,26 ECD spectra for a set of enantiomers of 1 obtained by geometry optimization were generated using time-dependent density functional theory (TD-DFT) calculations at B3LYP/AUG-cc-pVDZ level. The general features of the experimental ECD spectrum of 1 agreed well with that calculated for the 4R,5R,9R,10S,13R isomer though the simulated ECD spectra, but were blue-shifted by 35 nm (Figure 6). These absolute configurations were identical to those obtained from the X-ray crystal structure described above. Consequently, the absolute configurations in 1 were firmly determined as 4R,5R,9R,10S,13R.
Figure 5.

X-ray crystal structure for 1. Thermal displacement ellipsoids depicted at 50% probability with chloroform and the water of crystallization omitted for clarity.
Figure 6.

Experimental (A) and calculated (B) ECD spectra of 1.
On the other hand, three possible tautomers (I, II and III) based on the conjugated diketo-enol moiety were considered in solution state, though tautomer I was originally assigned as the structure of 1 using NMR techniques as well as X-ray crystal structure analysis (Figure 7). A literature search for this class of compounds implied that the type III tautomer was the most favored form among the three tautomers. However, analysis of the published carbon chemical shift values for the four conjugated sp2 carbons in each tautomeric form indicated that it was impossible to assign an appropriate form using these carbon chemical shifts because the ranges of these chemical shifts in the three tautomers overlapped one another. (Table S1). Consequently, the structure of 1 in solution remaines undetermined.
Figure 7.

Three possible tautomers for 1.
Enzyme inhibitory activities of streptosetin A (1) were assessed using the yeast screening described above and SIRT1/SIRT2 fluorescent assays. In these assays, sirtinol was selected as a positive control. In the yeast screening, compound 1 showed weak cytotoxicity at 10 mM and a minimum inhibitory concentration (MIC) at 2.5 mM based on Sir2p inhibition whereas sirtinol showed an MIC at 12.5 μM (Figure S4). The yeast assay results for 1 and sirtinol mirrored human SIRT inhibitory activities. Compound 1 showed IC50 values of 3.7 mM (SIRT1) and 4.5 mM (SIRT2), while sirtinol inhibited SIRT1 and SIRT2 with IC50 values of 40.1 μM and 45.8 μM, respectively (Figure S5).
Conclusions
The genetically modified yeast strain used in this study proved to be a useful screening tool for identifying SIRT inhibitors from an actinomycete-derived chemical library. Though the new compound streptosetin A (1) obtained through this yeast screening turned out to be a weak SIRT inhibitor, this compound showed clear dose-response curves in the SIRT1 and SIRT2 enzyme assays (Figure S5). In addition, MIC values defined by the yeast assay appear to be parallel to the IC50 values determined by SIRT1 and SIRT2 enzyme assays. Structurally, streptosetin A (1) is categorized as a polyketide tetramic acid derivative produced by an NRPS-PKS hybrid pathway, as reported for equisetin (2) isolated from a Fusarium equiseti.27 The structure elucidation of this class of compounds appears to be challenging due to proton and/or carbon signal broadening caused by the dynamic tautomeric system. It is even more challenging to assign the correct tautomer for this class of compounds, which is often overlooked in the process of structure elucidation. More importantly, this compound may be applied to a chemical epigenetic approach to alter the chemical profile for microorganisms as epigenetic enzyme inhibitors like SAHA (class I HDAC inhibitor) and 5-AZT (DNA methyl transferase inhibitor) are known to induce new compounds based on activating silent biosynthetic genes in fungi.28,29 We will continue to search for new and/or potent SIRT inhibitors from other active strains identified through this study.
Experimental Section
General Experimental Procedures
Melting points were recorded on a Barnstead International Mel-Temp Electrothermal melting point apparatus 1101D, and are uncorrected. Optical rotations were obtained on a JASCO P-2000 polarimeter. The UV spectrum was measured on an Agilent 8453 UV/vis diode array spectrophotometer. The CD spectrum was collected with a JASCO J-820 CD spectrometer. 1D and 2D NMR spectra were recorded on a Varian Inova spectrometer operated at 500 MHz (1H) and 125.7 MHz (13C) and a Bruker Avance spectrometer operated at 300 MHz (1H) and 75.4 MHz (13C). The chemical shifts were calibrated using the internal solvent peak (δH 3.31 and δC 49.0). High resolution MS data were obtained on a Marinar ESI-TOFMS. HPLC was performed on an Agilent 1200 LC system equipped with a diode array detector.
