

Abstract
Irreversible translation arrest occurs in reperfused neurons that will die by delayed neuronal death. It is now recognized that suppression of protein synthesis is a general response of eukaryotic cells to exogenous stressors. Indeed, stress-induced translation arrest can be viewed as a component of cell stress responses, and consists of initiation, maintenance, and termination phases that work in concert with stress-induced transcriptional mechanisms. Within this framework, we review translation arrest in reperfused neurons. This framework provides a basis to recognize that phosphorylation of the alpha subunit of eukaryotic initiation factor 2 is the initiator of translation arrest, and a key marker indicating activation of neuronal stress responses. However, eIF2 alpha phosphorylation is reversible. Other phases of stress-induced translation arrest appear to contribute to irreversible translation arrest specifically in ischemic vulnerable neuron populations. We detail two lines of evidence supporting this view. First, ischemia, as a stress stimulus, induces irreversible co-translational protein misfolding and aggregation after 4 to 6 h of reperfusion, trapping protein synthesis machinery into functionally inactive protein aggregates. Second, ischemia and reperfusion leads to modifications of stress granules (SGs) that sequester functionally inactive 48S preinitiation complexes to maintain translation arrest. At later reperfusion durations, these mechanisms may converge such that SGs become sequestered in protein aggregates. These mechanisms result in elimination of functionally active ribosomes and preclude recovery of protein synthesis in selectively vulnerable neurons. Thus, recognizing translation arrest as a component of endogenous cellular stress response pathways will aid in making sense of the complexities of postischemic translation arrest.
Keywords: brain ischemia and reperfusion, CA1, cellular stress, cotranslational aggregation, stress granules, translation arrest or protein synthesis inhibition
Introduction
Transient brain ischemia results in the selective death of specific neuronal populations. After brief global ischemia, the pyramidal neurons in the cornu ammonis 1 (CA1) sector die after several days of reperfusion, by a phenomenon termed delayed neuronal death (DND; Ito et al, 1975; Kirino, 1982; Pulsinelli et al, 1982). After transient focal ischemia, penumbral neurons display a similar delayed type death (Siesjo, 1988). The mechanisms of DND are not fully understood.
Ischemia and reperfusion (I/R) also alter cellular regulatory systems that control neuronal protein synthesis. A striking correlate of the delayed death of postischemic neurons is that they show a persistent inhibition of protein synthesis (or translation arrest) until they die (Kleihues and Hossmann, 1971; Dienel et al, 1980; Bodsch et al, 1985; Thilmann et al, 1986; Araki et al, 1990). This observation lead Hossmann (1993), as well as many others, to suggest that lack of recovery of protein synthesis is a causal component of DND. Based on mechanisms reviewed below, we now know that postischemic neuronal translation arrest is biphasic (DeGracia, 2004): an initial, reversible translation arrest occurs in all postischemic neurons, followed by an irreversible translation arrest that persists in vulnerable neurons (Dienel et al, 1980; Yoshimine et al, 1987; Mies et al, 1991; Hu and Wieloch, 1993; Hu et al, 2004). As detailed below, irreversible translation arrest in vulnerable neurons is caused by different mechanisms than the reversible translation arrest that occurs in all postischemic neurons (Liu et al, 2005a, b; Kayali et al, 2005; Zhang et al, 2006).
Here we will bring together evidence pertaining to irreversible translation arrest after brain I/R. We will do so within the current general understanding of the role of protein synthesis inhibition in the context of cellular stress pathways. This will provide a basis to recognize that translation arrest after brain I/R can now be viewed as one example among many of stress-induced translation arrest. In the context of activation of endogenous cellular stress pathways, we will offer a model that recognizes the time evolution of processes involved in translation arrest. Using this model, we aim to show that multiple points of translational control are altered in postischemic neurons, some of which relate to reversible and others to irreversible translation arrest. We thus suggest that recognizing translation arrest as a component of endogenous cellular stress response pathways will aid in making sense of the complexities of postischemic translation arrest.
Summary of Translation Arrest After Ischemia and Reperfusion
As previous reviews have summarized in detail studies of translation arrest after I/R (Hossmann, 1993; Wieloch et al, 1993, 1996; DeGracia et al, 2002; DeGracia, 2004; Hu et al, 2004; Hu, 2006), here we will only outline the salient issues. The initial work of Kleihues and Hossmann (1971) established that inhibition of protein synthesis occurred in neurons after brain I/R, likely involving a defect in translation initiation. Chavko et al (1987) and Burda and Chavko (1991) showed that excesses of either eIF2 or GTP could recover in vitro translation of reperfused spinal cord homogenates, implicating changes in eIF2 after I/R.
The role of eIF2 during translation initiation is to deliver the methionyl-charged initiator tRNA to the 40S subunit, via an eIF2 · GTP · met–RNAi ternary complex. Binding of ternary complex, along with the mRNA, leads to the 48S translation preinitiation complex, followed by the formation of an 80S initiation complex and an inactive eIF2 · GDP. Ternary complex formation is blocked after phosphorylation of the alpha subunit of eIF2 (eIF2α; phosphorylated form, eIF2α(P)). eIF2α(P) restricts the activity of the guanine nuclear exchange factor eIF2B, thus preventing recycling of eIF2 · GDP to active eIF2 · GTP · met–RNAi ternary complex after initiation (Proud, 2005).
Hu and Wieloch (1993) reported the first detailed molecular study of eIF2 activity by showing that eIF2 · GTP · met–RNAi ternary complex formation and eIF2B activity were inhibited persistently in CA1 neurons undergoing delayed cell death, but transiently in dentate gyrus (DG) surviving neurons after transient cerebral ischemia. Subsequently, Burda et al (1994) demonstrated a large increase in eIF2α phosphorylation after brain I/R. DeGracia et al (1997) showed by immunohistochemical mapping that eIF2α phosphorylation occurred in the principle neurons of postischemic hippocampus and cerebral cortex. In fact, eIF2α phosphorylation occurs in all postischemic neurons (Figure 1) and, therefore, regionalization of eIF2α phosphorylation does not correlate with selectively vulnerable neuron populations.
