

Graphical abstract

Keywords: Malaria, Kinase, Cellular assay, Cyclic amines, Pharmacokinetics, Resistant strains
Abstract
Screening our in-house compound collection using a cell based Plasmodium falciparum proliferation assay we discovered a known pan-kinase inhibitor scaffold as a hit. Further optimization of this series led us to a novel benzamide scaffold which was devoid of human kinase activity while retaining its antiplasmodial activity. The evolution of this compound series leading to optimized candidates with good cellular potency against multiple strains as well as decent in vivo profile is described in this Letter.
Malaria is an infectious disease that profoundly affects many developing countries. With hundreds of million cases and one million deaths each year, malaria poses a tremendous health and economic burden to the affected regions.1 There is still no effective antimalarial vaccine available and we still heavily depend on low molecular weight entities to treat the affected population. Quinine, chloroquine, mefloquine and artemisinin derivatives have play an important role in the treatment of malaria. However, widespread drug resistance has made many of these compounds less effective. Artemisinin is the only anti-malarial for which there are yet no reported cases of clinical resistance. However, parasite tolerance to artemisinin has been observed recently2 and it seems likely that resistance will emerge soon. Therefore, it is important to discover new chemotherapies that are effective against the multi-drug resistant parasite strains.3 In this Letter, we discuss an effort4 to find and optimize novel antimalarial entities using a cell-based screening strategy.
Currently there is a need for novel chemical scaffolds with different mechanisms of action, since most of the current approved antimalarial drugs belong to the aminoquinoline family. In order to find new chemical scaffolds, we initiated a compound screen using our in-house kinase inhibitor collection and subjected them to a cell-based of Plasmodium falciparum proliferation assay5,6 We envisioned that the hits arising from this screen can be rapidly optimized by leveraging our past experiences with these compounds series in alternative target-classes/indications. Furthermore, we thought that it would be prudent to remove the human kinase activity early on during the compound optimization phase to negate the possibility of toxicity arising from host-related off-target activities (Scheme 1).
Scheme 1.

Proprietary kinase scaffolds offered hits: piperidine benzamides.
Our starting point is compound 1, which was originally designed as a pan-kinase Bcr-Abl inhibitor7,8 Compound 1 shows a moderate EC50 of 200 nM against the chloroquine sensitive 3D7 P. falciparum parasite strain. By switching the solubility enhancing group, compound 2 exhibits a ∼3-fold improvement in potency. When the 2-methyl group in the left phenyl ring of 2 is replaced with a 3-methoxy group, compound 3 is obtained which is equipotent on malaria parasite. In addition, 3 no longer has any human kinase activities of 2, as measured in a Ba/F3 transformed cell-line RTK panel9 Since preserving the pharmocophore necessary for inhibiting human kinases is not necessary, we speculated that the benzamide portion of the molecule might be responsible for the antimalarial activity of 3 and decided initiate a broad SAR investigation.
Schemes 2–4 describe the synthetic strategies used to study the three key portions of compound 3. Scheme 2 outlines the synthesis of the amide connectivity of 3. The synthesis starts from 3-fluoro-5-(trifluoromethyl)benzonitrile. A SNAr reaction followed with a H2SO4 mediated hydrolysis provides the acid in good yield. The amide bond formation is executed using various amines and HATU as the activating agent. We were satisfied with the straightforward synthesis for these compounds given the need for low cost of goods is one of the essential criteria for the antimalarial target product profile (TPP).12
Scheme 2.
