

Abstract
Aldosterone contributes to the endocrine basis of heart failure and studies on cardiac aldosterone signaling have reinforced its value as a therapeutic target. Recent focus has shifted to new roles of aldosterone that appear to depend on co-existing pathologic stimuli, cell type, and disease etiology. This review evaluates recent advances in mechanisms underlying aldosterone-induced cardiac disease and highlights the interplay between aldosterone and Ca2+ and calmodulin dependent protein kinase II, whose hyperactivity during heart failure contributes to disease progression. Increasing evidence implicates aldosterone in diastolic dysfunction, and there is need to develop more targeted therapeutics such as aldosterone synthase inhibitors and molecularly specific anti-oxidants. Despite accumulating knowledge, many questions still persist and will likely dictate areas of future research.
Keywords: aldosterone, CaMKII, heart failure, signaling
The endocrine basis of heart failure
Ischemic heart disease is a leading cause of death in the world [1], and myocardial infarction (MI, see Glossary) is a commonly presenting manifestation of ischemic heart disease. The post-MI myocardium undergoes molecular and cellular mechanisms of remodeling with fetal gene reprogramming, collectively termed pathological remodeling. While initially compensatory, this remodeling process eventually jeopardizes the structural and functional integrity of the myocardium leading to congestive heart failure (CHF). CHF and the accompanied peripheral edema take place in the setting of upregulated neurohormonal stimuli, including adrenergic signaling and activation of the renin-angiotensin-aldosterone system (RAAS). It is this neurohormonal milieu that constitutes the known endocrine basis of heart failure.
As the terminal effector of the RAAS cascade, aldosterone has been well studied in the context of renal physiology. Clear evidence for the therapeutic value of aldosterone blockade in patients with severe heart failure was provided by the RALES and EPHESUS trials [2], [3] as well as in heart failure patients with mild symptoms [4]. Aldosterone antagonist drugs, namely spironolactone and the more selective MR antagonist eplerenone, are now frontline drugs for heart failure, along with other agents targeting the neurohormonal imbalance driving pathologic remodeling after MI (reviewed in [5]). Multiple new agents are available or under investigation to target various steps in aldosterone action and biosynthesis (Figure 1 and Box 2).
Figure 1. RAAS activation and levels of pharmacologic intervention.
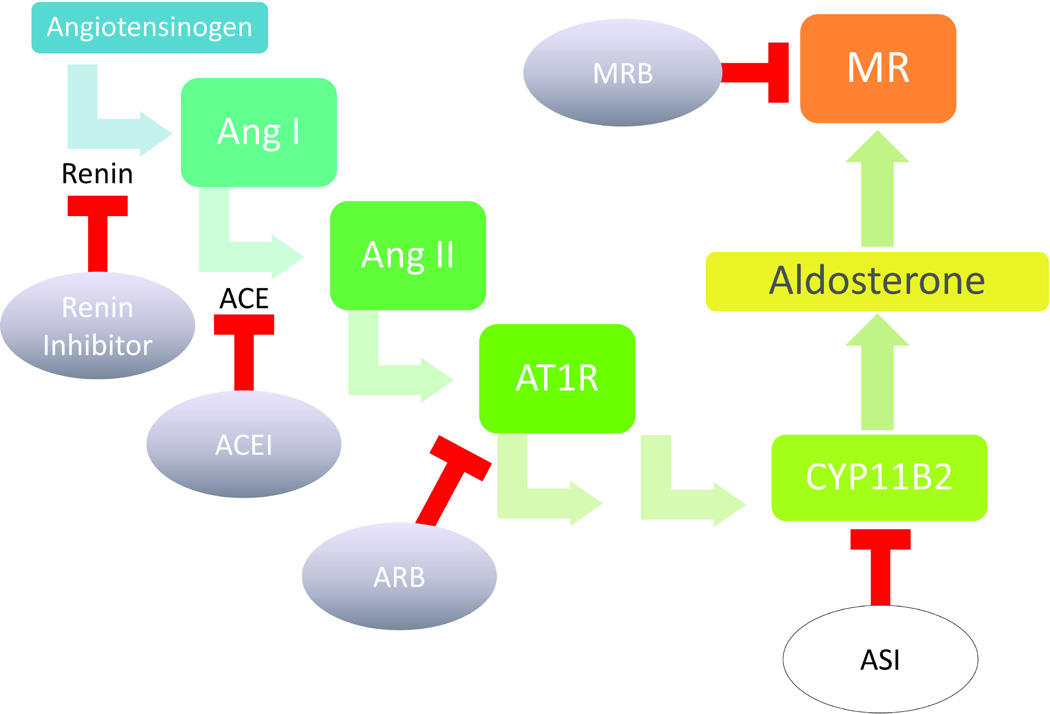
The renin-angiotensin-aldosterone system (RAAS) is a multisystem, endocrine cascade that regulates electrolyte homeostasis and blood pressure through actions on the kidney and heart. RAAS overactivation is pathologic, and multiple pharmacologic agents have been devised, both in experimental phase (white ovals) and in clinical use (shaded ovals), to target various levels of RAAS activation. Heart failure treatment is multimodal with combinatorial pharmacologic agents including inhibitors of RAAS as well as of other neuroendocrine systems that become pathologically overactivated in heart disease. Aldosterone is the terminal effector of the RAAS pathway, and advantages of targeting aldosterone synthesis or its receptor activation include specificity and avoidance of the aldosterone escape phenomenon. While the two mineralocorticoid receptor blockers (MRBs), spironolactone and eplerenone, have emerged to the front-line of heart failure therapy, the aldosterone synthase inhibitors (ASIs) such as FAD286 and LCI699 are still currently in experimental phase (Box 2). Other abbreviations: ACEI=angiotensin converting enzyme inhibitor, ARB=angiotensin receptor blocker, AT1R=angiotensin II receptor, CYP11B2=aldosterone synthase.
BOX 2: Aldosterone synthase inhibitors.
An alternative approach to distinguishing MR activation from aldosterone excess is the comparison of the aldosterone synthase inhibitors to MR antagonist drugs. Interest in aldosterone synthase inhibitors has also been fueled by recognition of nongenomic, MR-independent actions of aldosterone, and by adverse effects of MR antagonist drugs (e.g. hyperkalemia, aldosterone rebound). LCI699 is the first and only aldosterone synthase inhibitor tested clinically in patients with essential hypertension in a recent double-blinded, randomized clinical trial. Results show that LCI699 can significantly and safely reduce blood pressure compared to placebo, but this reduction does not appear to be superior to eplerenone. LCI699 at all tested doses had some degree of blood pressure reduction but only twice daily dosing had significant reduction in plasma aldosterone levels [81], consistent with observations from a prior trial of LCI699 in patients with primary aldosteronism [82]. A preclinical study documented anti-atherosclerotic and anti-inflammatory effects of the aldosterone synthase inhibitor FAD286 in spontaneous atherosclerotic apolipoprotein E-deficient mice [83]. In a direct comparison of FAD286 versus spironolactone treatment in rats after myocardial infarction, both drugs improved left ventricular hemodynamics and contractility [84]. However, FAD286 resulted in more sustained reduction in left ventricular oxidant signaling as well as increased protection from heart failure-induced gene expression alteration. The mechanisms for this additional protection require further investigation. In addition, it is not known whether survival will differ with these different approaches of aldosterone blockade.
Of note, from the methods reported by these earlier studies on aldosterone synthase inhibitors [81–83], it appears that a daily dosing regimen does not translate into measurable reduction in the plasma aldosterone levels, leading some authors to question whether these agents work through reduction of aldosterone excess or through alternative mechanisms [85]. For instance, FAD286 may inhibit CYP11B1, which catalyzes the final synthetic step for cortisol [86]. To what degree this action mediates the anti-atherosclerotic and anti-inflammatory effects of FAD286 remains unknown. Further research is necessary to dissect the mechanisms of aldosterone synthase inhibitors and to identify possible off-target effects. Ongoing work to develop more selective inhibitors of CYP11B2 [87, 88] may be help to resolve this issue.
Aldosterone is known to promote cardiac fibrosis, hypertrophy, and cell death with concurrent elevation of inflammatory and oxidant signaling [6–8]. While these cellular responses have been repeatedly observed, the underlying mechanisms for aldosterone-induced cardiac pathology remain under active research. Recent studies suggest that the multifunctional Ca2+/calmodulin (CaM) dependent protein kinase II (CaMKII) is a downstream signal activated by aldosterone-induced increases in myocardial reactive oxygen species (ROS) (Box 1) [9]. CaMKII is activated by increases in cellular Ca2+ to enable rate-dependent increases in myocardial contraction [10] and fight or flight increases in heart rate [11]. However, CaMKII transitions into a Ca2+ and CaM-independent enzyme, by oxidation of methionines 281 and 282 in its regulatory domain [12]. The molecular physiology of CaMKII in relationship to ROS was recently reviewed [13]. Other findings suggest that autonomously activated CaMKII promotes adverse outcomes in myocardial injury models mimicking conditions of increased catecholamine [14] and angiotensin II (Ang II) signaling [12]. CaMKII is required for aldosterone induced post-MI matrix remodeling and cardiac rupture in mice [9]. A pathway has been proposed where aldosterone stimulates CaMKII activation through direct oxidation by NADPH oxidase-derived ROS, and activated CaMKII in turn stimulates an increase in matrix metalloproteinase 9 (MMP9) to promote cardiac rupture. Reduction of CaMKII activity either through direct inhibition or reversal of its oxidation protects against aldosterone induced rupture and early death after MI. These studies advocate CaMKII inhibition and/or targeted anti-oxidant therapy as useful modalities in preventing early post-MI myocardial remodeling. This pathway may also be operative in conditions marked by elevated ROS and RAAS, such as hypertension-evoked atrial dilation, an important risk factor for the transition from paroxysmal to permanent atrial fibrillation, an atrial arrhythmia significantly associated with primary aldosteronism [15], and in some forms of dilated cardiomyopathy. The purpose of this review is to highlight recent advances in the understanding of the signaling pathways involved in aldosterone stimulated cardiac disease with focus on how the heart participates as an endocrine target while exhibiting characteristics of paracrine behavior.