Figure 1.

Phosphorylation of eIF2α does not correlate with selectively vulnerable neuron populations. Immunostaining for eIF2α(P) after 10 mins cardiac arrest-induced brain ischemia and 10 mins reperfusion. (A) Brainstem, at the level of the pons where the trigeminal nerve exits, −9.8 mm posterior to bregma. The stained cell bodies are motoneurons of the facial motor nucleus of VII. (B) Sagittal section through the cerebellum, 1.2 mm lateral to midline, showing intense perinuclear eIF2α(P) staining of Purkinje cells; there was also intense staining in the neurons of the deep nuclei (not shown). (C) Posterior hypothalamic nucleus, taken from same section as in panel (D). Thalamus, 3.6 mm posterior to bregma. Polymorphic principle neurons of the posterior nucleus of the thalamus showed intense perinuclear eIF2α(P) staining. (E) Striatum, 0.7 mm anterior to bregma, showed intense perinuclear eIF2α(P) staining in the medium spiny neurons of striatum. (F) Lateral cerebral cortex, 0.3 mm posterior to bregma, showed intense eIF2α(P) staining of pyramidal neurons of the olfactory piriform cortex. All scale bars are 50 μm. Immunostaining was as previously described (DeGracia et al, 1997).
Additionally, several laboratories have shown that the phosphorylation of eIF2α is transient, abating completely after >4 h reperfusion (DeGracia et al, 1997; Althausen et al, 2001; Martin de la Vega et al, 2001; Kumar et al, 2003). Of significance to the issue of whether eIF2α(P) contributes to cell death, it has been shown that transient translation arrest (Nakagomi et al, 1993) and eIF2α phosphorylation (Garcia et al, 2004) occur in animals exposed to ischemic preconditioning (IPC, in which a prior sublethal ischemic insult subsequently protects the brain from a later, lethal insult). As IPC is well known to prevent cell death (Kirino, 2002), this result demonstrates a type of double dissociation between reversible and irreversible translation arrest in reperfused neurons, and argues that cell death is independent of eIF2α phosphorylation. Thus, it is reasonable to conclude that eIF2α phosphorylation represents a general acute response of all postischemic neurons to I/R stress, is transient and reversible, and does not correlate with selective neuronal vulnerability. Below, we will discuss other facets of eIF2α phosphorylation relevant to prolonged translation arrest.
The other regulator of translation initiation known to undergo substantial change after brain I/R is eIF4G. eIF4G is a component of the eIF4F complex that delivers the mRNA to the 40S subunit during translation initiation (Gingras et al, 1999). During ischemia, eIF4G undergoes partial, μ-cal-pain-mediated proteolysis that increases as a function of ischemia duration (DeGracia et al, 1996; Neumar et al, 1998; Martin de la Vega et al, 2001; Mengesdorf et al, 2002; Garcia et al, 2004). Additionally, Garcia et al (2004) showed increased eIF4G phosphorylation during reperfusion, which DeGracia et al (2006) showed to be confined to interneurons of the supragranular layer of the DG. Potential roles for eIF4G alterations in irreversible translation arrest will also be discussed below.
Other regulators of protein synthesis that have been investigated include eIF2B, eIF4E, the eIF4E binding proteins (4EBPs), eEF2, S6 kinase (as reviewed in DeGracia, 2004), and small ribosomal subunit (40S) protein S6 (Kayali et al, 2005; Zhang et al, 2006). While the roles of biochemical changes associated with other regulators of protein synthesis in I/R-induced translational arrest remain to be fully defined, their contributions to initial translation arrest are not as dramatic as that of eIF2α phosphorylation and some do not contribute at all (DeGracia, 2004). In general, translational regulators other than eIF2 and eIF4F have not been intensively studied, and existing studies do not rule out their participation in mechanisms of irreversible translation arrest, as will be discussed more fully below. We now turn attention to the current general understanding of the role of protein synthesis inhibition when cells experience exogenous stress.
Cellular Stress Elicits Translation Arrest
It is now widely recognized that a large variety of exogenous stressors cause cells to transiently suppress protein synthesis as a means of coping with the stress (Brostrom and Brostrom, 1998). That is, reversible translation arrest is now recognized as an essential ‘subroutine’ of several endogenous intracellular stress response programs. Examples of stresses that induce translation arrest are hyperthermia (heat shock) (Panniers, 1994), heavy metal poisoning (e.g., arsenite) (McEwen et al, 2005), stress to the endoplasmic reticulum (ER; e.g., by depleting ER Ca2+ stores or inhibiting ER-mediated post-translation modifications) (Ron, 2002), viral infection (Gale et al, 2000), nutrient (amino acid or glucose) deprivation (Halford et al, 2004), excessive free radical production (Kaufman, 1999), and ethanol intoxication (Ron, 2004), among others.
Cells have evolved a variety of stress response pathways to cope with exogenous stresses, including the heat shock response (HSR), the unfolded protein response (UPR), the integrated stress response (ISR; Harding et al, 2003), the interferon response, and the ER overload response (Kaufman, 1999), among others. This group of stress response pathways are activated in response to damage of intracellular protein systems, and need to be distinguished from stress pathways related to DNA damage, the latter of which will not be discussed here.
It is important to be aware where translation arrest fits into endogenous cellular stress response programs. Cells experience application of exogenous stress, and, in a reflex-like fashion, initiate endogenous stress pathways. Figure 2 diagrammatically portrays what is emerging as the underlying ‘algorithm’ behind several cellular defense pathways. Cells experience application of exogenous stress as damage to intracellular components. Specific molecular mechanisms sense cellular damage, which in turn activate effectors of the stress response pathways. Generally, stress responses include two major parallel pathways of activity. First, there is a transient suppression of protein synthesis. Note, in Figure 2, that we have shown translation arrest to be divided into initiation, maintenance and termination phases, and these will be explained below. Second, there is activation of transcriptional inducers that will up-regulate transcription of mRNAs that code for stress proteins.