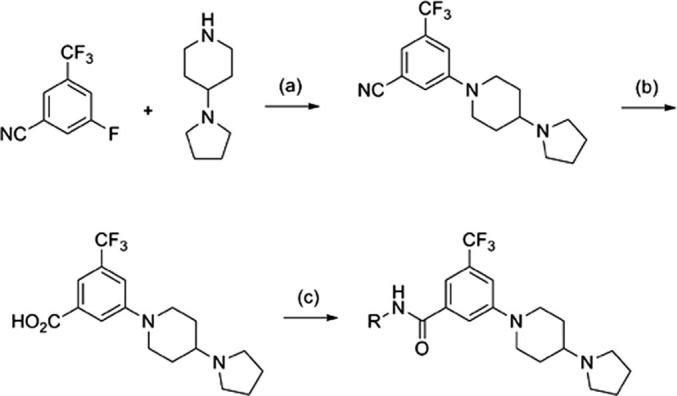
General synthesis scheme of piperidyl benzamides amides. Reagents and conditions: (a) K2CO3, DMSO, 80 °C; (b) 50% H2SO4, reflux, 78%, 2 steps; (c) RNH2, HATU, DIEA, DMF, 23 °C, 60–70%.
Scheme 3.
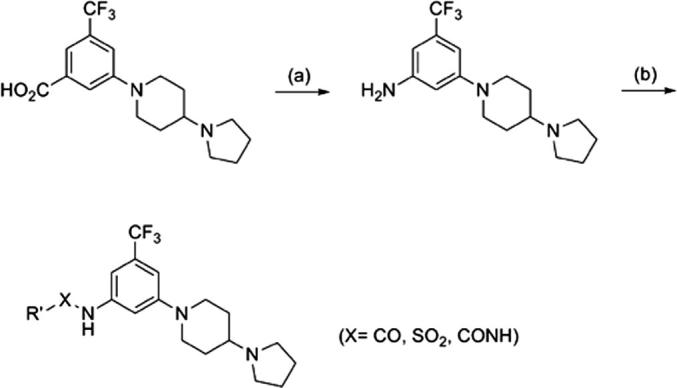
General synthesis scheme of piperidyl benzamides reverse amides, sulfonamides and ureas. Reagents and conditions: (a) (i) DPPA, Et3N, t-BuOH, reflux; (ii) TFA, DCM, 23 °C, 33%; (b) R′CO2H, HATU, DIEA, DMF, 23 °C; or R′SO2Cl, DIEA, CH2Cl2; or R′NCO, toluene, 80 °C, 40–80%.
Scheme 4.

General synthesis scheme of piperidyl benzamides amine SAR. Reagents and conditions: (a) (i) SOCl2, CHCl3, reflux; (ii) aniline, pyridine, 72–79%; (b) Pd2(dba)3, BINAP, t-BuOK, toluene, 100 °C, 45–84%.
Scheme 3 outlines the synthesis for the reverse amides, sulfonamides and ureas. Curtius rearrangement of the corresponding benzoic acids illustrated in Scheme 2 serves as the key step in the aniline synthetic route.
Scheme 4 outlines the synthesis for determining the SAR on the 3-position on CF3 bearing phenyl ring. We started with commercially available 3-bromo-5-(trifluoromethyl)benzoic acid and carried out a palladium catalyzed amination reaction on a variety of substrates to afford the final compounds in moderate to good yields.
Table 1 outline our SAR determination on the amide portion of compound 3. While linear alkyl (compound 4) and pyrimidines (compound 5) are incompatible, a wide variety substituted phenyl groups are well tolerated. The substitution pattern favors meta- and para- mono-substitution, with 3,4-disubstitution (compounds 11) and 3,5-disubstitution (compound 13) modestly enhancing potency as well.
Table 1.