Isolation of Actinomycete Strains
A total of 506 actinobacteria were separated from marine samples collected from various locations; Santa Monica basin, CA [sediments (−900 m), July 2008], Hawaii, HI [sands (various depths, −10 – −300 m), November 2008], San Francisco Bay, CA [sediments (−1 m), August 2008], Moss Landing, CA [sediments (−1 m), August 2009] and Baja, Mexico [sands, (−15 – −30 m), May 2009]. These marine samples were stored at −20 °C until processed. Each marine sample (1 cm−3) was thoroughly dried at 50 °C, and then aseptically diluted in 9 mL of phosphate buffered saline (PBS). This suspension was heated at 60 °C for 30 min to afford a 10-fold diluted suspension, which was further diluted to a 100-fold suspension with PBS. These two suspensions (100 μL) were individually plated on starch-nitrate agar and ISP3 agar plates. These two agar plates were prepared using artificial seawater [NaCl (1%), KCl (0.2%), MgSO4 · 7H2O (0.4%) and CaCl2 · 2H2O (0.03%)]. The plates were incubated at 28 °C for two months. Each actinomycete colony on the plates was transferred onto a seawater-based ISP2 agar plate, and was passaged repeatedly until a pure strain was obtained.
Building the Extract Library
All of the separated strains were cultured in a seed medium (25 mL) composed of soluble starch (2%), D-glucose (1%), yeast extract (0.5%), peptone (0.5%) and CaCO3 (0.5%) in artificial seawater as described above with a trace element mix (25 μL) at 30 °C for 5 days at 200 rpm. The trace element mix is composed of the following inorganic salts: FeSO4 · 7H2O (4 g), MnSO4 · H2O (5 g), ZnSO4 · 7H2O (2.5 g), Na2B4O7 · 10H2O (1.4 g), CoCl2 · 6H2O (0.2 g), CuSO4 · 5H2O (0.5 g), Na2MoO4 (0.2 g) in ultrapure H2O (1 L). When the trace element mix was prepared, the pH was adjusted between 1 and 2, and then each ingredient was added in the order listed. The seed culture (1 mL) was inoculated into a production media (100 mL) at 30 °C for 7 days at 200 rpm. The composition of the production media was the same as that of the seed media described above with HP20 resin (5 mL). The bacterial pellet and HP-20 separated from the production culture were extracted with MeOH (50 mL) twice and the combined MeOH solution was dried to obtain a MeOH extract. The MeOH extract was dissolved in EtOAc (100 mL) and rinsed with H2O (50 mL) twice to remove salts and media components. The EtOAc solution was dried under reduced pressure to provide an organic extract. A library of the extracts was generated with a concentration of 10 mg/mL in DMSO in 96 deep-well plates. This library of extracts was stored at −20 °C before being screened for bioactivity against the strain.
Yeast Strains and URA3 Reporter Gene Screening
Two genetically modified Saccharomyces cerevisiae strains (DMY2843 and UCC1001) with a URA3 reporter gene embedded in the telomere region were used for screening. The genotypes of the two strains are as followed: DMY2843 (MATa ade2-1 ura3-1 his3-11 trp1-1 leu2-3 leu2-112 can1-100 TEL-VIIL::URA3) and UCC1001 (MATα ura3-52 lys-801 ade2-101 trpΔ63 his3D200 leu2-Δ1 TEL VIIL adh4::URA3). A loopful (10 μL) of a fresh yeast colony from a YPD agar plate was incubated in YPD media (40 mL) overnight at 27 °C and 180 rpm to furnish the yeast culture for screening. Yeast screening was performed in two different screening solutions (solutions A and B) using flat bottom clear 96-well plates.14 The master mix of solution A was composed of fresh YPD media (9.765 mL), 15% 5-FOA in DMSO (0.135 mL) and yeast culture (0.1 mL), while the master mix of solution B was composed of fresh YPD media (9.9 mL) and yeast culture (0.1 mL). The chemical library samples (2 μL) adjusted as 10 mg/mL in DMSO were mixed separately with each master mix (98 μL) in the well. DMSO and splitomicin (50 μM as final concentration) were used as negative and positive standard compounds, respectively. The screening results were evaluated within 24 to 36 h when the negative control well was saturated with yeast growth, which was determined by visual inspection. Active samples resulted in a visibly clear well due to the death of yeast cells whereas inactive samples resulted in a visibly cloudy well due to the growth of yeast cells. Sir2p inhibitory activity was defined as selective when the screened sample resulted in the death of yeast cells only in the presence of 5-FOA. Cytotoxic or non-selective activity was determined as the death of yeast cells in both the screening solutions A and B.