BOX 1: Redox regulation of aldosterone signaling.
Redox signaling is vital to cardiovascular physiology and disease, and oxidative stress promotes several cardiac disease processes, including atherosclerosis, vascular inflammation, endothelial dysfunction, pathologic hypertrophy, and myocardial fibrosis (recently reviewed elsewhere [60]). The heart is exposed to multiple sources of ROS including NADPH oxidases, mitochondrial enzymes, xanthine oxidase, and NO synthase (NOS). Surprisingly, clinical trials using global antioxidants such as vitamin C or vitamin E did not show significant improvement in major outcomes and, in some cases, resulted in adverse effects [61, 62]. Early evidence suggests that a targeted antioxidant approach may be beneficial without the adverse effects from global ROS reduction [63, 64]. It was recently shown that transgenic mice overexpressing cardiac-specific methionine sulfoxide reductase A (MsrA) are resistant to aldosterone induced CaMKII oxidation without reduction in aldosterone stimulated total ROS [9]. These mice performed better after MI surgery and were protected against aldosterone exacerbated cardiac rupture after MI. Further work will be needed to dissect how redox signaling contributes to different aldosterone-related disease phenotypes.
Aldosterone generates redox stress on multiple levels (Figure 2). For instance, cardiac-specific overexpression of human MR in mice leads to increased ROS production and expression of NADPH oxidase subunit gp91phox [65]. Cyclic stretching of vascular smooth muscle cells is known to increase ROS production, and aldosterone was recently found to play a role in this. The mechanism involves cyclic stretching-induced upregulation of the cytochrome P450 aldosterone synthase and therefore aldosterone synthesis and MR stimulation [66]. Aldosterone-stimulated EPC dysfunction occurs through PKA-dependent generation of ROS [22]. MR blockade with spironolactone treatment in the transgenic Ren2 rat, which overexpresses the mouse renin transgene, led to decreased ROS formation and NADPH oxidase subunit expression. Of note, these observations were not accompanied by reversal of blood pressure elevation characteristic of the Ren2 rat, supporting a role of aldosterone in myocardium independent of blood pressure effect [67]. Finally, aldosterone may trigger ROS release from the mitochondria, which may underlie aldosterone stimulated EGFR and NHE activation [33]. Improved understanding of the sources of aldosterone-stimulated ROS and how they contribute to pathology will provide better therapeutic options.
Aldosterone and heart failure
Aldosterone participates in the initiation and progression of CHF. During CHF, initial decline in cardiac output stimulates neurohormonal compensatory systems, such as the sympathetic nervous system and the RAAS, which in turn place additional stress on the heart. While the pathophysiology of CHF has been characterized in detail, less is known about aldosterone.s role in diastolic dysfunction.
Diastolic dysfunction or heart failure with preserved ejection fraction (HFpEF) has been proposed to lie on a continuum of heart failure progression, although some argue that it is a separate entity from systolic heart failure. Recent evidence supports a mechanistic role for aldosterone in the pathogenesis of HFpEF. For instance, in studies with adiponectin knockout mice, an adipose-derived plasma protein that participates in anti-inflammatory and anti-fibrotic signaling, uninephrectomy plus aldosterone treatment induced worse diastolic dysfunction compared to uninephrectomy alone [16]. Biochemical profiling studies identified enhanced matrix turnover in patients with diastolic dysfunction, in particular increased carboxy-terminal pro-peptide of collagen 1 (Col I) [17] and MMP9 [18]. Increased MMP9 is known to positively correlate with echocardiographic parameters of diastolic heart failure [19, 20]. There is strong evidence for aldosterone to increase matrix turnover in heart (discussed below). Studies in aldosterone-exacerbated cardiac rupture identified increased MMP9 expression as a downstream consequence of aldosterone-triggered CaMKII activation [9], suggesting that a CaMKII/MMP9 pathway may contribute to aldosterone stimulated diastolic dysfunction. Understanding the temporal profile of aldosterone stimulation in the context of disease onset may provide future therapeutic insights for HFpEF (Table 1).
Table 1.
Current clinical trials, outcomes and recommendations
| Preliminary clinical studies suggest the utility of aldosterone antagonist drugs in treatment of patients with symptomatic HFpEF: | |
| Randomized Aldosterone Antagonism in Heart Failure with Preserved Ejection Fraction (RAAM-PEF) | [97] |
| |
| Neurohormonal Blockade and Outcomes in Diastolic Heart Failure (ongoing) | [98] |
| |
| Treatment Of Preserved Cardiac function heart failure with an Aldosterone antagonist Trial (TOPCAT) (ongoing) | [99] |
| |
Aldosterone-stimulated signaling pathways
Aldosterone regulates matrix disposition and promotes vascular dysfunction. It is known that aldosterone-stimulated cellular inflammation progresses to cardiac fibrosis through upregulation of plasminogen activator inhibitor-1 (PAI-1), first shown in rat aortic smooth muscle cells [21]. In cultured human bone marrow derived endothelial progenitor cells (EPCs) and in studies using mice, aldosterone treatment resulted in impaired differentiation, migration, and proliferation of EPCs, shown through increased protein kinase A (PKA)-dependent generation of ROS. EPCs isolated from patients with primary aldosteronism also showed decreased migration, compared to normal control EPCs [22]. Aldosterone appears to stimulate cardiac CaMKII oxidation and activation through ROS, generated by NADPH oxidase in cultured cardiomyocytes [9]. Therefore, aldosterone might recruit various signal transduction molecules, in a cell type specific manner. Determining the contextual modulation of aldosterone-stimulated signaling pathways will be important to building a more complete understanding of the cardiotoxic effects of aldosterone.
Aldosterone and MR action
Aldosterone and MR regulate the expression of numerous genes implicated in pathologic cardiac remodeling [21, 23–27], which can be inhibited by pharmacologic blockers of translation and transcription and/or by MR antagonist drugs. For instance, eplerenone treatment reduces both hypertension and pathological remodeling in spontaneously hypertrophic mice lacking guanylyl cyclase-A, the receptor for atrial and brain natriuretic peptides (ANP, BNP) [28]. Eplerenone treatment also decreased expression of ANP, BNP, TGF-β, Col I and Col II, implicating these proteins as either direct or indirect targets of aldosterone/MR. Importantly, administration of the vasodilator hydralazine reduced blood pressure without reversal of pathological remodeling, suggesting that MR is involved in the genetic reprogramming of remodeling that is independent of blood pressure control. One caution is that not all MR mediated gene transcription is through classic hormone response elements (HREs), as illustrated for example by the interaction between MR and calcineurin [29]. Another example is aldosterone stimulated ANP expression in neonatal rat cardiomyocytes through MR.s interaction with the transcription factor p300 and subsequent binding to the GATA4 regulatory element in the promoter of ANP [24].
In addition to pro-hypertrophic genes such as ANP, aldosterone also stimulates inflammatory markers. In DOCA-salt treated and uninephrectomized mice, administration of a pharmacologic antagonist drug to the chemokine receptor CXCR4 improved cellular damage, suggesting that CXCR4 is another downstream mediator of mineralocorticoid excess [30]. It is unknown whether MR stimulated CXCR4 involves direct promoter regulation through HREs. Even in the presence of identified HREs, such as for example PAI-1 [21], epidermal growth factor receptor (EGFR) [25], endothelin-1[26], and osteopontin [27], the mechanism of binding and the differential stimulation by mineralocorticoids versus glucocorticoids remain unclear and will require more investigation. Overall, aldosterone transcriptionally regulates many pro-inflammatory and pro-hypertrophic genes in the heart, through mechanisms that are under active research (Figure 3).
Figure 3. Schematic of the genomic and nongenomic actions of MR activation.