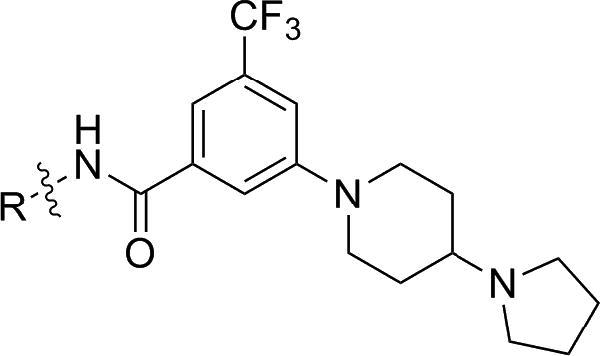
| Compds | R | P. falciparum 3D7 strain EC50, μMa |
|---|---|---|
| 4 |  |
6.48 |
| 5 | 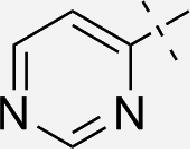 |
2.16 |
| 6 | 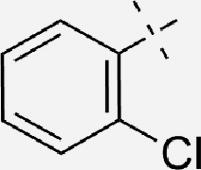 |
1.72 |
| 7 | 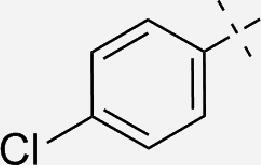 |
0.114 |
| 8 | 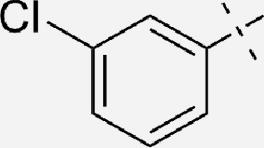 |
0.134 |
| 9 | 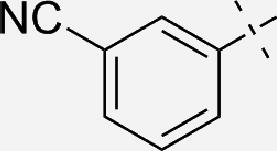 |
0.146 |
| 10 | 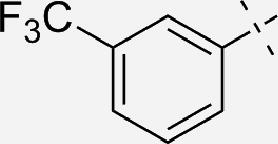 |
0.174 |
| 11 | 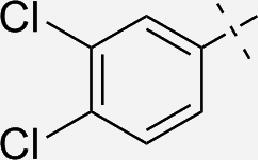 |
0.054 |
| 12 | 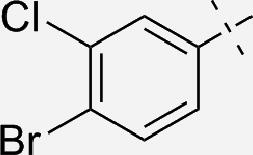 |
0.098 |
| 13 | 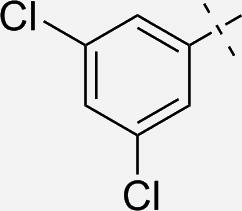 |
0.048 |
Values are means of two experiments. Each assay plate has mefloquine, sulfadoxine and artimesinin as internal standards. The EC50 values for standard compounds match the literature values.
Table 2 summarizes how varying the amide linkage affects the potency using compound 8 and 10 as our template since they were potent starting points in our early SAR. The sulfonamide (compound 16) was not tolerated nor was removal of the carbonyl and replacement with a methylene unit (compound 20). A free amide NH was essential for activity as the N-methyl compound loses potency by 20-fold (compound 17). Ureas do not seem to offer any advantage over amides, moreover both amide orientations (compound 10 vs 14) are tolerated and offer advantage in terms of physiochemical properties (e.g., improved solubility). Therefore we decided to explore additional structure–activity relationships with the amide in place.
Table 2.
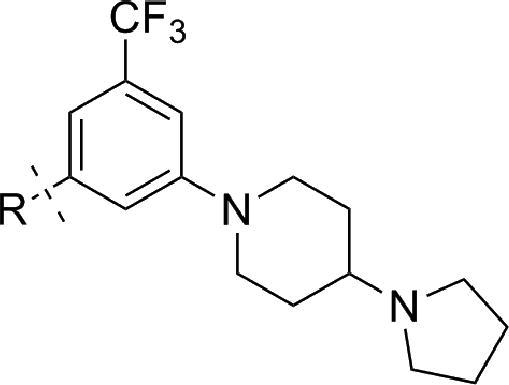
| Compds | R | P. falciparum 3D7 strain EC50, μMa |
|---|---|---|
| 14 | 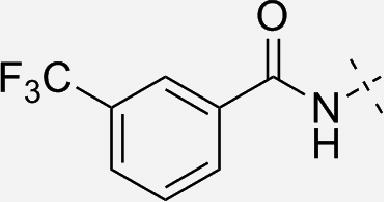 |