Figure 2.

Translation arrest is a ‘subroutine’ of certain classes of endogenous cellular stress responses. Exogenous stressors induce damage to intracellular protein systems. Cellular damage-detection mechanisms activate in response to accumulation of damaged cell components. Damage detection mechanisms either couple to or are effectors that, in parallel, lead to transient inhibition of protein synthesis and induce transcription of mRNAs coding for stress proteins. Stress-induced mRNAs escape translation arrest, and are translated by ‘nonclassical’ initiation mechanisms such as by-pass scanning or internal ribosome entry site initiation. Translation arrest is initiated, usually by eIF2α phosphorylation, maintained in stress granules, and terminated after successful execution of the stress response program. Success in execution of the stress response program leads to repair of cell damage and recovery of general protein synthesis. If cell damage overwhelms the capacity of the cell's stress response, cell death mechanisms are triggered, and general protein synthesis never fully recovers. Ischemia and reperfusion-induced translation arrest is thus a specific example of this more general cellular response.
The transient inhibition of cellular protein synthesis serves a dual role. First, it prevents further damage to cellular proteins by shutting off accumulation of newly synthesized proteins that could potentially be damaged by the stress (Harding et al, 2002). Second, shut off of the translation of constitutive (or ‘housekeeping’) cellular proteins allows the cell to exclusively translate the mRNAs coding for the stress proteins. Selective synthesis of stress proteins provides a mechanism for the cell to buffer and repair stress-induced damage. Additionally, the stress proteins can participate in feedback loops that induce recovery of overall protein synthesis as cell damage is repaired. A balance between the intensity of stress-induced damage and the activity of translated stress proteins sets a decision point (or flow control point), determining whether the outcome is cell survival or cell death (Oyadomari and Mori, 2004; Beere, 2005; Meriin and Sherman, 2005; Zhang and Kaufman, 2006).
We note this model is perhaps an idealization of known stress pathways. Specific pathways show nuances and additional complexities that may deviate from this general pattern. However, the utility of this model is that it provides an integrated view of translation arrest as a component of cellular stress response systems. As such it provides a more general view by which to understand translation arrest in pathological situations such as after brain I/R. This model provides the recognition that brain I/R is not simply triggering protein synthesis control pathways in vacuo, but is activating endogenous stress responses of which translation arrest is an integral subroutine.
The HSR and UPR are specific examples of cell stress pathways that have given rise to this general model (Figure 3). The HSR and the UPR activate after abnormal folding or denaturation of proteins in the cytoplasmic and ER compartments, respectively. Stress sensors for both pathways involve chaperone-mediated inhibition of latent transcription factors and/or eIF2α kinases. By mass action, chaperones dissociate from stress sensors to bind accumulated denatured proteins, in the cytoplasm for the HSR (Voellmy, 2004), and in the ER lumen for the UPR (Ron, 2002). Relief of chaperone-mediated inhibition activates the stress sensors: heat shock factor 1 (Voellmy, 2004) (HSF1) in the case of the HSR, and PERK, IRE1, and ATF6 in the case of the UPR (Zhang and Kaufman, 2006). Both the HSR and UPR lead to a transient inhibition of protein synthesis. During the UPR, translation arrest is due to activation of the ER localized eIF2α kinase PERK and subsequent phosphorylation of eIF2α (Harding et al, 1999). The mechanisms of translation arrest during the HSR are less well understood and appear to be cell type specific, but involve a transient phosphorylation of eIF2α and modification of eIF4F (Panniers, 1994; Scheper et al, 2001; Kim and Jang, 2002). HSF1 is a transcription factor that upregulates the genes coding for heat shock proteins (HSPs). The UPR induces transcription of genes coding for ER chaperones and enzymes via the transcription factors ATF6 (Haze et al, 1999; Yoshida et al, 2000) and XBP-1 (Shen et al, 2001; Calfon et al, 2002). The HSR leads to selective translation of HSPs (Panniers, 1994), and the UPR leads to selective translation of proteins such as ATF4 (Harding et al, 2000) and GADD34 (Novoa et al, 2001), while overall cellular protein synthesis is inhibited. Heat shock proteins are chaperones that bind and stabilize denatured cytoplasmic proteins (Latchman, 2004) and also lead to recovery of overall cellular protein synthesis (Gilks et al, 2004). Increased synthesis of ER chaperones and enzymes during the UPR expands the processing capacity of the ER to abate ER stress (Ron, 2002), and restores general protein synthesis via GADD34, a protein phosphatase I subunit that targets eIF2α(P) for dephosphorylation (Novoa et al, 2001).
Figure 3.

The HSR and UPR as examples of endogenous cellular stress systems typical of the model in Figure 2. Accumulation of denatured or misfolded proteins in the cytoplasm or ER, activate the HSR and UPR, respectively. HSF and ATF6 and XBP-1 are the transcriptional effectors for the HSR and UPR, respectively. Both the HSR and the UPR lead to transient inhibition of protein synthesis initiated by at least transient phosphorylation of eIF2α. Both systems lead to increased translation of stress-induced mRNAs. These mRNAs escape translation arrest and lead to production of stress proteins. Heat shock proteins catalyze proper protein folding and prevent protein aggregation, thereby abating cell damage. Heat shock proteins also dissolve stress granules, leading to recovery of protein synthesis. Upregulation of ER enzymes and chaperones increase the processing capacity of the ER lumen, thereby abating ER damage. Translation of GADD34 during the UPR catalyzes dephosphorylation of eIF2α(P), allowing recovery of general protein synthesis.