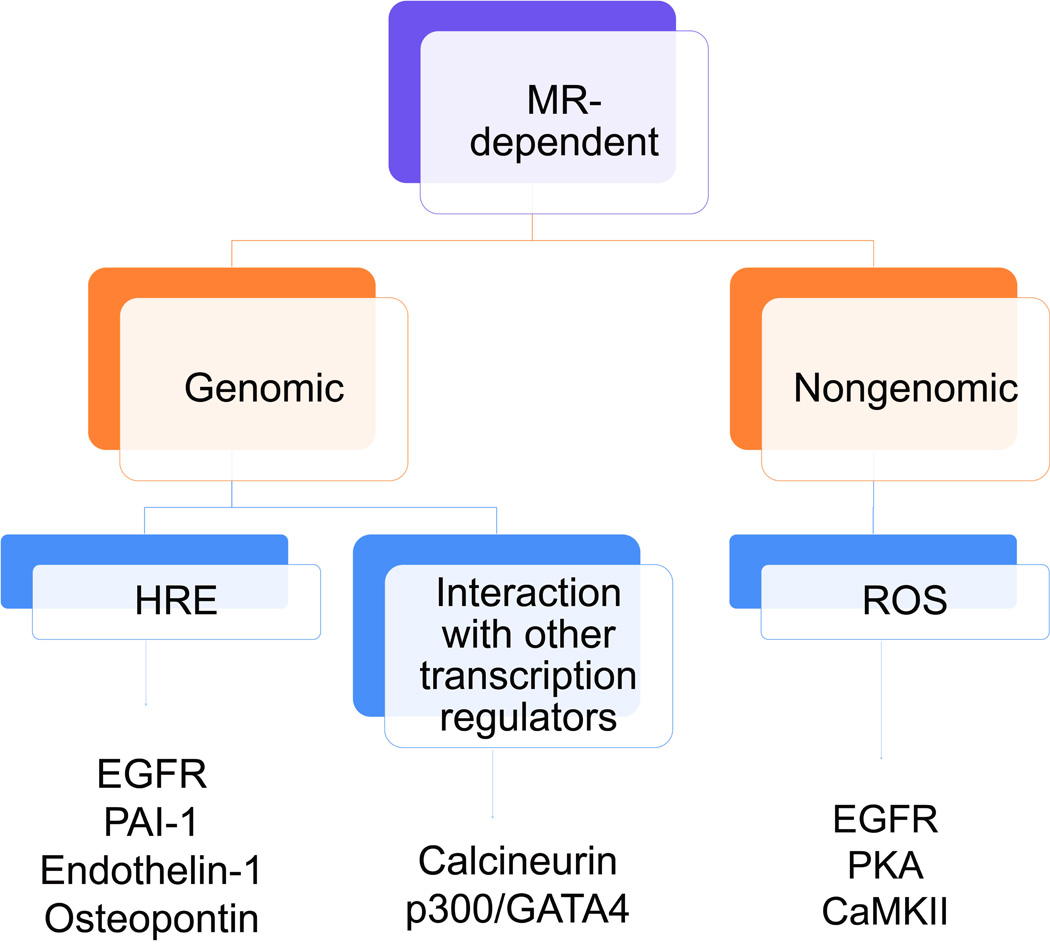
MR activation may lead to either genomic or nongenomic mechanisms of action. Genomic actions of MR may be further subdivided into binding to hormone response elements (HREs) in the promoter regions of target genes or to other transcription regulators. Shown here is subset of recently identified HRE-dependent genomic targets of MR: epidermal growth factor receptor (EGFR), plasminogen activator inhibitor-1 (PAI-1), endothelin-1, and osteopontin. Activated MR can associate with the transcription factor complex p300/GATA4 to drive upregulation of atrial natriuretic peptide [24]. Recent evidence shows that activated MR can also interact with calcineurin to promote its nuclear translocation and activation of the transcription factor nuclear factor of activated T cells [29]. Finally, highlighted here under nongenomic actions of MR is ROS production. Examples of ROS dependent signaling molecules that have been recently identified to be responsive to MR-generated ROS include EGFR, PKA, and CaMKII.
Regardless of mechanisms of action, MR blockade is cardiac protective, and there is increasing support for early MR antagonism after MI. Immediate MR blockade by administering eplerenone in the first post-operative week after MI in rats promoted early revascularization by mobilizing inflammatory cells, leading to reduced pathologic remodeling [31]. Aldosterone infusion in mice after MI led to cardiac rupture [9], an acute phenotype after MI that may benefit from early intervention with MR blockade. A multicenter, randomized clinical trial is underway to evaluate the utility of aldosterone receptor blockade in the immediate post-MI phase [32]. Therefore, aldosterone receptor blockade is cardiac protective in chronic CHF and may be important in the acute remodeling events after MI.
Aldosterone and the cardiac Na+/H+ exchanger
Another recently recognized aldosterone target is the cardiac Na+/H+ exchanger (NHE). Aldosterone increases NHE through MR-mediated, nongenomic transactivation of EGFR in rat ventricular myocytes (Figure 3). Interestingly, the mechanism for aldosterone stimulated NHE activity requires ROS (Box 1). NHE hyperactivity contributes to cardiac hypertrophy and heart failure through increases in intracellular calcium and sodium. Modulating NHE hyperactivity through EGFR inhibition may be another pharmaceutical approach to treating aldosterone-related cardiac disease [33]. The intermediary steps between MR activation and EGFR transactivation remain uncertain but seem to involve src-kinase phosphorylation and metalloproteinase recruitment. MR can also directly regulate EGFR transcription via promoter stimulation [25]. More details of MR and EGFR crosstalk can be found in a recent review [34]. The potential involvement of other intermediary ROS-dependent kinases, such as CaMKII or PKA, will require future research.
Lessons from the MR transcriptome
Recent studies have taken a more comprehensive approach to identifying aldosterone/MR regulated genes in cardiac cells. Latouche et al, in an elegantly designed study, compared the transcriptomes of MR versus GR activation in vitro and in vivo [35]. MR specific genes were confirmed to be upregulated either by aldosterone exposure in cultured cardiac H9C2 cells or by chronic MR overexpression in conditional, cardiomyocytes-specific transgenic mice. Interestingly, three genes are involved in matrix remodeling: Adamts1, a zinc metalloprotease involved in the acute phase response to experimental MI [36]; PAI-1, a well-known mediator of aldosterone stimulated fibrinolysis and inflammation [37] and Serpina-3, a serine protease inhibitor that modulates MMP9 activation [38]. The authors noted that while these and other pro-fibrotic genes are upregulated in the MR overexpressing mice, the phenotype remains mild until the addition of Ang II. The requirement for a second 'hit' is consistent with other studies [21, 23, 39–42] and our recent finding of aldosterone-worsened cardiac rupture in mice after MI [9]. The identification of these MR selective targets indicates that aldosterone/MR recruits multiple pathways to regulate the extracellular matrix and that phenotypic outcome is context dependent.
A follow-up study to this transcriptome analysis focused on one MR target, the neutrophil gelatinase-associated lipocalin 2 (NGAL/lipocalin 2) [43]. Aldosterone exposure increases NGAL/lipocalin 2 levels both in cultured cardiomyocytes and in uninephrectomized, sodium challenged mice. While NGAL/lipocalin 2 levels are elevated in heart failure, the source and consequence of this elevation remain unclear and will require more research. Collectively, it appears that aldosterone, through MR activation, regulates the expression of numerous genes related to heart failure and pathologic remodeling. The relative contribution of these targets to disease remains an active area of research.
Effects of moderate aldosterone elevation
The aforementioned models utilized extreme MR activation either through direct aldosterone exposure or MR overexpression. In contrast, moderate aldosterone elevation through transgenic overexpression of cardiomyocyte-specific aldosterone synthase (CYP11B2) in mice leads to coronary vessel dysfunction by decreasing the expression of the repolarizing calcium activated potassium channel BKCa, in coronary smooth muscle cells [44, 45]. Recently, moderate aldosterone excess in hypertensive mice was associated with a decrease in BNP [40] and ANP [41] expression. While the intermediary steps remain unclear, this process seems to require activated MR. BNP downregulation appears to underlie the pro-fibrotic effect of aldosterone excess in this model, which exhibits concurrent downregulation of bone morphogenic protein 4 (BMP-4) and increased inflammatory markers CD68 and galectin 3 [40]. ANP downregulation, in conjunction with downregulated β-myosin heavy chain, suggests suppressed fetal gene reprogramming, which is traditionally associated with cardiac hypertrophy and pathologic remodeling [41]. While these findings appear contradictory to previous observations, it is important to note that the model used in these studies is a double transgenic mouse with both aldosterone synthase and renin overexpression and therefore has moderate aldosterone as well as moderate Ang II excess. Aldosterone excess in this model retains its pro-fibrotic and pro-hypertrophic effects, despite natriuretic peptide downregulation. Interestingly, Lopez et al recently found that β-myosin heavy chain expression is limited to only a small subset of myocytes that resist hypertrophy after pressure overload [46]. Therefore, aldosterone stimulated hypertrophy may occur through a separate pathway that negatively feeds back to downregulate β-myosin heavy chain. These studies also reinforce the urgency of some unanswered questions in the field: what is the pathologically relevant dose of aldosterone exposure and what are the interactions of aldosterone with pre-existing, maladaptive conditions such as elevated Ang II?
We can glean insights into the first question from a study that showed biphasic, dose-dependent properties of aldosterone. Following pretreatment with aldosterone, human coronary microarteries exhibited a biphasic response to Ang II-induced vasoconstriction. Aldosterone at nanomolar levels potentiated Ang II-induced vasoconstriction, whereas this effect was absent at micromolar levels with concurrent eNOS phosphorylation and activation [47]. Whether this biphasic response applies to other situations of aldosterone excess requires further research.