Bacterial Identification
The actinomycete strain (CP13-10) was separated from a sediment sample collected at the depth of 1m in San Francisco Bay in August 2008. The genomic DNA of this strain was extracted using a DNA release kit (MicroLysis, Gel Company). The 16s rRNA (ca. 1400 bp) was amplified using a hot start TAQ PCR mix (MegaMix Royal, Gel Company) employing two universal primers, 27F and 1492R.30 The PCR was performed under the following thermal condition: 95 °C for 5 min, 35 cycles of denaturation at 94 °C for 1 min, annealing at 64 °C for 1 min, extension at 72 °C for 1.5 min and additional extortion at 72 °C for 10 min. The PCR product was purified with a purification kit (MicroElute DNA clean up kit, Omega Bio-tek). The purified PCR product was sequenced at Saga Gene, Inc. using the same primers described above and four additional universal primers including 514F, 530R, 936R, and 1114F.30 This strain was identified as a Streptomyces sp. on the basis of 100% sequence similarity to Streptomyces violaceusniger Tu 4113 (GenBank accession number CP002994). The 16s rRNA sequence of the CP13-10 strain has been deposited to GenBank with the accession number JX235443.
Isolation of Streptosetin A (1)
The seed culture (10 mL) of the strain CP13-10 prepared as described above was inoculated to the production media (1L × 6). The production media was cultured at 30 °C for 7 days at 200 rpm. The culture was centrifuged (6000g, 30 min) to separate the culture broth from the actinomycete pellet. To the culture broth, HP-20 resin (50 mL/L) was added and the suspension was stirred for 30 min. This resin was filtered and rinsed thoroughly with H2O. Then, organic materials were extracted from the HP-20 resin using MeOH. The pellet was soaked in MeOH overnight three times. The combined MeOH solution from the HP-20 resin and pellet was dried under reduced pressure. The residue that dissolved in EtOAc (200 mL) was rinsed with H2O (200 mL) three times and the organic layer was dried under reduced pressure to provide an extract (261 mg). This extract was subjected to reversed-phase HPLC using a linear gradient system (10% – 100% CH3CN in H2O over 20 min). The semipure fraction obtained around 15 min was further purified using another gradient system (40% to 70% CH3CN in H2O over 20 min) to afford 1 (6.5 mg).
Streptosetin A (1)
Colorless crystals; mp 99–100°C; [α]24D-115 (c 1.0 MeOH); UV (MeOH) λmax (log ε) 243 nm (2.53), 283 nm (3.12); CD (EtOH) Δε (nm) 228 (−1.70), 250 (−0.27), 285 (−4.24), 317 (+2.22); 1H and 13C NMR data, see Table 1; HRESI-TOFMS m/z 370.1622 [M+Na]+ (calcd for C19H25NO5Na, 370.1625).