During periods of stress-induced translation arrest, there are mechanisms by which the stress-induced mRNAs escape translation arrest. These mechanisms involve special sequences in the 5′ untranslated region (5′UTR) of stress-induced mRNAs (Pickering and Willis, 2005). Two such sequences are upstream open reading frames (uORF) (Meijer and Thomas, 2002) and internal ribosome entry sites (IRES) (Kim and Jang, 2002). Both uORFs and IRES allow ribosomes to initiate on the mRNAs when ‘housekeeping’ protein synthesis is inhibited. mRNAs containing uORFs are preferentially translated when eIF2α is phosphorylated via a process termed ‘by-pass scanning’ (Morris and Geballe, 2000). Internal ribosome entry site containing mRNAs are preferentially translated when eIF4F is inactivated (Perales et al, 2003; Nevins et al, 2003). Some of the implication of such ‘nonclassical’ forms of translation initiation for I/R brain injury have been previously discussed (DeGracia et al, 2002).
Maintainance of Translation Arrest: Stress Granules
Originally identified in cell culture after heat shock (Nover et al, 1983), punctate (i.e., granular) structures form in the cytoplasm on exposure to a large variety of exogenous stresses. These structures are called stress granules (SGs). Formation of SGs is now recognized as a general response that occurs during stress-induced translation arrest (Anderson and Kedersha, 2006). SGs form on stress induction and persist for the duration of the stress; as the stress abates, the number of SGs decreases (Kedersha et al, 1999). Stress granules are ribonucleic acid–protein complexes that sequester the ‘housekeeping’ poly-A mRNAs (Nover et al, 1989; Kedersha et al, 2000), 40S ribosomal subunits, several initiation factors and RNA metabolism enzymes, but do not contain the 60S ribosomal subunit (Kedersha et al, 2002; Kimball et al, 2003; Anderson and Kedersha, 2006).
Anderson's group has shown that two related RNA-binding proteins, TIA-1 (T-cell internal antigen) (Tian et al, 1991) and TIAR (TIA-1 related protein) (Kawakami et al, 1992), form part of the matrix of SGs (Anderson and Kedersha, 2002; Kedersha and Anderson, 2002). When cells are unstressed, TIA-1 and TIAR are predominantly nuclear RNA binding proteins involved in splicing certain pre-mRNAs (Dember et al, 1996; Gueydan et al, 1999; Le Guiner et al, 2001). On cell stress, TIA-1 and TIAR export from the nucleus and contribute to the formation of SGs (Zhang et al, 2005). Both TIA-1 and TIAR contain polyglutamine repeat regions that are homologous to prion protein, and both have been shown to self-aggregate (Gilks et al, 2004). Thus, SG formation involves binding of mRNA by TIA-1 and TIAR while they simultaneously self-aggregate, thereby trapping mRNA-bound 40S subunits (Figure 4). However, there is a growing list of protein components found in SGs, many of which are RNA binding proteins involved in various facets of RNA metabolism (Anderson and Kedersha, 2006).
Figure 4.

Polysomes and stress granules are in equilibrium. Initiation of translation arrest under physiological conditions is often due to phosphorylation of eIF2α, which causes dissaggregation of polysomes. Dissaggregation of polysomes results in accumulation of 48S preinitiation complexes (depicted here by the 40S subunit bound to an mRNA). The mRNA in the 48S preinitiation complex is bound by TIA-1 and/or TIAR, which is believed to set up an equilibrium situation leading to further efflux of TIA-1 from the nucleus to the cytoplasm. TIA-1 and TIAR self-aggregate, thereby trapping 48S preinitiation complexes within a matrix of TIA-1 and TIAR, forming stress granules. HSP70 dissociates TIA-1/TIA-1 interactions, causing dissolution of stress granules, and a shift in the equilibrium back towards polysomes. This figure is based on ideas developed in Anderson and Kedersha (2002) and Kedersha and Anderson (2002). At least a dozen other proteins participate in SG formation (indicated by circled X), many of which are proteins involved in RNA binding and metabolism (Anderson and Kedersha, 2006).
A general model has emerged in which SGs are in equilibrium with polysomes (Figure 4) (Anderson and Kedersha, 2002; Kedersha and Anderson, 2002). Under nonstress conditions, the majority of ribosomes are actively synthesizing proteins in the form of polysomes. With cell stress, polysomes dissociate and push the equilibrium towards SG formation. However, mRNAs coding for stress proteins escape sequestration by SGs. For example, mRNAs coding for HSPs are not present in SGs (Nover et al, 1989). Hence, SGs provide an environment that allows for the general suppression of ‘housekeeping’ mRNA translation, but allows the selective translation of mRNAs coding stress proteins. Rather than being static, SGs are dynamic structures with their protein components entering and exiting at rates ranging from milliseconds to seconds (Kedersha et al, 2000).
As well as representing morphological correlates of inhibited protein synthesis, SGs are intimately involved in cytoplasmic mRNA trafficking and metabolism (Anderson and Kedersha, 2006). Stress granules persist as long as there is cell stress. In reversible models of cell stress (e.g., arsenite treatment of cultured cells followed by application of fresh media), the SGs decrease in the cytoplasm as the stress abates (Kedersha et al, 2000). In cells treated with lethal stressors, SGs persist until the cells die (Kedersha et al, 2000).
The Phases of Stress-Induced Translation Arrest
We can summarize the foregoing by recognizing that stress-induced translation arrest follows a classical pattern of possessing initiation, maintenance, and termination phases.
Initiation
Cell stress triggers inhibition of protein synthesis. For a large variety of stresses, the initiating event is phosphorylation of eIF2α (Clemens, 2001). In fact, all four known eIF2α kinases are activated by specific stresses (Kaufman, 1999; Clemens, 2001; Ron, 2002). Note that eIF2α phosphorylation is readily reversible (via phosphatase activation); hence, underscoring the functionally transient character of stress-induced translation arrest.