Emerging evidence supports the existence of crosstalk between Ang II and aldosterone, for instance through aldosterone- and MR-dependent modulation of the Ang II receptor (AT1) expression [48] and through aldosterone-mediated AT1 transglutamination and potential dimerization [49]. Ang II infusion in mice with conditional, cardiomyocyte-delimited overexpression of human MR have worsened diastolic dysfunction compared to control mice, suggesting that the detrimental effects of Ang II and MR activation are additive [39]. This mouse model showed enhanced matrix turnover and fibrosis, indicated by increased activities of MMP2 and MMP9 with concurrently elevated NADPH oxidase-derived ROS. In an elegant study using siRNA mediated knockdown in vascular smooth muscle cells, Lemarie et al showed that aldosterone-stimulated activation of ERK1/2, NF-κB, and JNK, required AT1 [50], again arguing for the combined therapeutic value of dual Ang II and aldosterone blockade. The interrelationship between aldosterone and Ang II is reviewed in more detail elsewhere [51].
A role of aldosterone/MR in cardiac conduction
Increasing evidence supports an important role for aldosterone/MR in the generation of both atrial [52] and ventricular arrhythmias [53]. Aldosterone enhances the automaticity of cardiomyocytes through increases in transcription of the voltage gated T-type calcium channel α1H [54]. Treatment of neonatal rat cardiomyocytes with aldosterone upregulates the expression of the hyperpolarization activated I(f) channels, leading to increased excitability and predisposition to arrhythmias. These effects appear genomic and were blocked by the MR antagonists spironolactone and eplerenone, but were unaffected by the GR receptor antagonist RU-38486 [55]. MR activation has previously been shown to increase calcium influx in ventricular myocytes [56] and in vivo [53], prolonging repolarization and predisposing to early afterdepolarizations, a trigger for arrhythmias. MR blockade normalized post-MI pathological electrical remodeling in rat ventricular myocytes [57]. More recently, a relationship between aldosterone/MR and delayed afterdepolarizations was established in a study that showed aldosterone/MR can increase diastolic calcium leak from ryanodine receptors (RyRs) by down regulating the RyR stabilizing proteins FKBP12 and 12.6 [58]. The molecular steps between aldosterone/MR and FKBP downregulation will require more research.
Aldosterone and microRNA expression
Finally, aldosterone can regulate microRNA expression. Aldosterone exposure in neonatal rat cardiomyocytes increased expression of miR-23a, and knockdown of miR-23a prevented aldosterone stimulated cardiomyocyte hypertrophy [59]. This pioneering study expands the role of aldosterone stimulated pathways to include microRNAs and their targets. How aldosterone regulates the expression of this and other microRNAs requires further research. Interestingly, aldosterone downregulation of β-myosin heavy chain was found in association with decreased miR-208b, encoded intronically by the same gene [41]. MiR-208b deficiency led to increased levels of the transcriptional repressor Sox 6, further repressing β-myosin heavy chain transcription. This example resembles a feed forward mechanism of transcriptional control and suggests a previously underappreciated complexity of aldosterone- and MR-regulated gene expression.
MR activation versus aldosterone excess
A persistent question is with regards to MR activation versus aldosterone excess in patients. Low dose MR antagonist drugs are therapeutically valuable despite what have been proposed as mostly normal plasma aldosterone concentrations. In the absence of measured aldosterone levels in RALES and EPHESUS trials, the exact degree of aldosterone excess in these patients remains unknown. Nonetheless, there is clear prognostic value to elevated plasma aldosterone concentration in patients after MI. In a study of plasma aldosterone concentration and risk stratification in patients presenting with ST segment elevation myocardial infarction (STEMI), Beygui et al observed a positive correlation between aldosterone levels on admission and the risk of death [68]. In a separate study that includes a longer follow-up period, plasma aldosterone concentrations in patients presenting with either STEMI or non-STEMI positively correlated with mortality or heart failure-related hospitalization within a 5 year follow-up [69]. It is important to note that in both studies, the majority of patients had plasma aldosterone levels within normal ranges. The Endocrine Society has not recommended a formal cutoff value for plasma aldosterone in the diagnosis of primary aldosteronism [70] given the variability across laboratories and the heterogeneity of aldosterone levels in patients. In the absence of a baseline, it is uncertain if there is a relative increase in patients with poor prognosis after MI. Nonetheless, the stepwise increase in mortality in patients with relatively high plasma aldosterone concentrations suggest that 1) there may be an increased susceptibility to MR activation in these patients and/or 2) there may be enhanced sensitivity to basal MR activity. In either scenario, MR blockade would be beneficial.
Finally, it is well known that both aldosterone and glucocorticoids activate MR [71]. In a recent study, both cortisol and aldosterone increased infarct size and apoptotic index in a rat model of ischemia and reperfusion [72]. Spironolactone administration reduced infarct size and cell death under either excess cortisol or aldosterone, suggesting that blocking MR activation is sufficient to confer cardiac protection. The role of the spironolactone as an inverse agonist in this study further supports the importance of basal MR activity in pathologic states, but what is the principle driver of this basal MR activity? Given that circulating cortisol levels surpass aldosterone by several orders of magnitude, a premise is that aldosterone activation of MR requires the glucocorticoid inactivating enzyme 11β-hydroxysteroid dehydrogenase type 2 (11β-HSD2), whose expression in renal epithelium allows for selective mineralocorticoid driven MR activation in the kidney. 11β-HSD2 expression is present in endothelial cells and vascular smooth muscle cells, but the existence of cardiac 11β-HSD2 has been controversial with conflicting reports. In the original study that outlined the cloning and tissue distribution of human 11β-HSD2, northern blot analysis showed virtually no detectable signal from heart despite high expression in kidney, colon, and pancreas [73]. However, Lombes et al later reported11β-HSD2 expression in human heart both at the mRNA level by in situ hybridization and at the protein level by immunodetection [74]. The presence of 11β-HSD2 in cardiomyocytes remains an active area of debate, and a conservative interpretation of the current evidence is that 11β-HSD2 is expressed in cardiomyocytes but at low levels, permitting basal glucocorticoid occupancy of MR.
The next question is with regards to selective aldosterone activation of MR under stress. It has been previously reported that transgenic mice overexpressing cardiac-specific 11β-HSD2 spontaneously develop hypertrophy, fibrosis, heart failure, and premature death, arguing for a pathologic role of inappropriate aldosterone activation of MR through increased 11β-HSD2 activity [6]. Of note, 11β-HSD2 is redox sensitive due to coupling to NAD+/NADH conversion. Perturbed redox balance from increases in oxidant signaling reflected by increased NAD+ may trigger increased 11β-HSD2 activity. More recent evidence provides additional support for selective aldosterone activation of MR under stress. For instance, in a rat model of myocardial infarction, cardiac MR and 11β-HSD2 were found to be elevated by real time PCR compared to sham controls. Spironolactone treatment prevented MI-induced increase in MR and 11β-HSD2 [75]. In rat hearts subject to chronic intermittent hypoxia, there is upregulation of 11β-HSD2 mRNA [76]. In human atrial tissue from subjects with atrial fibrillation, there is increased expression of both MR and 11β-HSD2 compared to samples from subjects with normal sinus rhythm [77, 78]. Cardiac MR transcripts were also elevated in human subjects with end-stage heart failure [79]. Finally, concurrent upregulation of MR and 11β-HSD2 transcripts in subjects with hypertrophic cardiomyopathy [79] has also been reported. Additional strategies that allow for ligand specificity and for MR to distinguish between different ligands recently underwent extensive review [80]. Because different etiologies of heart disease seem to trigger different patterns of MR activation and/or 11β-HSD2 upregulation, better understanding of these patterns will be important for development of targeted therapeutics.
Paracrine actions of the heart
The heart is not a classic aldosterone/MR “target organ” like the kidney; however, the presence of the cardiac MR renders the heart a target organ of the endocrine system in general, regardless of ligand. Perhaps more appropriate terminology would be to designate the heart as an MR responsive organ. But is cardiac MR limited to stimulation from endocrine signals? Recent evidence suggests that cardiac cells can have paracrine effects.
More than a decade ago, the mRNA of CYP11B2, the cytochrome P450 enzyme that catalyzes the final three steps of aldosterone synthesis, was detected in the rat heart, setting the stage for local, cardiac steroidogenesis [89]. Later, Hayashi et al detected an increase in aldosterone gradient from the coronary sinus to the aortic root of patients after acute MI, suggesting local aldosterone production by the heart [90]. The concept of local aldosterone synthesis creating a microdomain of concentrated hormonal release, especially under pathologic conditions, seems attractive because it provides an explanation for aldosterone activated MR, despite low levels of cardiac 11β-HSD2. However, recent evidence suggests that the human heart is incapable of synthesizing either cortisol or aldosterone at baseline or under stress because of near-to-complete absence of key synthetic enzymes [79]. One explanation for these seemingly conflicting observations is that aldosterone is retained in the myocardium under certain conditions, and its release is triggered by pathologic stimuli resulting in an apparent local aldosterone increase. Another possible explanation is that aldosterone is delivered to the heart by circulating inflammatory cells, such as monocytes and macrophages, known to mediate cardiac fibrosis and hypertension, in animal studies, through MR activation [91]. Whether aldosterone is bound to the MR or another, yet-to-be identified receptor or a third binding partner, will require future research.