X-Ray Crystallography of 1
Suitable crystals were grown at 4 °C by vapor diffusion of pentane into a CHCl3 solution of 1. A representative crystal (0.51 × 0.22 × 0.13 mm) was selected and mounted on a Bruker APEX diffractometer31 under a cold nitrogen stream at 100(2) K. An arbitrary hemisphere of data was recorded (3600 frames, 0.5° per frame with a combination of ω- and ϕ-scans, exposure time 20s/frame, 2θmax = 135.92°) with graphite monochromated Cu-Kα radiation (1.54178 Å). Data were integrated with Bruker SAINT v7.66A31 yielding 19805 reflections, of which 4334 were independent (Rint = 3.53 %) and 3843 with I > 2σ(I). Data were corrected for absorption, Lorentz and polarization effects with SADABS32 (Tmin/Tmax = 0.4396/0.8361). The structure was solved by direct methods (SHELXS) and refined by full-matrix least-square techniques on F2 (SHELXL32). All non-hydrogen atoms, except the disordered water of crystallization, were refined with anisotropic thermal displacement parameters. Hydrogen atoms on the molecule of interest and chloroform of crystallization were included in geometrically calculated positions. Hydrogen atoms on the waters of crystallization could not be reliably located and were not included in the model but were included in the chemical formula. The structure refined to R1 = 9.42 % and wR2 = 0.2665 for all 3846 data I>2σ(I) and R1 = 10.32 % and wR2 = 27.93% for all 4334 data (GooF = 1.023) for 297 parameters. The absolute configuration was determined based on a Flack x parameter of 0.06(6) and a Hooft y parameter of 0.060(9).
Crystal Data for 1
C19H25NO5 · CHCl3 · H2O; Mr = 484.78; trigonal; space group P3221; a = 11.1313(3) Å; b = 11.1313(3) Å; c = 33.9045(9) Å; α = 90°; β = 90°; γ = 120°; V = 3638.14(17) Å3; Z = 6; T = 100(2) K; λ (Cu-Kα) = 1.54178 Å; μ(Cu-Kα) = 3.716 mm−1; dcalc = 1.328 g.cm−3.
ECD Calculations for 1
All the DFT calculations were performed using Gaussian 09.33 Geometry optimization employing the B3LYP function (AUG-cc-pVDZ level) was performed in the gas phase for 1. A set of enantiomers of 1 generated from geometry optimization was subjected to the TD-DFT method with the B3LYP/AUG-cc-pVZD basis set using SCRF (self-consistent reaction field) to calculate the excitation energies and oscillator and rotational strengths of the lowest 50 electronic excitations. These calculation results were simulated into ECD spectra with a Gaussian function (a half-bandwidth of 0.3 eV) using GaussView 5.0.9.
SIRT1 and SIRT2 Inhibition Assays
SIRT1 and SIRT2 inhibitory activities were measured using SIRT1 and SIRT2 Direct Fluorescent Screening Assay kits (Cayman Chemical Company). The assays were performed in a 96-well plate based on the supplier’s protocol. To the 50 mM Tris-HCl buffer (25 μL), human recombinant SIRT or SIRT2 (5 μL), the substrate [Arg-His-Lys-Lys(ε-acetyl)-AMC] (15 μL, 125 μM), and various concentrations of the samples (5 μL in DMSO) were added and the assay plate was incubated on a shaker at room temperature for 45 min (SIRT1) or 37 °C for 45 min (SIRT2). Then, to each well, the stop/developer solution containing nicotinamide (50 μL) was added to stop deacetylation. Thirty minutes after adding this stop/developer solution, the fluorescence was measured using a fluorometric reader (Molecular Device, SpectraMax M2) at 350 nm for excitation and 450 nm for emission. The values of % inhibition against SIRT1 and SIRT2 were calculated from the fluorescence readings of the sample wells relative to those of 100% initial activity wells [50 mM Tris-HCl (25 μL), SIRT1/SIRT2 (5 μL), DMSO (5 μL), the substrate (15 μL) and the stop/developer solution (50 μL)]. The IC50 values of the samples were determined using IDBS XL fit5.
Supplementary Material
Figure 1.

Yeast screening result (24 h) for the peak library of the CP13-10 strain; (A) YPD media with 5-FOA, (B) YPD media, (C) HPLC chromatogram (UV at 280 nm) of the extract peak library (10% to 100% CH3CN in H2O, linear gradient over 20 min). The number of fractions in A and B corresponds to the retention time in the HPLC chromatogram. Selective activity was observed for fractions: F14 (14 min), F15 (15 min) and F16 (16 min).