Maintenance
Kedershaet al (1999) have suggested that SGs serve to maintain translation arrest, even after dephosphorylation of eIF2α. Maintenance of the translation arrest allows the cell time to elicit genetic repair programs via increased transcription of mRNAs coding for stress response proteins. The SGs sequester ‘house keeping’ mRNAs, but not stress-induced mRNAs, allowing for selective translation of stress proteins via by-pass scanning, IRES initiation, or other unidentified mechanisms, while general protein synthesis is maintained in a quiescent state.
Termination
The selective translation of stress proteins serves to initiate repair of the stress-induced damage. The stress proteins also act to terminate the general translation arrest. In the case of heat shock, HSP70 dissociates SGs by disaggregating TIA-1/TIAR (Gilks et al, 2004), thereby freeing protein synthesis machinery for resumption of normal cellular protein synthesis. In the UPR and ISR, synthesis of GADD34 accelerates dephosphorylation of eIF2α(P) (Novoa et al, 2001), thereby shifting the equilibrium back towards polysome formation (Figure 4).
The above model provides an integrated picture of the role of translation arrest in general cell stress responses. As such, this model provides a theoretical basis by which to state that I/R-induced translation arrest can be viewed as one example among many of stress-induced translation arrest. Like many other stresses, the initiating event for translation arrest during postischemic brain reperfusion is due predominantly to eIF2α phosphorylation. However, in selectively vulnerable neurons, the transient character of the translation arrest is compromised. The key question is: by what mechanisms? The above model provides a general focus by which to determine key mechanisms. Do alterations occur with initiation, maintenance, or termination phases of translation arrest? Additionally, how do other known I/R-induced damage mechanisms, such as oxidative damage (Hayashi et al, 2003, 2005) or excessive proteolysis (Neumar et al, 1998), interface with the phases of stress-induced translation arrest?
We now turn attention to recent studies suggesting that alterations in the phases of translation arrest underlie the irreversible translation arrest associated with DND after brain I/R. We will see that such studies require a substantial focus on sub-cellular localization, and point to the need to combine biochemical and morphological forms of analysis for a fuller understanding of the mechanisms of irreversible translation arrest. We first review evidence of what may be the main stimulus activating endogenous stress response pathways after brain I/R: the denaturation and aggregation of nascent peptides that are in the process of being synthesized, a process that has been termed ‘co-translational protein aggregation’ (Hu et al, 2004; Liu et al, 2005a, b; Zhang et al, 2006; Hu, 2006). We then review recent studies of SG behavior after brain I/R (Kayali et al, 2005; DeGracia et al, 2006).
Co-Translational Protein Folding and Aggregation
Recently, aberrant protein homeostasis or proteo-toxicity has been demonstrated to play a causal role in irreversible inhibition of protein synthesis and DND after brain ischemia (Hu et al, 2000, 2001, 2004; Ouyang and Hu, 2001; Liu et al, 2004, 2005a, b; Liu and Hu, 2004; Zhang et al, 2006). The importance of protein misfolding and aggregation or proteotoxicity in DND after brain ischemia is accentuated by two lines of studies: (1) introduction of molecular chaperones by transgenic gene expression or viral vector gene delivery protects neurons from ischemia (Sharp et al, 1999; Yenari et al, 2001); and (2) the acquisition of stress tolerance via IPC has been widely viewed to be brought about by means of induction of molecular chaperones and of the ubiquitin–proteasomal systems for protecting misfolded proteins from protein aggregation (Liu et al, 2005b).
In an early electron microscopic (EM) study, increases in electron-dense deposits were observed in CA1 pyramidal neurons during the later periods of reperfusion in a rat transient forebrain ischemia model, and were termed ‘dark substances’ (Kirino et al, 1984; Kirino and Sano, 1984). This observation was recapitulated later and described as ‘electron-dense fluffy dark material’ in an ultrastructural study of programmed cell death after brain ischemia (Deshpande et al, 1992). Deshpande et al (1992) proposed that DND is probably not a form of programmed cell death, and the dark materials might represent protein complexes or internalized plasma membrane fragments. However, no study had been conducted to identify the nature and molecular composition of these dark deposits in postischemic neurons.
Hu and co-workers performed a series of morphological, biochemical, and molecular studies demonstrating that these dark deposits are, in fact, protein aggregates made of misfolded proteins overproduced in postischemic neurons of rat forebrain and focal ischemia models (Hu et al, 1998, 2000, 2001, 2004; Liu et al, 2004, 2005a, b; Liu and Hu, 2004; Zhang et al, 2006; Hu, 2006). Identification of these intracellular dark deposits was begun with applying ethanolic phosphotungstic acid to study postsynaptic density alterations by EM after brain ischemia (Hu et al, 1998; Martone et al, 1999). In those studies, ethanolic phosphotungstic acid stains only postsynaptic density structures and nuclear histones in nonischemic control neurons under EM, but, unexpectedly, it additionally labeled intracellular dark deposits only in CA1 pyramidal neurons before DND occurred after ischemia. Ethanolic phosphotungstic acid is known to stain protein complexes under EM (Burry and Lasher, 1978; Martone et al, 1999; Hu et al, 1998, 2000, 2001). This provided a clue indicating that these dark deposits were protein aggregates and likely composed of misfolded proteins. This view was further confirmed by studies showing that the dark deposits were strongly labeled with ubiquitin-immunogold under EM, suggesting that ubi-protein conjugates were major components of the dark deposits (Hu et al, 2000, 2001). Furthermore, a host of biochemical studies revealed that molecular chaperones and folding enzymes, as well as proteosomes, are clumped into inactive protein aggregates, thus demolishing the protein quality control system in postischemic neurons (Liu et al, 2004, 2005a, b; Liu and Hu, 2004; Zhang et al, 2006; Hu, 2006). As demonstrated below, without a functional protein quality control system, cellular unfolded proteins are unable to fold, or be protected or degraded properly, and thus they eventually become protein aggregates deposited in neurons after brain ischemia.