Supposing that aldosterone production occurs in heart, there is a potential for CaM kinases to regulate steroidogenesis. Increase in intracellular calcium, either induced by AT1 signaling or potassium influx, regulates aldosterone synthesis in the adrenal gland and CaM kinases have been implicated in this pathway. For instance, CaMKI overexpression in human adrenocortical cells increases CYP11B2 transcription, through phosphorylation of the CREB family of transcription factors [92]. More recently, the PPARγ agonist pioglitzone was found to suppress CYP11B2 expression, and therefore aldosterone synthesis by disrupting the CaMKI driven enhancement of CYP11B2 transcription in human adrenocortical carcinoma H295R cells [93]. Excess aldosterone in aldosterone producing adenomas involves increased CaMKIIβ expression [94], suggesting that CaMKII may also be an upstream trigger for aldosterone biosynthesis, in certain pathologic states. CaMKII participates in a positive feedback loop to increase Ang II stimulated aldosterone production through phosphorylation and activation of T-type Ca2+ channels [95]. In addition to transcriptional regulation of synthetic enzymes, CaMKII can also regulate aldosterone synthesis farther downstream. Older pharmacologic studies show that CaMKII enhances transport of cholesterol and aldosterone precursors to the mitochondria where aldosterone synthesis occurs [96]. While these studies are all in non-cardiac cells, the enzymatic machinery for CaM kinase regulated aldosterone synthesis appears to be intact in heart. This observation combined with studies outlining the activation of CaMKII by aldosterone [9] suggests that CaMKII may regulate aldosterone production in heart via a feed-forward mechanism.
Concluding Remarks and Future Perspectives
Accumulating evidence supports a concept where aldosterone plays a critical role in the pathogenesis of cardiovascular disease, including direct toxic actions on cardiomyocytes. There is recent interest in the role of aldosterone in arrhythmias and in systolic as well as diastolic forms of heart failure. The specific etiology of heart failure may be important in determining the therapeutic value of aldosterone and MR blockade. Aldosterone drives heart failure by recruiting different molecular pathways in mechanisms both MR-dependent and MR-independent. The activation of these pathways also depends on context and differs based on cell type and presence of co-morbidities such as hypertension. One of these MR-dependent, but nongenomic pathways is activation of CaMKII by ROS (Figures 2 and 3). Improved understanding of the molecular actions of aldosterone will feed into new, improved therapeutics including MR blockade in patients with HFpEF (TOPCAT trial, Table 1) or selective MR blockade based on comorbidities, aldosterone synthase inhibitors (Box 2) and targeted anti-oxidant therapies. In turn, lessons from the clinical studies will likely direct future research to address the mechanistic gaps.
Figure 2. Overview of aldosterone stimulated ROS generation.
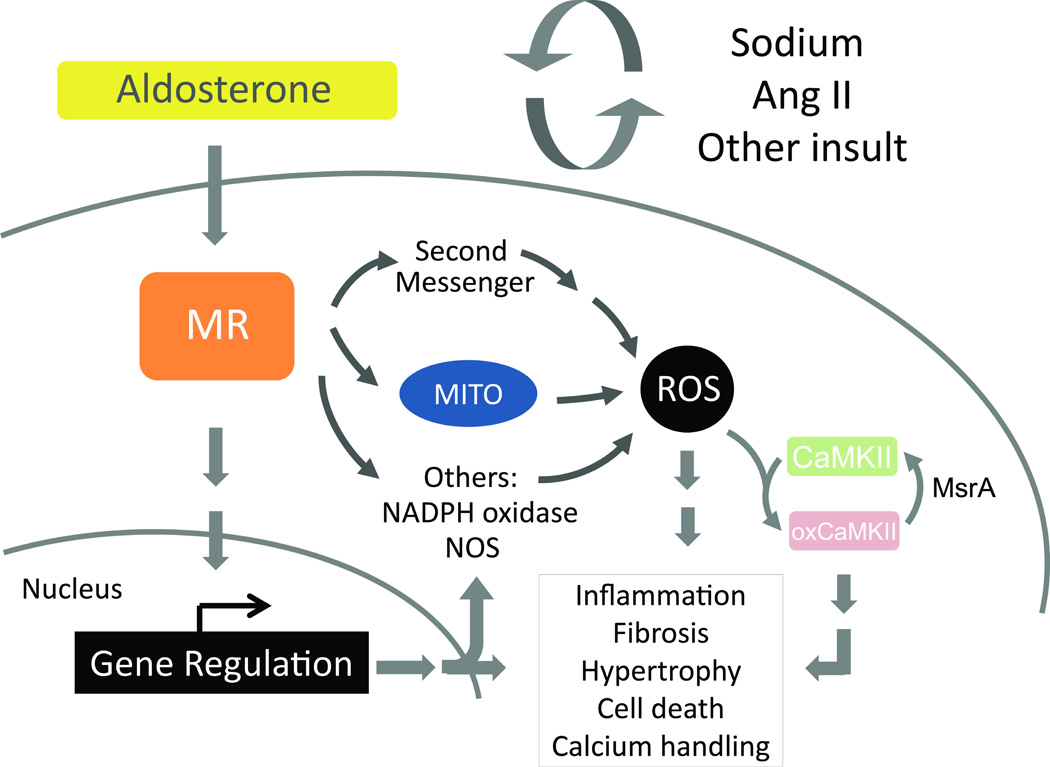
Aldosterone modulates multiple cellular processes through both genomic and nongenomic mechanisms. Highlighted here are aldosterone stimulated actions through reactive oxygen species (ROS) generation. Aldosterone/MR can stimulate ROS generation by recruiting second messenger signal transduction molecules such as PKA [22]. Aldosterone may also stimulate ROS production by the mitochondria (MITO) or by upregulation of subunits of NADPH oxidase and of nitric oxide synthase (NOS) [33, 65]. The cellular response to heightened aldosterone/MR signaling includes inflammation, fibrosis, pathologic cellular hypertrophy, cell death, and alterations to calcium handling, predisposing cardiomyocytes to arrhythmogenic firing. Also highlighted upstream here is the involvement of a second “hit,” where aldosterone/MR signaling is potentiated by enhanced sodium or Ang II or an additional pathologic stimulus. Finally, one downstream signaling switch activated by aldosterone/MR/ROS is the oxidation of CaMKII [9], which contributes to maladaptive cellular phenotypes. Reversal of CaMKII oxidation by methionine sulfoxide reductase A (MsrA) reflects one dynamic way of regulating excess ROS.
OUTSTANDING QUESTIONS BOX: Many questions remain on the mechanisms of aldosterone actions on cardiac tissue and will likely influence future work in the field.
What are the genomic versus nongenomic and sodium versus no sodium actions?
Do these actions depend on MR?
What is more important: MR activation or aldosterone excess?
What is the temporal profile of aldosterone-activated pathways in post-MI remodeling and progression to heart failure?
What are the interactions of aldosterone with pre-existing, maladaptive conditions such as elevated Ang II?
Acknowledgements
We acknowledge support by the US National Institutes of Health (R01HL70250, R01HL079031, R01HL113001 and R01HL096652 to M.E.A.) as well as a grant (08CVD01) from the Fondation Leducq as part of the ‘Alliance for CaMKII Signaling in Heart’.
GLOSSARY
- Aldosterone
a steroid hormone and a mineralocorticoid receptor (MR) agonist. Binding of aldosterone to MR induces MR nuclear translocation and binding of the receptor to promoter sequences on MR target genes that regulate fluid and electrolyte homeostasis, resulting in sodium retention and potassium excretion.
- Aldosterone antagonist drugs
MR antagonists, which include spironolactone and eplerenone
- Aldosterone escape phenomenon
used here to refer to a persistently elevated level of plasma aldosterone despite therapies to block upstream angiotensin
- Angiotensin II receptors (AT1)
G protein-coupled receptors that have angiotensin II as their ligands. AT1 mediate the signal transduction of the vasoconstricting property of angiotensin II.
- Ca2+/calmodulin-dependent protein kinase II (CaMKII)
a serine/threonine-specific protein kinase that is regulated by the Ca2+/calmodulin complex. CaMKII regulates excitation and contraction coupling in cardiomyocytes.
- Congestive heart failure (CHF)
CHF is a clinical syndrome of fluid overload exhibited by a constellation of symptoms including dyspnea with mild exertion or at rest, pulmonary congestion, and peripheral edema
- Heart failure with preserved ejection fraction (HFpEF)
a subset of CHF in the context of normal left ventricular volume and preserved ejection fraction. HFpEF may account for nearly 50% of CHF [100] and is associated with high rates of mortality and morbidity. The diagnosis, risk stratification, and treatment of HFpEF remain clinically challenging due to a lack of molecular understanding.
- Myocardial infarction (MI)
also referred to as heart attack, is a condition whereby interruption of blood supply to a region of the heart results in ischemia and subsequent cell death.
- Matrix metalloproteinase 9 (MMP9)
an enzyme involved in the breakdown of extracellular matrix in normal physiological and pathologic processes and a known culprit in cardiac rupture.
- Renin-angiotensin-aldosterone system (RAAS)
a hormone system that regulates blood pressure and fluid balance. Decreased perfusion pressure triggers the juxtaglomerular cells of the kidney to release renin into the blood stream. Circulating renin then cleaves angiotensinogen, a liver-derived serum protein, to angiotensin I, which is then cleaved into angiotensin II by angiotensin converting enzyme found in vascular endothelium. Finally, circulating angiotensin II triggers aldosterone synthesis and release primarily by the adrenal cortex.