Acknowledgments
This work was supported by Grant Number SC2GM088057 from the National Institute of General Medical Sciences. The Cell and Molecular Image Center (CMIC) of the College of Science and Engineering, SF State University was funded by Grant Number P20MD000544 from the National Center on Minority Health and Health Disparities. We are grateful for the gift of yeast strains from Dr. D. Moazed (Harvard Medical School) and Dr. D. E. Gottschling (Fred Hutchinson Cancer Research Center). We thank Dr. A. Chan (SFSU, CMIC) for assistance with microscopy, Dr. T. Komada (SFSU) for providing sediment samples, and Dr. T. Yamada (OUPS) for experimental ECD measurement.
Footnotes
Dedicated to the memory of the late Dr. Atsushi Numata of Osaka University of Pharmaceutical Sciences for his effort on discovering anticancer lead compounds.
Supporting Information Available.
Sequence data of the Streptomyces sp. CP13-10, 1D and 2D NMR data (1H, 13C NMR, COSY, HSQC, HMBC and NOESY), and crystal data for 1. This information is free of charge via the Internet at http://pubs.acs.org.
References and Notes
- 1.Yamamoto H, Schoonjans K, Auwerx J. Mol Endocrinol. 2007;21:1745–1755. doi: 10.1210/me.2007-0079. [DOI] [PubMed] [Google Scholar]
- 2.Jung-Hynes B, Nihal M, Zhong W, Ahmad N. J Biol Chem. 2009;284:3823–3832. doi: 10.1074/jbc.M807869200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Inoue T, Hiratsuka M, Osaki M, Oshimura M. Cell Cycle. 2007;6:1011–1018. doi: 10.4161/cc.6.9.4219. [DOI] [PubMed] [Google Scholar]
- 4.Yi J, Luo J. Biochim Biophys Acta. 2010;1804:1684–1689. doi: 10.1016/j.bbapap.2010.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.North BJ, Marshall BL, Borra MT, Denu JM, Verdin E. Mol Cell. 2003;11:437–444. doi: 10.1016/s1097-2765(03)00038-8. [DOI] [PubMed] [Google Scholar]
- 6.Wang J, Kim TH, Ahn MY, Lee J, Jung JH, Choi WS, Lee BM, Yoon KS, Yoon S, Kim HS. Int J Oncol. 2012;41:1101–1109. doi: 10.3892/ijo.2012.1534. [DOI] [PubMed] [Google Scholar]
- 7.Hymes KB. Clin Lymphoma Myeloma Leuk. 2010;10:98–109. doi: 10.3816/CLML.2010.n.014. [DOI] [PubMed] [Google Scholar]
- 8.Gutierrez M, Andrianasolo EH, Shin WK, Goeger DE, Yokochi A, Schemies J, Jung M, France D, Cornell-Kennon S, Lee E, Gerwick WH. J Org Chem. 2009;74:5267–5275. doi: 10.1021/jo900578j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gey C, Kyrylenko S, Hennig L, Nguyen LH, Buttner A, Pham HD, Giannis A. Angew Chem Int Ed Engl. 2007;46:5219–5222. doi: 10.1002/anie.200605207. [DOI] [PubMed] [Google Scholar]
- 10.Oh WK, Cho KB, Hien TT, Kim TH, Kim HS, Dao TT, Han HK, Kwon SM, Ahn SG, Yoon JH, Kim TH, Kim YG, Kang KW. Mol Pharmacol. 2010;78:855–864. doi: 10.1124/mol.110.065961. [DOI] [PubMed] [Google Scholar]
- 11.Napper AD, Hixon J, McDonagh T, Keavey K, Pons JF, Barker J, Yau WT, Amouzegh P, Flegg A, Hamelin E, Thomas RJ, Kates M, Jones S, Navia MA, Saunders JO, DiStefano PS, Curtis R. J Med Chem. 2005;48:8045–8054. doi: 10.1021/jm050522v. [DOI] [PubMed] [Google Scholar]
- 12.Brachmann CB, Sherman JM, Devine SE, Cameron EE, Pillus L, Boeke JD. Genes Dev. 1995;9:2888–2902. doi: 10.1101/gad.9.23.2888. [DOI] [PubMed] [Google Scholar]
- 13.Frye RA. Biochem Biophys Res Commun. 2000;273:793–798. doi: 10.1006/bbrc.2000.3000. [DOI] [PubMed] [Google Scholar]