On Western blots, free ubiquitin is essentially depleted in CA1 neurons as early as at 30 mins of reperfusion, and this persists until DND takes place in CA1 neurons at 2 to 3 days after brain ischemia. Concomitantly, high molecular weight ubi-proteins are highly accumulated in neurons destined to die after brain ischemia (Magnusson and Wieloch, 1989; Hayashi et al, 1993; Dewar et al, 1994; Morimoto et al, 1996; Ouyang and Hu, 2001; Hu et al, 2000, 2001; Liu et al, 2004, 2005a, b; Zhang et al, 2006). When misfolded ubi-proteins are irreversibly aggregated, they become insoluble in a Triton X-100 detergent solution (Hu et al, 2000, 2001; Liu et al, 2005a, b; Zhang et al, 2006). Most ubi-proteins during 0.5 to 1 h of reperfusion after ischemia can still be dissolved by a Triton X-100 solution, suggesting that they are not yet irreversibly aggregated during the early period of reperfusion (Hu et al, 1998, 2000, 2001; Liu et al, 2004; Liu and Hu, 2004). Ubi-proteins gradually become detergent-insoluble in the CA1 region from 4 h of reperfusion onward after ischemia, implying that they are irreversibly aggregated (Hu et al, 2000; Liu et al, 2004). Emerging evidence suggests that depletion of free ubiquitin reflects an overproduction of misfolded or damaged proteins after brain ischemia. Although all available free ubiquitins in neurons are utilized, they can conjugate only a subfraction of the misfolded proteins overproduced after brain ischemia. These escaping misfolded proteins, together with undegraded ubi-proteins, eventually become protein aggregates visible by EM from 4 h of reperfusion onward in vulnerable neurons after ischemia (Hu et al, 2000, 2001; Ouyang and Hu, 2001; Liu et al, 2004, 2005a,b; Liu and Hu, 2004; Zhang et al, 2006; Hu, 2006).
Transient cerebral ischemia also induces a morphologically identifiable clustering of ubi-proteins during the post-ischemic phase. The size of ubi-protein clusters was approximately 2 to 5mm, and thus were visualized by immunofluorescence (IF) confocal microscopy (Hu et al, 2000, 2001). Ubiquitin immunostaining in control neurons was relatively evenly distributed. Following 0.5 to 4 h reperfusion after 15 mins ischemia, ubi-protein immunoclusters were observed in all postischemic neurons, including hippocampal CA1, CA3, DG, and neocortical neurons. At 24 h of reperfusion, ubi-protein immunostaining returned to the pattern of even distribution in most ischemic resistant neurons of CA3, DG, and cerebral cortex. In vulnerable CA1 neurons from 4 h of reperfusion onward, however, ubi-protein immunostaining are transformed into a patchy pattern concentrated around the nucleus and along dendritic membranes (Hu et al, 2000, 2001; Ouyang and Hu, 2001; Liu et al, 2004, 2005a, b).
Do ubi-protein immunoclusters observed by confocal microscopy represent the EM-visible protein aggregates described above? Several lines of evidence suggest that this is not exactly the case: (1) The size of EM-visible protein aggregates in vulnerable CA1 neurons after ischemia is approximately 50 to 200 nm, much smaller than mitochondria (500 to 1000 nm). Therefore, EM-visible protein aggregates cannot be seen directly by light microscopy; (2) The regional distribution is different: ubi-protein immunoclusters are observed virtually in all post-ischemic neurons, whereas EM-visible protein aggregates are found only in vulnerable neurons before DND occurs after brain ischemia; and (3) The time course is also different: ubi-protein clusters appear as early as 0.5 to 1 h of reperfusion in most postischemic neurons, whereas EM-visible protein aggregates are observed only in CA1 vulnerable neurons from 4 h of reperfusion onward. The different sizes, as well as different regional and temporal distributions, suggest that confocal microscopically viewed ubi-protein immunoclusters are not equivalent to EM-visible protein aggregates, per se, during early reperfusion, but they can becomes irreversible protein aggregates. Therefore, a subset of ubi-protein conjugates are trapped into misfolded protein aggregates visible by EM in vulnerable neurons at > 4 h reperfusion (Hu et al, 2000, 2001; Liu et al, 2004; Hu, 2006).
From the above studies, it has become clear that transient cerebral ischemia generates excessive amounts of unfolded or misfolded proteins during ischemia and the early period of reperfusion. These abnormal proteins are immediately ubiquitinated on resuming cellular ATP during reperfusion (Hu, 2006). This is indicated by complete depletion of free ubiquitin and accumulation of ubiquitin-conjugated proteins (ubi-proteins) immediately after ischemia. However, before 4 to 6 h of reperfusion, ubi-proteins are not irreversibly aggregated because they are still detergent-soluble. After 4 to 6 h of reperfusion, ubi-proteins become Triton X-100 insoluble irreversible protein aggregates detectable by conventional and ethanolic phosphotungstic acid electron microscopy (Hu et al, 2000, 2001; Liu et al, 2004, 2005a; Zhang et al, 2006; Hu, 2006).
Newly synthesized polypeptides, also referred to as nascent polypeptides, are the major sources of unfolded protein in normal cells (Bukau et al, 1996; Frydman, 2001; Kusmierczyk and Martin, 2001; Hartl and Hayer-Hartl, 2002). Are translating nascent polypeptides irreversibly aggregated after ischemia? The polyribosome is the protein synthesis machine of the cell, and consists of an mRNA chain, at least one 48S preinitiation complex with initiation factors, and multiple 80S ribosomes with translating nascent polypeptides, protein translational elongation and termination factors, co-translational chaperones, and protein degradation components. To become a nontoxic functional protein, a translating nascent polypeptide on a ribosome has to overcome a key rate-limiting hurdle: folding into a unique three-dimensional conformation (Bukau et al, 1996; Frydman, 2001; Kusmierczyk and Martin, 2001; Hartl and Hayer-Hartl, 2002). Folding of a nascent polypeptide occurs at the levels of protein domain or multidomains; folding may not take place until a whole protein domain has been synthesized on the ribosome. This folding process during polypeptide elongation on each ribosome is referred to as co-translational folding (Hardesty et al, 1999; Frydman, 2001; Hartl and Hayer-Hartl, 2002). Polyribosomes are highly abundant in cells, and numerous nascent polypeptides are emerging from them at any given moment. Nascent polypep-tides expose their hydrophobic segments and are highly prone to intramolecular misfolding and intermolecular aggregation driven by hydrophobic forces (Hartl and Hayer-Hartl, 2002). Therefore, co-translational folding is generally assisted and protected by (1) molecular chaperone proteins; (2) their co-chaperones; and (3) the cellular energy supply necessary to support chaperone activity.