- ST segment elevation myocardial infarction (STEMI)
a type of MI where there is occlusion of a coronary vessel with corresponding changes on electrocardiogram, typically defined as elevation of the ST-segment of greater than 0.1mV in at least two contiguous leads. Can also be a new left bundle branch block of the cardiac conduction system or a true posterior wall MI.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lopez AD, et al. Global and regional burden of disease and risk factors, 2001: systematic analysis of population health data. Lancet. 2006;367:1747–1757. doi: 10.1016/S0140-6736(06)68770-9. [DOI] [PubMed] [Google Scholar]
- 2.Pitt B, et al. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized Aldactone Evaluation Study Investigators. The New England journal of medicine. 1999;341:709–717. doi: 10.1056/NEJM199909023411001. [DOI] [PubMed] [Google Scholar]
- 3.Pitt B, et al. Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction. The New England journal of medicine. 2003;348:1309–1321. doi: 10.1056/NEJMoa030207. [DOI] [PubMed] [Google Scholar]
- 4.Zannad F, et al. Eplerenone in patients with systolic heart failure and mild symptoms. The New England journal of medicine. 2011;364:11–21. doi: 10.1056/NEJMoa1009492. [DOI] [PubMed] [Google Scholar]
- 5.Dorn GW., 2nd Novel pharmacotherapies to abrogate postinfarction ventricular remodeling. Nature reviews. Cardiology. 2009;6:283–291. doi: 10.1038/nrcardio.2009.12. [DOI] [PubMed] [Google Scholar]
- 6.Qin W, et al. Transgenic model of aldosterone-driven cardiac hypertrophy and heart failure. Circulation research. 2003;93:69–76. doi: 10.1161/01.RES.0000080521.15238.E5. [DOI] [PubMed] [Google Scholar]
- 7.Tsybouleva N, et al. Aldosterone, through novel signaling proteins, is a fundamental molecular bridge between the genetic defect and the cardiac phenotype of hypertrophic cardiomyopathy. Circulation. 2004;109:1284–1291. doi: 10.1161/01.CIR.0000121426.43044.2B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hayashi H, et al. Aldosterone nongenomically produces NADPH oxidase-dependent reactive oxygen species and induces myocyte apoptosis. Hypertension research : official journal of the Japanese Society of Hypertension. 2008;31:363–375. doi: 10.1291/hypres.31.363. [DOI] [PubMed] [Google Scholar]
- 9.He BJ, et al. Oxidation of CaMKII determines the cardiotoxic effects of aldosterone. Nature medicine. 2011;17:1610–1618. doi: 10.1038/nm.2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kushnir A, et al. Role of CaMKIIdelta phosphorylation of the cardiac ryanodine receptor in the force frequency relationship and heart failure. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:10274–10279. doi: 10.1073/pnas.1005843107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wu Y, et al. Calmodulin kinase II is required for fight or flight sinoatrial node physiology. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:5972–5977. doi: 10.1073/pnas.0806422106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Erickson JR, et al. A dynamic pathway for calcium-independent activation of CaMKII by methionine oxidation. Cell. 2008;133:462–474. doi: 10.1016/j.cell.2008.02.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Erickson JR, et al. CaMKII in the cardiovascular system: sensing redox states. Physiological reviews. 2011;91:889–915. doi: 10.1152/physrev.00018.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang R, et al. Calmodulin kinase II inhibition protects against structural heart disease. Nature medicine. 2005;11:409–417. doi: 10.1038/nm1215. [DOI] [PubMed] [Google Scholar]
- 15.Milliez P, et al. Evidence for an increased rate of cardiovascular events in patients with primary aldosteronism. Journal of the American College of Cardiology. 2005;45:1243–1248. doi: 10.1016/j.jacc.2005.01.015. [DOI] [PubMed] [Google Scholar]
- 16.Sam F, et al. Adiponectin deficiency, diastolic dysfunction, and diastolic heart failure. Endocrinology. 2010;151:322–331. doi: 10.1210/en.2009-0806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Martos R, et al. Diagnosis of heart failure with preserved ejection fraction: improved accuracy with the use of markers of collagen turnover. European journal of heart failure. 2009;11:191–197. doi: 10.1093/eurjhf/hfn036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Collier P, et al. Can emerging biomarkers of myocardial remodelling identify asymptomatic hypertensive patients at risk for diastolic dysfunction and diastolic heart failure? European journal of heart failure. 2011;13:1087–1095. doi: 10.1093/eurjhf/hfr079. [DOI] [PubMed] [Google Scholar]
- 19.Martos R, et al. Diastolic heart failure: evidence of increased myocardial collagen turnover linked to diastolic dysfunction. Circulation. 2007;115:888–895. doi: 10.1161/CIRCULATIONAHA.106.638569. [DOI] [PubMed] [Google Scholar]
- 20.Buralli S, et al. Circulating matrix metalloproteinase-3 and metalloproteinase-9 and tissue Doppler measures of diastolic dysfunction to risk stratify patients with systolic heart failure. The American journal of cardiology. 2010;105:853–856. doi: 10.1016/j.amjcard.2009.11.038. [DOI] [PubMed] [Google Scholar]
- 21.Brown NJ, et al. Synergistic effect of adrenal steroids and angiotensin II on plasminogen activator inhibitor-1 production. The Journal of clinical endocrinology and metabolism. 2000;85:336–344. doi: 10.1210/jcem.85.1.6305. [DOI] [PubMed] [Google Scholar]
- 22.Thum T, et al. Impairment of endothelial progenitor cell function and vascularization capacity by aldosterone in mice and humans. European heart journal. 2011;32:1275–1286. doi: 10.1093/eurheartj/ehq254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Peters J, et al. Lack of cardiac fibrosis in a new model of high prorenin hyperaldosteronism. American journal of physiology. Heart and circulatory physiology. 2009;297:H1845–H1852. doi: 10.1152/ajpheart.01135.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yoshida Y, et al. Aldosterone signaling associates with p300/GATA4 transcriptional pathway during the hypertrophic response of cardiomyocytes. Circulation journal : official journal of the Japanese Circulation Society. 2010;74:156–162. doi: 10.1253/circj.cj-09-0050. [DOI] [PubMed] [Google Scholar]
- 25.Grossmann C, et al. Aldosterone-induced EGFR expression: interaction between the human mineralocorticoid receptor and the human EGFR promoter. American journal of physiology. Endocrinology and metabolism. 2007;292:E1790–E1800. doi: 10.1152/ajpendo.00708.2006. [DOI] [PubMed] [Google Scholar]
- 26.Stow LR, et al. Aldosterone modulates steroid receptor binding to the endothelin-1 gene (edn1) The Journal of biological chemistry. 2009;284:30087–30096. doi: 10.1074/jbc.M109.030718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kiyosue A, et al. Aldosterone-induced osteopontin gene transcription in vascular smooth muscle cells involves glucocorticoid response element. Hypertension research : official journal of the Japanese Society of Hypertension. 2011;34:1283–1287. doi: 10.1038/hr.2011.119. [DOI] [PubMed] [Google Scholar]
- 28.Zhang Q, et al. The specific mineralocorticoid receptor blocker eplerenone attenuates left ventricular remodeling in mice lacking the gene encoding guanylyl cyclase-A. Hypertension research : official journal of the Japanese Society of Hypertension. 2008;31:1251–1256. doi: 10.1291/hypres.31.1251. [DOI] [PubMed] [Google Scholar]
- 29.Seiferth A, et al. The phosphatase calcineurin PP2BAbeta mediates part of mineralocorticoid receptor transcriptional activity. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2012;26:2327–2337. doi: 10.1096/fj.11-199976. [DOI] [PubMed] [Google Scholar]
- 30.Chu PY, et al. CXCR4 antagonism attenuates the cardiorenal consequences of mineralocorticoid excess. Circulation. Heart failure. 2011;4:651–658. doi: 10.1161/CIRCHEARTFAILURE.110.960831. [DOI] [PubMed] [Google Scholar]
- 31.Fraccarollo D, et al. Immediate mineralocorticoid receptor blockade improves myocardial infarct healing by modulation of the inflammatory response. Hypertension. 2008;51:905–914. doi: 10.1161/HYPERTENSIONAHA.107.100941. [DOI] [PubMed] [Google Scholar]
- 32.Beygui F, et al. Rationale for an early aldosterone blockade in acute myocardial infarction and design of the ALBATROSS trial. American heart journal. 2010;160:642–648. doi: 10.1016/j.ahj.2010.06.049. [DOI] [PubMed] [Google Scholar]
- 33.De Giusti VC, et al. Aldosterone stimulates the cardiac Na(+)/H(+) exchanger via transactivation of the epidermal growth factor receptor. Hypertension. 2011;58:912–919. doi: 10.1161/HYPERTENSIONAHA.111.176024. [DOI] [PubMed] [Google Scholar]