- 14.Grozinger CM, Chao ED, Blackwell HE, Moazed D, Schreiber SL. J Biol Chem. 2001;276:38837–38843. doi: 10.1074/jbc.M106779200. [DOI] [PubMed] [Google Scholar]
- 15.Bedalov A, Gatbonton T, Irvine WP, Gottschling DE, Simon JA. Proc Natl Acad Sci U S A. 2001;98:15113–15118. doi: 10.1073/pnas.261574398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Silar P, Thiele DJ. Genes Dev. 1991;104:99–102. doi: 10.1016/0378-1119(91)90472-n. [DOI] [PubMed] [Google Scholar]
- 17.Tanny JC, Kirkpatrick DS, Gerber SA, Gygi SP, Moazed D. Mol Cell Biol. 2004;24:6931–6946. doi: 10.1128/MCB.24.16.6931-6946.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ondeyka J, Harris G, Zink D, Basilio A, Vicente F, Bills G, Platas G, Collado J, Gonzalez A, de la Cruz M, Martin J, Kahn JN, Galuska S, Giacobbe R, Abruzzo G, Hickey E, Liberator P, Jiang B, Xu D, Roemer T, Singh SB. J Nat Prod. 2009;72:136–141. doi: 10.1021/np800511r. [DOI] [PubMed] [Google Scholar]
- 19.Phillips NJ, Goodwin JT, Fraiman A, Cole RJ, Lynn DG. J Am Chem Soc. 1989;111:8223–8231. [Google Scholar]
- 20.Lang G, Blunt JW, Cummings NJ, Cole ALJ, Munro MHG. J Nat Prod. 2005;68:810–811. doi: 10.1021/np0500979. [DOI] [PubMed] [Google Scholar]
- 21.Crystallographic data for streptosetin A (1) have been deposited at the Cambridge Crystallographic Data Center (deposition number: CCDC894421). Copies of the data can be obtained, free of charge, on application to the Director, CCDC, 12 Union Road, Cambridge CB21EZ, UK [fax: +44-(0)1223–33603.3 or e-mail: deposit@ccdc.cam.ac.uk or http://www.ccdc.cam.ac.uk.
- 22.Flack HD. Acta Crystallogr Sect A. 1983;39:876–881. [Google Scholar]
- 23.Hooft RWW, Straver LH, Spek AL. J Appl Crystallogr. 2008;41:96–103. doi: 10.1107/S0021889807059870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ohtani I, Kusumi T, Kashman Y, Kakisawa H. J Am Chem Soc. 1991;113:4092–4096. [Google Scholar]
- 25.Li XC, Ferreira D, Ding Y. Curr Org Chem. 2010;14:1678–1697. doi: 10.2174/138527210792927717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li L, Si YK. J Pharm Biomed Anal. 2011;56:465–470. doi: 10.1016/j.jpba.2011.07.002. [DOI] [PubMed] [Google Scholar]
- 27.Sims JW, Fillmore JP, Warner DD, Schmidt EW. Chem Commun. 2005:186–188. doi: 10.1039/b413523g. [DOI] [PubMed] [Google Scholar]
- 28.Henrikson JC, Hoover AR, Joyner PM, Cichewicz RH. Org Biomol Chem. 2009;7:435–438. doi: 10.1039/b819208a. [DOI] [PubMed] [Google Scholar]
- 29.Wang XR, Sena JG, Hoover AR, King JB, Ellis TK, Powell DR, Cichewicz RH. J Nat Prod. 2010;73:942–948. doi: 10.1021/np100142h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mincer TJ, Fenical W, Jensen PR. Appl Environ Microbiol. 2005;71:7019–7028. doi: 10.1128/AEM.71.11.7019-7028.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bruker APEX-II v2010-7 (SAINT v7.66A) Bruker AXS Inc; Madison WI: 2010. [Google Scholar]
- 32.Sheldrick GM. Acta Crystallogr, Sect A. 2008;64:112–122. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- 33.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA, Jr, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas Ö, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ. Gaussian 09, Revision A.1. Gaussian, Inc; Wallingford CT: 2009. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.