The following evidence suggests that overload of unprotected nascent polypeptides, and aggregated molecular chaperones and proteosomes on polyribosomes after brain ischemia, may lead to aggregation and irreversible damage to the protein synthesis machinery(Hu et al, 2004; Liu et al, 2005a, b; Zhang et al,2006; Hu, 2006): (1) EM shows that ribosomes are abnormally aggregated only in neurons undergoing DND after ischemia; (2) ribosomal proteins, translational initiation factors, co-translational chaperones and folding enzymes, and components of the ubiquitin–proteosomal system are highly aggregated in ischemic vulnerable neurons during the postischemic phase; (3) IPC reduces co-translational aggregation of molecular chaperones (Liu et al, 2005a). Thus, emerging evidence suggests that irreversible inhibition of protein synthesis in vulnerable neurons is caused, at least in part, by aggregation of translational complexes into inactive protein aggregates after ischemia (Hu et al, 2004; Liu et al, 2005a, b; Zhang et al, 2006; Hu, 2006).
Co-translational protein aggregation in ischemic vulnerable neurons thus represents a new mechanism underlying irreversible translation arrest after an episode of ischemia (Liu et al, 2005a, b; Zhang et al, 2006; Hu, 2006). An ischemia-induced cascade of energy failure, intracellular calcium overload, overproduction of reactive oxygen species, and acidosis cumulatively damages ATP-dependent protein quality control machinery for co-translational folding, co-translational chaperoning, and translation-coupled protein degradation after brain ischemia (Liu et al, 2004; Hu et al, 2004; Hu, 2006). As a result, nascent polypeptides cannot fold efficiently. Consequently, ribosomes, together with their associated molecular chaperones, aberrant nascent polypeptides, and protein translational initiation, elongation, and termination factors are irreversibly aggregated with each other, or with other subcellular structures (Liu et al, 2005a; Zhang et al, 2006). Co-translational protein aggregation essentially demolishes protein synthesis machinery in ischemic vulnerable neurons. Co-translational protein aggregates accumulate over time, and would be expected to cause irreversible protein synthesis inhibition and contribute to DND in ischemic vulnerable neurons after ischemia.
This model appears consistent with several established facts of ischemic damage (Hu, 2006). (1) Energy failure and loss of ionic homeostasis induce neuronal damage after ischemia. Both depletion of ATP and loss of ionic homeostasis disable co-translational folding machinery and cause overproduction of toxic abnormal proteins in cells (Hartl and Hayer-Hartl, 2002). (2) Permanent inhibition of protein synthesis is one of the most accurate indicators for DND after ischemia (Cooper et al, 1977; Mies et al, 1991; Hossmann, 1993; Hu and Wieloch, 1993; DeGracia, 2004; Hu et al, 2004; Hu, 2006). Abnormal aggregation of ribosomal proteins results in permanent inhibition of protein synthesis after brain ischemia (Liu et al, 2005a; Zhang et al, 2006; Hu, 2006). (3) Free ubiquitin is permanently depleted only in ischemic vulnerable neurons (Magnusson and Wieloch, 1989; Morimoto et al, 1996; Hu et al, 2000, 2001). Abnormal proteins accumulating in neurons deplete intracel-lular free ubiquitin to form ubi-proteins after ischemia. (4) Expression of molecular chaperones before ischemia protects neurons against ischemia (Nowak, 1985; Angelidis et al, 1999; Sharp et al, 1999; Rajdev et al, 2000; Yenari et al, 2001; Giffard and Yenari, 2004). Molecular chaperones can directly neutralize accumulated unfolded or misfolded proteins and assist in protein degradation. However, HSP70 cannot disaggregate protein aggregates that are already formed, and protein aggregation is virtually irreversible (Hu et al, 2000, 2004; Hu, 2006).
Endogenous Stress Pathways and Brain Ischemia and Reperfusion
The above discussion of cotranslational protein aggregation indicates that ischemia leads to dysfunction of protein folding machinery during the postischemia phase (Hu et al, 2000, 2001; Liu et al, 2004, 2005a, b; Zhang et al, 2006). As a result, postischemic cells elicit endogenous stress response pathways. In fact, the cellular stress responses of the brain to ischemia were experimentally established before the dysfunction of protein folding was well understood. By the early 1980s, it was clearly established that the HSR was strongly activated after I/R (for reviews, see Nowak and Jacewicz, 1994; Sharp et al, 1999). Presently, there is evidence for activation of the HSR and the UPR. Although not our intent to review this evidence in great detail, it is important in the present context to (1) establish the activation of these pathways, (2) have some sense of their relative degree of activation, and (3) understand the implications of irreversible translation arrest for the success or failure of the postischemic cells in executing these pathways.
Heat Shock
The first endogenous stress pathway to receive substantial attention after brain I/R was the HSR. Heat shock response-induced transcription is complex, consisting of several families of HSPs including HSP110, HSP90, HSP70, HSP60, HSP47, HSP32, HSP27, and HSP10 (Sharp et al, 1999). There is a large increase in the mRNAs coding for HSPs in brain cells after I/R (Nowak, 1990; Simon et al, 1991; Sharp et al, 1999). Patterns of HSP induction are complex and dependent on: (1) the specific transcript, (2) the ischemia duration, (3) cell type specific expression, (4) induction time course, (5) the species studied, and (6) whether the transcript is translated. In CA1 of gerbil brain, HSP70 was not translated in neurons after 10 mins global ischemia (Vass et al, 1988; Giffard and Yenari, 2004), but it is was translated in rat (Simon et al, 1991), and the latter study showed the complex dependence of HSP70 translation on ischemia duration. The specific details of time course, cell type, species and transcription, and translation products are clearly important for understanding the role of HSR expression after reperfusion. However, it is unequivocal that I/R strongly activates the HSR in post-ischemic neurons.