- 34.Grossmann C, Gekle M. Interaction between mineralocorticoid receptor and epidermal growth factor receptor signaling. Molecular and cellular endocrinology. 2012;350:235–241. doi: 10.1016/j.mce.2011.07.045. [DOI] [PubMed] [Google Scholar]
- 35.Latouche C, et al. Molecular signature of mineralocorticoid receptor signaling in cardiomyocytes: from cultured cells to mouse heart. Endocrinology. 2010;151:4467–4476. doi: 10.1210/en.2010-0237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nakamura K, et al. Dynamic induction of ADAMTS1 gene in the early phase of acute myocardial infarction. Journal of biochemistry. 2004;136:439–446. doi: 10.1093/jb/mvh138. [DOI] [PubMed] [Google Scholar]
- 37.Sawathiparnich P, et al. Spironolactone abolishes the relationship between aldosterone and plasminogen activator inhibitor-1 in humans. The Journal of clinical endocrinology and metabolism. 2002;87:448–452. doi: 10.1210/jcem.87.2.7980. [DOI] [PubMed] [Google Scholar]
- 38.Han YP, et al. Proteolytic activation of matrix metalloproteinase-9 in skin wound healing is inhibited by alpha-1-antichymotrypsin. The Journal of investigative dermatology. 2008;128:2334–2342. doi: 10.1038/jid.2008.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Di Zhang A, et al. Cross-talk between mineralocorticoid and angiotensin II signaling for cardiac remodeling. Hypertension. 2008;52:1060–1067. doi: 10.1161/HYPERTENSIONAHA.108.117531. [DOI] [PubMed] [Google Scholar]
- 40.Azibani F, et al. Aldosterone inhibits antifibrotic factors in mouse hypertensive heart. Hypertension. 2012;59:1179–1187. doi: 10.1161/HYPERTENSIONAHA.111.190512. [DOI] [PubMed] [Google Scholar]
- 41.Azibani F, et al. Aldosterone inhibits the fetal program and increases hypertrophy in the heart of hypertensive mice. PloS one. 2012;7:e38197. doi: 10.1371/journal.pone.0038197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ruhs S, et al. Modulation of transcriptional mineralocorticoid receptor activity by nitrosative stress. Free radical biology & medicine. 2012;53:1088–1100. doi: 10.1016/j.freeradbiomed.2012.06.028. [DOI] [PubMed] [Google Scholar]
- 43.Latouche C, et al. Neutrophil gelatinase-associated lipocalin is a novel mineralocorticoid target in the cardiovascular system. Hypertension. 2012;59:966–972. doi: 10.1161/HYPERTENSIONAHA.111.187872. [DOI] [PubMed] [Google Scholar]
- 44.Ambroisine ML, et al. Aldosterone-induced coronary dysfunction in transgenic mice involves the calcium-activated potassium (BKCa) channels of vascular smooth muscle cells. Circulation. 2007;116:2435–2443. doi: 10.1161/CIRCULATIONAHA.107.722009. [DOI] [PubMed] [Google Scholar]
- 45.Benard L, et al. Effects of aldosterone on coronary function. Pharmacological reports : PR. 2009;61:58–66. doi: 10.1016/s1734-1140(09)70007-6. [DOI] [PubMed] [Google Scholar]
- 46.Lopez JE, et al. beta-myosin heavy chain is induced by pressure overload in a minor subpopulation of smaller mouse cardiac myocytes. Circulation research. 2011;109:629–638. doi: 10.1161/CIRCRESAHA.111.243410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Batenburg WW, et al. Angiotensin II-aldosterone interaction in human coronary microarteries involves GPR30, EGFR, and endothelial NO synthase. Cardiovascular research. 2012;94:136–143. doi: 10.1093/cvr/cvs016. [DOI] [PubMed] [Google Scholar]
- 48.Bogdarina IG, et al. Characterization of the angiotensin (AT1b) receptor promoter and its regulation by glucocorticoids. Journal of molecular endocrinology. 2009;43:73–80. doi: 10.1677/JME-09-0036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yamada M, et al. Vasoconstrictor effect of aldosterone via angiotensin II type 1 (AT1) receptor: possible role of AT1 receptor dimerization. Cardiovascular research. 2008;79:169–178. doi: 10.1093/cvr/cvn064. [DOI] [PubMed] [Google Scholar]
- 50.Lemarie CA, et al. Aldosterone-induced activation of signaling pathways requires activity of angiotensin type 1a receptors. Circulation research. 2009;105:852–859. doi: 10.1161/CIRCRESAHA.109.196576. [DOI] [PubMed] [Google Scholar]
- 51.Lemarie CA, et al. New insights on signaling cascades induced by cross-talk between angiotensin II and aldosterone. Journal of molecular medicine. 2008;86:673–678. doi: 10.1007/s00109-008-0323-5. [DOI] [PubMed] [Google Scholar]
- 52.Reil JC, et al. Aldosterone promotes atrial fibrillation. European heart journal. 2012;33:2098–2108. doi: 10.1093/eurheartj/ehr266. [DOI] [PubMed] [Google Scholar]
- 53.Ouvrard-Pascaud A, et al. Conditional mineralocorticoid receptor expression in the heart leads to life-threatening arrhythmias. Circulation. 2005;111:3025–3033. doi: 10.1161/CIRCULATIONAHA.104.503706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lalevee N, et al. Aldosterone increases T-type calcium channel expression and in vitro beating frequency in neonatal rat cardiomyocytes. Cardiovascular research. 2005;67:216–224. doi: 10.1016/j.cardiores.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 55.Muto T, et al. Aldosterone modulates I(f) current through gene expression in cultured neonatal rat ventricular myocytes. American journal of physiology. Heart and circulatory physiology. 2007;293:H2710–H2718. doi: 10.1152/ajpheart.01399.2006. [DOI] [PubMed] [Google Scholar]
- 56.Benitah JP, Vassort G. Aldosterone upregulates Ca(2+) current in adult rat cardiomyocytes. Circulation research. 1999;85:1139–1145. doi: 10.1161/01.res.85.12.1139. [DOI] [PubMed] [Google Scholar]
- 57.Perrier E, et al. Mineralocorticoid receptor antagonism prevents the electrical remodeling that precedes cellular hypertrophy after myocardial infarction. Circulation. 2004;110:776–783. doi: 10.1161/01.CIR.0000138973.55605.38. [DOI] [PubMed] [Google Scholar]
- 58.Gomez AM, et al. Mineralocorticoid modulation of cardiac ryanodine receptor activity is associated with downregulation of FK506-binding proteins. Circulation. 2009;119:2179–2187. doi: 10.1161/CIRCULATIONAHA.108.805804. [DOI] [PubMed] [Google Scholar]
- 59.Lin Z, et al. miR-23a functions downstream of NFATc3 to regulate cardiac hypertrophy. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:12103–12108. doi: 10.1073/pnas.0811371106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tsutsui H, et al. Oxidative stress and heart failure. American journal of physiology. Heart and circulatory physiology. 2011;301:H2181–H2190. doi: 10.1152/ajpheart.00554.2011. [DOI] [PubMed] [Google Scholar]
- 61.Lonn E, et al. Effects of long-term vitamin E supplementation on cardiovascular events and cancer: a randomized controlled trial. JAMA : the journal of the American Medical Association. 2005;293:1338–1347. doi: 10.1001/jama.293.11.1338. [DOI] [PubMed] [Google Scholar]
- 62.Heart Protection Study Collaborative G. MRC/BHF Heart Protection Study of antioxidant vitamin supplementation in 20,536 high-risk individuals: a randomised placebo-controlled trial. Lancet. 2002;360:23–33. doi: 10.1016/S0140-6736(02)09328-5. [DOI] [PubMed] [Google Scholar]
- 63.Jauslin ML, et al. Mitochondria-targeted antioxidants protect Friedreich Ataxia fibroblasts from endogenous oxidative stress more effectively than untargeted antioxidants. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2003;17:1972–1974. doi: 10.1096/fj.03-0240fje. [DOI] [PubMed] [Google Scholar]
- 64.Chen CH, et al. Activation of aldehyde dehydrogenase-2 reduces ischemic damage to the heart. Science. 2008;321:1493–1495. doi: 10.1126/science.1158554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Favre J, et al. Coronary endothelial dysfunction after cardiomyocyte-specific mineralocorticoid receptor overexpression. American journal of physiology. Heart and circulatory physiology. 2011;300:H2035–H2043. doi: 10.1152/ajpheart.00552.2010. [DOI] [PubMed] [Google Scholar]
- 66.Ohmine T, et al. The involvement of aldosterone in cyclic stretch-mediated activation of NADPH oxidase in vascular smooth muscle cells. Hypertension research : official journal of the Japanese Society of Hypertension. 2009;32:690–699. doi: 10.1038/hr.2009.76. [DOI] [PubMed] [Google Scholar]
- 67.Stas S, et al. Mineralocorticoid receptor blockade attenuates chronic overexpression of the renin-angiotensin-aldosterone system stimulation of reduced nicotinamide adenine dinucleotide phosphate oxidase and cardiac remodeling. Endocrinology. 2007;148:3773–3780. doi: 10.1210/en.2006-1691. [DOI] [PubMed] [Google Scholar]
- 68.Beygui F, et al. High plasma aldosterone levels on admission are associated with death in patients presenting with acute ST-elevation myocardial infarction. Circulation. 2006;114:2604–2610. doi: 10.1161/CIRCULATIONAHA.106.634626. [DOI] [PubMed] [Google Scholar]