0.201 |
| 15 |  |
0.402 |
| 16 | 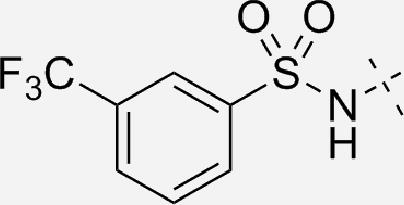 |
8.25 |
| 17 | 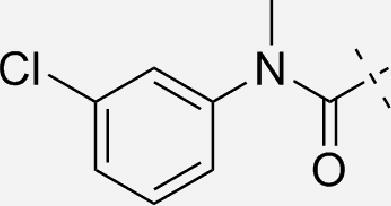 |
4.68 |
| 18 | 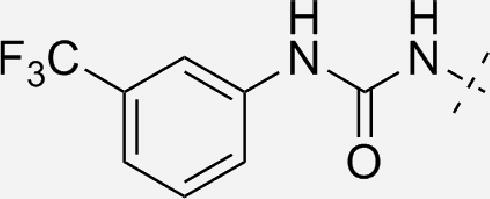 |
0.452 |
| 19 | 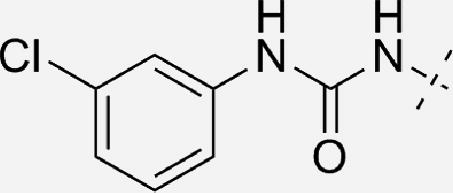 |
0.328 |
| 20 | 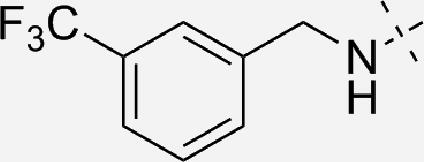 |
1.98 |
| 21 | 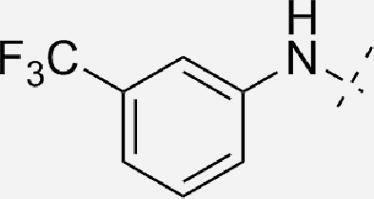 |
0.638 |
| 22 | 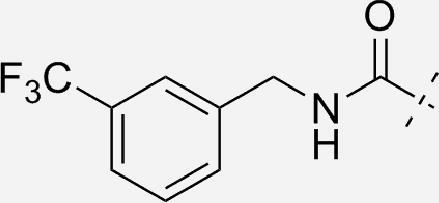 |
0.734 |
| 23 | 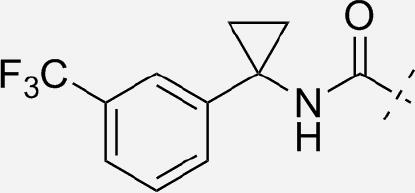 |
0.423 |
Values are means of two experiments. Each assay plate has mefloquine, sulfadoxine and artimesinin as internal standards. The EC50 values for standard compounds match the literature values.
We next turned our attention to the center benzamide ring to explore the possibility of incorporating heterocycles in that ring. As depicted in Table 3, attempts to make substituent changes (compound 24 and 25) or incorporate heteroatoms (compound 26, 27, 28 and 29) failed to enhance the potency of the compounds demonstrating tight SAR in that region of the pharmocophore.
Table 3.
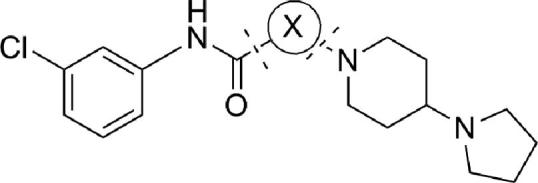
| Compds | X | P. falciparum 3D7 strain EC50, μMa |
|---|---|---|
| 24 |  |
0.766 |
| 25 | 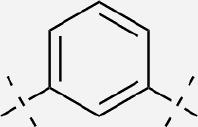 |
5.88 |
| 26 | 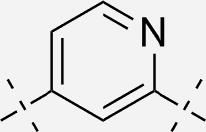 |
0.674 |
| 27 | 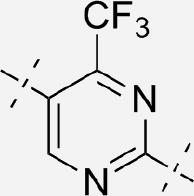 |
1.67 |
| 28 | 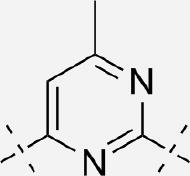 |
2.98 |
| 29 | 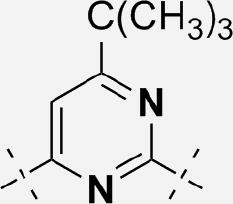 |
2.08 |
Values are means of two experiments. Each assay plate has mefloquine, sulfadoxine and artimesinin as internal standards. The EC50 values for standard compounds match the literature values.