- 69.Palmer BR, et al. Plasma aldosterone levels during hospitalization are predictive of survival post-myocardial infarction. European heart journal. 2008;29:2489–2496. doi: 10.1093/eurheartj/ehn383. [DOI] [PubMed] [Google Scholar]
- 70.Funder JW, et al. Case detection, diagnosis, and treatment of patients with primary aldosteronism: an endocrine society clinical practice guideline. The Journal of clinical endocrinology and metabolism. 2008;93:3266–3281. doi: 10.1210/jc.2008-0104. [DOI] [PubMed] [Google Scholar]
- 71.Sheppard KE, Funder JW. Equivalent affinity of aldosterone and corticosterone for type I receptors in kidney and hippocampus: direct binding studies. Journal of steroid biochemistry. 1987;28:737–742. doi: 10.1016/0022-4731(87)90406-7. [DOI] [PubMed] [Google Scholar]
- 72.Mihailidou AS, et al. Glucocorticoids activate cardiac mineralocorticoid receptors during experimental myocardial infarction. Hypertension. 2009;54:1306–1312. doi: 10.1161/HYPERTENSIONAHA.109.136242. [DOI] [PubMed] [Google Scholar]
- 73.Albiston AL, et al. Cloning and tissue distribution of the human 11 beta-hydroxysteroid dehydrogenase type 2 enzyme. Molecular and cellular endocrinology. 1994;105:R11–R17. doi: 10.1016/0303-7207(94)90176-7. [DOI] [PubMed] [Google Scholar]
- 74.Lombes M, et al. Prerequisite for cardiac aldosterone action. Mineralocorticoid receptor and 11 beta-hydroxysteroid dehydrogenase in the human heart. Circulation. 1995;92:175–182. doi: 10.1161/01.cir.92.2.175. [DOI] [PubMed] [Google Scholar]
- 75.Takeda M, et al. Spironolactone modulates expressions of cardiac mineralocorticoid receptor and 11beta-hydroxysteroid dehydrogenase 2 and prevents ventricular remodeling in post-infarct rat hearts. Hypertension research : official journal of the Japanese Society of Hypertension. 2007;30:427–437. doi: 10.1291/hypres.30.427. [DOI] [PubMed] [Google Scholar]
- 76.Klusonova P, et al. Chronic intermittent hypoxia induces 11beta-hydroxysteroid dehydrogenase in rat heart. Endocrinology. 2009;150:4270–4277. doi: 10.1210/en.2008-1493. [DOI] [PubMed] [Google Scholar]
- 77.De-An P, et al. Increased expression of mineralocorticoid receptor and 11beta-hydroxysteroid dehydrogenase type 2 in human atria during atrial fibrillation. Clinical cardiology. 2010;33:23–29. doi: 10.1002/clc.20689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tsai CT, et al. Increased expression of mineralocorticoid receptor in human atrial fibrillation and a cellular model of atrial fibrillation. Journal of the American College of Cardiology. 2010;55:758–770. doi: 10.1016/j.jacc.2009.09.045. [DOI] [PubMed] [Google Scholar]
- 79.Chai W, et al. Steroidogenesis vs. steroid uptake in the heart: do corticosteroids mediate effects via cardiac mineralocorticoid receptors? Journal of hypertension. 2010;28:1044–1053. doi: 10.1097/HJH.0b013e328335c381. [DOI] [PubMed] [Google Scholar]
- 80.Fuller PJ, et al. Mechanisms of ligand specificity of the mineralocorticoid receptor. The Journal of endocrinology. 2012;213:15–24. doi: 10.1530/JOE-11-0372. [DOI] [PubMed] [Google Scholar]
- 81.Calhoun DA, et al. Effects of a novel aldosterone synthase inhibitor for treatment of primary hypertension: results of a randomized, double-blind, placebo- and active-controlled phase 2 trial. Circulation. 2011;124:1945–1955. doi: 10.1161/CIRCULATIONAHA.111.029892. [DOI] [PubMed] [Google Scholar]
- 82.Amar L, et al. Aldosterone synthase inhibition with LCI699: a proof-of-concept study in patients with primary aldosteronism. Hypertension. 2010;56:831–838. doi: 10.1161/HYPERTENSIONAHA.110.157271. [DOI] [PubMed] [Google Scholar]
- 83.Gamliel-Lazarovich A, et al. FAD286, an aldosterone synthase inhibitor, reduced atherosclerosis and inflammation in apolipoprotein E-deficient mice. Journal of hypertension. 2010;28:1900–1907. doi: 10.1097/HJH.0b013e32833c2197. [DOI] [PubMed] [Google Scholar]
- 84.Mulder P, et al. Aldosterone synthase inhibition improves cardiovascular function and structure in rats with heart failure: a comparison with spironolactone. European heart journal. 2008;29:2171–2179. doi: 10.1093/eurheartj/ehn277. [DOI] [PubMed] [Google Scholar]
- 85.Keidar S, Gamliel-Lazarovich A. Letter by keidar and gamliel-lazarovich regarding article, "effects of a novel aldosterone synthase inhibitor for treatment of primary hypertension: results of a randomized, double-blind, placebo- and active-controlled phase 2 trial". Circulation. 2012;125:e653. doi: 10.1161/CIRCULATIONAHA.111.080341. [DOI] [PubMed] [Google Scholar]
- 86.LaSala D, et al. Coexpression of CYP11B2 or CYP11B1 with adrenodoxin and adrenodoxin reductase for assessing the potency and selectivity of aldosterone synthase inhibitors. Analytical biochemistry. 2009;394:56–61. doi: 10.1016/j.ab.2009.07.025. [DOI] [PubMed] [Google Scholar]
- 87.Adams CM, et al. The discovery of potent inhibitors of aldosterone synthase that exhibit selectivity over 11-beta-hydroxylase. Bioorganic & medicinal chemistry letters. 2010;20:4324–4327. doi: 10.1016/j.bmcl.2010.06.086. [DOI] [PubMed] [Google Scholar]
- 88.Roumen L, et al. Synthesis, biological evaluation, and molecular modeling of 1-benzyl-1H-imidazoles as selective inhibitors of aldosterone synthase (CYP11B2) Journal of medicinal chemistry. 2010;53:1712–1725. doi: 10.1021/jm901356d. [DOI] [PubMed] [Google Scholar]
- 89.Silvestre JS, et al. Myocardial production of aldosterone and corticosterone in the rat. Physiological regulation. The Journal of biological chemistry. 1998;273:4883–4891. doi: 10.1074/jbc.273.9.4883. [DOI] [PubMed] [Google Scholar]
- 90.Hayashi M, et al. Relationship between transcardiac extraction of aldosterone and left ventricular remodeling in patients with first acute myocardial infarction: extracting aldosterone through the heart promotes ventricular remodeling after acute myocardial infarction. Journal of the American College of Cardiology. 2001;38:1375–1382. doi: 10.1016/s0735-1097(01)01539-x. [DOI] [PubMed] [Google Scholar]
- 91.Usher MG, et al. Myeloid mineralocorticoid receptor controls macrophage polarization and cardiovascular hypertrophy and remodeling in mice. The Journal of clinical investigation. 2010;120:3350–3364. doi: 10.1172/JCI41080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Condon JC, et al. Calmodulin-dependent kinase I regulates adrenal cell expression of aldosterone synthase. Endocrinology. 2002;143:3651–3657. doi: 10.1210/en.2001-211359. [DOI] [PubMed] [Google Scholar]
- 93.Uruno A, et al. Peroxisome proliferator-activated receptor-{gamma} suppresses CYP11B2 expression and aldosterone production. Journal of molecular endocrinology. 2011;46:37–49. doi: 10.1677/JME-10-0088. [DOI] [PubMed] [Google Scholar]
- 94.Lenzini L, et al. Heterogeneity of aldosterone-producing adenomas revealed by a whole transcriptome analysis. Hypertension. 2007;50:1106–1113. doi: 10.1161/HYPERTENSIONAHA.107.100438. [DOI] [PubMed] [Google Scholar]
- 95.Yao J, et al. Molecular basis for the modulation of native T-type Ca2+ channels in vivo by Ca2+/calmodulin-dependent protein kinase II. The Journal of clinical investigation. 2006;116:2403–2412. doi: 10.1172/JCI27918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Pezzi V, et al. Role of calmodulin-dependent protein kinase II in the acute stimulation of aldosterone production. The Journal of steroid biochemistry and molecular biology. 1996;58:417–424. doi: 10.1016/0960-0760(96)00052-0. [DOI] [PubMed] [Google Scholar]
- 97.Deswal A, et al. Results of the Randomized Aldosterone Antagonism in Heart Failure with Preserved Ejection Fraction trial (RAAM-PEF) Journal of cardiac failure. 2011;17:634–642. doi: 10.1016/j.cardfail.2011.04.007. [DOI] [PubMed] [Google Scholar]
- 98.Zhang Y, et al. Design and rationale of studies of neurohormonal blockade and outcomes in diastolic heart failure using OPTIMIZE-HF registry linked to Medicare data. International journal of cardiology. 2011 doi: 10.1016/j.ijcard.2011.10.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Desai AS, et al. Rationale and design of the treatment of preserved cardiac function heart failure with an aldosterone antagonist trial: a randomized, controlled study of spironolactone in patients with symptomatic heart failure and preserved ejection fraction. American heart journal. 2011;162:966–972. e910. doi: 10.1016/j.ahj.2011.09.007. [DOI] [PubMed] [Google Scholar]
- 100.Maeder MT, Kaye DM. Heart failure with normal left ventricular ejection fraction. Journal of the American College of Cardiology. 2009;53:905–918. doi: 10.1016/j.jacc.2008.12.007. [DOI] [PubMed] [Google Scholar]