One important aspect of antimalarial drug discovery is to identify agents that are active against the drug-resistant parasites. Table 4 outlines our results on chloroquine sensitive 3D7 strain and multidrug resistant W2 strain which is resistant to chloroquine, quinine, pyrimethamine, cycloguanil, and sulfadoxine. While most of these resistance phenotypes are due to point mutations in the drug target, the W2 strain also contains an amplification of the P. falciparum multidrug resistance transporter.10 Interestingly, while compound 8 show a ∼6-fold shift in potency between 3D7 and W2, replacement of the bicyclic amine part pyrrolidinyl piperidine to bipiperidine (compound 30, 31, and 32) led to much improved potency against W2 strain. Although 34 was the most potent compound, it was deprioritized since the introduction of a stereocenter in the molecule increases the complexity of the synthesis and cost of goods.
Table 4.
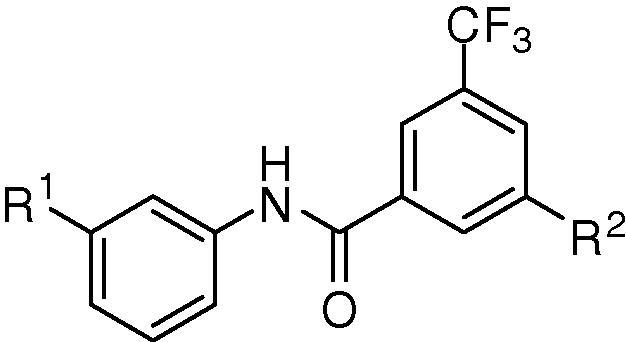
| Compds | R1 | R2 | P. falciparum 3D7 strain EC50, μMa | P. falciparum W2 strain EC50, μMa |
|---|---|---|---|---|
| 8 | Cl | 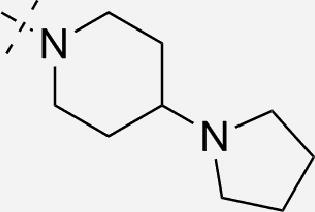 |
0.134 | 0.888 |
| 30 | Cl | 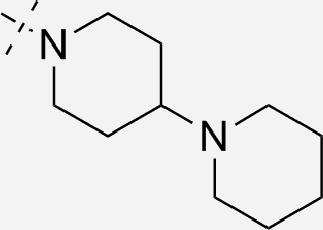 |
0.082 | 0.296 |
| 31 | CF3 | 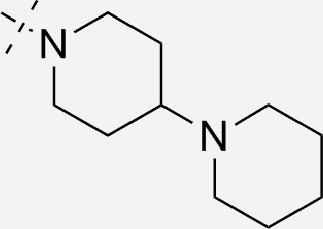 |
0.149 | 0.299 |
| 32 | Cl | 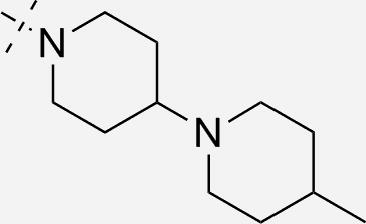 |
0.140 | 0.323 |
| 33 | CF3 | 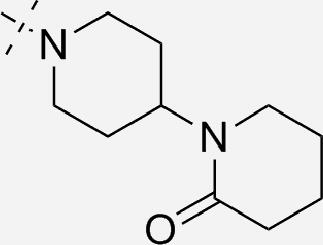 |
1.49 | 1.41 |
| 34 | CF3 | 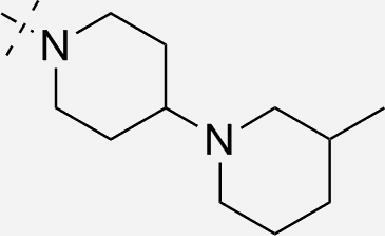 |
0.058 | 0.211 |
| 35 | CF3 | 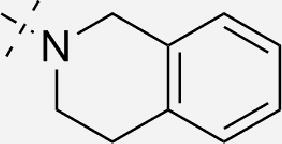 |
0.175 | 0.635 |
Values are means of two experiments. Each assay plate has mefloquine, sulfadoxine and artimesinin as internal standards. The EC50 values for standard compounds match the literature values.
Compound 32 was profiled in an extended panel of 15 drug resistant strains (Table 5) and against a 6-cell line toxicity panel. We were delighted to find that all the potencies are within 3-fold of each other. The observed cytotoxicity TC50s (293T, Ba/F3, CHO, HEp2, HeLa, Huh7) were greater than 8 μM which translates to a good selectivity index (SI >20).
Table 5.
| P. falciparum strain | EC50, μM |
|---|---|
| 3BAG | 0.268 |
| 7G8 | 0.309 |
| C188 | 0.083 |
| D10 | 0.094 |
| D6 | 0.081 |
| Dd2 | 0.141 |
| Camp R | 0.138 |
| FCB | 0.306 |
| FCR3 | 0.218 |
| HB3 | 0.146 |
| K1 | 0.150 |
| NF54 | 0.104 |
| 3D7 | 0.140 |
| TM91C235 | 0.120 |
| W2 | 0.323 |
a Values are means of two experiments. Each assay plate has mefloquine, sulfadoxine and artimesinin as internal standards. The EC50 values for standard compounds match the literature values.
Some of the more potent compounds against both 3D7 and W2 strains were selected to assess their in vivo pharmacokinetic profiles in mice. Mice were dosed a single dose of 20 mg/kg orally and their exposure levels were monitored over a period of 5 h and results are summarized in Table 6.11
Table 6.
Pharmacokinetic profiles of selected compounds
| Compds | AUC(0–5 h) (h nM) | Cmax (nM) | Tmax (h) | AUC(0–5h)/dose [(min ug/mL)/(mg/kg)] |
|---|---|---|---|---|
| 8 | 3916 | 1355 | 1.00 | 5.31 |
| 13 | 4806 | 1643 | 5.00 | 7.01 |
| 14 | 4766 | 1170 | 3.00 | 6.94 |
| 30 | 4365 | 1368 | 1.00 | 6.10 |
| 32 | 7932 | 2279 | 5.00 | 11.42 |
| 35 | 1672 | 672 | 0.50 | 2.33 |
Dose (mg/kg): 20; strain: mice balb/c.
Formulation: 2.5 mg/mL in: PEG300 /D5 W, 3:1, solution.
Salt form: free base.
Most compound demonstrated good oral exposure, with compound 32 exhibited the highest AUC(0–5h) as well as a high Cmax (∼13-fold above the 3D7 potency). We also performed preliminary solubility and metabolic stability measurements on compound 32. Compound 32 exhibits moderate solubility (HT-thermodynamic solubility at pH6.8 0.026 mg/mL free base) and modest metabolic stability (extraction ratio in mouse microsomes = 0.383 ± 0.04 and CLint (μL/min/mg) = 21.399) that are consistent with the in vivo pharmacokinetic results.
In conclusion, we have successfully transformed a pan human kinase inhibitor scaffold (compound 2) into a selective anti-malarial series. SAR studies revealed that the amide linkage is preferred for the antimalarial activity. Switching the bicyclic amine moiety from a 6,5 system to a substituted 6,6 system significantly improves the potency shift across the drug-resistant strains. The selected lead compound 32 exhibited good potency across different strains, favorable physiological properties and good in vivo pharmacokinetic profile. The further studies on this scaffold are warranted and would be reported in due course.
Acknowledgments
This work was supported by a joint grant to Genomics Institute of the Novartis Research Foundation, Biomedical Primate Research Center, Swiss Tropical Institute and Novartis Institute for the Tropical Diseases from the Wellcome Trust and Medicines for Malaria Venture.
References and notes
- 1.WHO ‘World Malaria Report 2008’, June 5, 2009, http://apps.who.int/malaria/wmr2008/malaria2008.pdf.
- 2.Noedl H., Se Y., Schaecher K., Smith B.L., Socheat D., Fukuda M. New Eng. J. Med. 2008;359:2619. doi: 10.1056/NEJMc0805011. [DOI] [PubMed] [Google Scholar]
- 3.Gelb M. Curr. Opin. Chem. Biol. 2007;11:440. doi: 10.1016/j.cbpa.2007.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wu, T.; Nagle, A.; Sakata, T.; Deng, X.; Chen, Z.; Henson, K.; Chang, J.; Plouffe, D.; Tuntland, T.; Kuhen, K.; Winzeler, E.; Gray, N.; Chatterjee, A. K. Abstract of Papers, 236th National Meeting of the American Chemical Society, Philadelphia, PA, August 2008. MEDI-339.
- 5.Plouffe D., Brinker A., McNamara C., Henson K., Kato N., Kuhen K., Nagle A., Adrian F., Matzen J.T., Anderson P., Nam T., Gray N.S., Chatterjee A., Janes J., Yan S.F., Trager R., Caldwell J.S., Schultz P.G., Zhou Y., Winzeler E.A. Proc. Natl. Acad. Sci. U.S.A. 2008;105:9059. doi: 10.1073/pnas.0802982105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kato N., Sakata T., Breton G., Le Roch K.G., Nagle A., Andersen C., Bursulaya B., Henson K., Johnson J., Kumar K.A., Marr F., Mason D., McNamara C., Plouffe D., Ramachandran V., Spooner M., Tuntland T., Zhou Y., Peters E.C., Chatterjee A., Schultz P.G., Ward G.E., Gray N., Harper J., Winzeler E.A. Nat. Chem. Biol. 2008;4:347. doi: 10.1038/nchembio.87. [DOI] [PubMed] [Google Scholar]
- 7.Ren, P.; Wang, X.; Zhang, G.; Ding, Q.; You, S.; Zhang, Q.; Chopiuk, G.; Albaugh, P. A.; Sim, T.; Gray, N. S. WO 2005123719; Chem. Abstr. 2005, 144, 88305.
- 8.Ren, P.; Gray, N. S.; Wang, X.; Zhang, G. WO 2006101783; Chem. Abstr. 2006, 145, 377368.
- 9.Melnick J.S., Janes J., Kim S., Chang J.Y., Sipes D.G., Gunderson D., Jarnes L., Matzen J.T., Garcia M.E., Hood T.L., Beigi R., Xia G., Harig R.A., Asatryan H., Yan S.F., Zhou Y., Gu X., Saadat A., Zhou V., King F.J., Shaw C.M., Su A.I., Downs R., Gray N.S., Schultz P.G., Warmuth M., Caldwell J.S. Proc. Natl. Acad. Sci. U.S.A. 2006;103:3153. doi: 10.1073/pnas.0511292103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kidgell C., Winzeler E.A. Chromosome Res. 2005;13:225. doi: 10.1007/s10577-005-1503-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Snapshot PK is developed and used by GNF pharmacology group as a rapid pharmacokinetic screening approach. The pharmacokinetic parameters of the compounds are studied in a single mode of administration (generally PO arm) over a period of 5 h. The study is carried out on two animals and the samples are pooled together at each time point. This high throughput approach offers exposure levels with reasonable confidence without committing to extensive resources. For details, see: Liu, B.; Chang, J.; Gordon, W. P.; Isbell, J.; Zhou, Yi.; Tuntland, T. Drug Discovery Today2008, 13, 360–367.
- 12.Medicines for Malaria Venture ‘Selection and Review: MMV Compound Progression Criteria’, June 17, 2009, http://www.mmv.org/article.php3?id_article=503.