Generally, it is thought that expression of the HSR is protective, although the functional roles of individual HSR transcription products require further clarification (Sharp et al, 1999). In the context of irreversible translation arrest, upregula-tion of the HSR is significant from two points of view: protecting against I/R-induced protein aggregation and contributing to the dissolution of SGs. Dissolution of SGs requires HSP70 (Gilks et al, 2004). Thus, as elaborated below, the possibility of dysfunctional SGs coupled to the late expression of HSP70 in vulnerable neurons may play a role in SG dysfunction, and highlights an additional aspect of HSP neuroprotection.
With regard to co-translational protein aggregation, recent evidence suggests that unfolded proteins are already irreversibly aggregated from 2 to 4 h of reperfusion onward, but HSP70 is not induced until 4 to 24 h of reperfusion in vulnerable neurons. Therefore, induction of HSP70 by ischemia may be too late to prevent protein aggregation after ischemia (Hu et al, 2000, 2001; Liu et al, 2005a, b; Zhang et al, 2006). Recent studies support the idea that clumping of protein aggregates in protein synthesis machinery in living neurons for an extended period of time will eventually contribute to DND after brain ischemia (Hu et al, 2004; Liu et al, 2005a, b; Zhang et al, 2006; Hu, 2006).
The Unfolded Protein Response
Although eIF2α phosphorylation itself is transient and unlikely to be directly related to cell death, it was important to discover its cause. It has now been established that the eIF2α kinase PERK is activated by reperfusion, and responsible for the large increase in eIF2α(P) during early reperfusion (Kumar et al, 2001, 2003; Hayashi et al, 2003; Owen et al, 2005; reviewed in DeGracia and Montie, 2004). As PERK is one of the upstream effectors of the UPR, its activation has supported the suggestion that I/R damage also involved ER stress (Paschen, 1996).
Several studies of the UPR in the reperfused brain have now been performed (reviewed in DeGracia and Montie, 2004; Paschen, 2003). Unexpectedly, significant aspects of the UPR are not expressed after reperfusion. The transcription factor ATF6 is not activated during early reperfusion (Kumar et al, 2003). Although there is evidence for the processing of xbp-1 mRNA (Paschen et al, 2003; Tajiri et al, 2004), only low levels of the functionally active translation product, 55kDa XBP-1, were detected in a focal ischemia model, and not at all after global ischemia (Paschen et al, 2003). Paschen et al (2003) showed an approximately 50% increase in grp78 mRNA and a doubling of chop mRNA, as opposed to a 550-fold increase in hsp70 mRNA in a focal ischemia model. In comparison, treatment of cultured primary neurons with thapsigargin (a specific inducer of ER stress) led to 200-fold increases in the UPR induction of grp94, chop, and gadd34 mRNAs (Mengesdorf et al, 2001). Increased ATF4 protein was not detected after cardiac arrest (Kumar et al, 2003), but was detected after occlusive global ischemia (Hayashi et al, 2005). Multiple studies have shown an increase in both the mRNA and protein for GADD34 (Doutheil et al, 1999; Imai et al, 2002; Paschen et al, 2004; McCaig et al, 2005), the phosphatase 1 subunit that accelerates the dephosphorylation of eIF2α(P) after I/R. Increased GADD34 is consistent with the short duration (within 4 to 8h) of eIF2α(P) in the reperfused brain.
Hence, the PERK response is strong with initial reperfusion, but increases in UPR-induced transcripts are unexpectedly low. This could be because of (1) PERK being more sensitive to ER-stress in brain than ATF6 and IRE1, and/or (2) concurrent damage mechanisms—such as free radical damage to ER membranes (Hayashi et al, 2003)—may attenuate the activity of the UPR transcriptional effectors IRE1 and ATF6. In this regard, Shang and Lehrman (2004) showed that there was no correlation between the degree of UPR effector activation and the resulting levels of UPR-induced mRNAs. This can provide one type of explanation as to why a large activation of PERK could occur in reperfused brain, but be followed by low levels of UPR-induced transcripts such as grp78 or chop. Additionally, as elaborated in Paschen (2003), the irreversible translation arrest will prevent synthesis of transcriptionally active XBP-1 protein, which, in conjunction with lack of activation of ATF6 (Kumar et al, 2003) will further preclude increases in UPR-induced transcripts.
However this issue eventually resolves, when looked at from the view of in vivo induction of stress-induced mRNAs, the UPR is orders of magnitude weaker than the HSR. This is vividly illustrated in Figure 5, in which, after global ischemia, hsp70 mRNA expression is the strongest among inducible chaperone genes, whereas grp78 transcription is only moderately expressed in vulnerable CA1 neurons (BR Hu, unpublished data). The massive increase in hsp70, and only moderate increase in grp78, is consistent with high levels of damage to cytoplasmic proteins, as discussed above.
Figure 5.

Induction of cytosolic and ER stress genes after transient cerebral ischemia. Brain sections were obtained from sham-operated control rats and rats subjected to 15 mins of ischemia followed by 4, 24, and 48 h of reperfusion. Ribo-probes were used for in situ hybridization. Cytosolic stress gene hsp70 is dramatically upregulated, whereas ER chaperone grp78 is mildly or moderately induced in 24 h of reperfusion in CA1 neurons undergoing delayed neuronal death after ischemia. At 48 h of reperfusion, however, both HSP70 and GRP78 mRNAs decline because delayed neuronal death occurred in CA1 neurons after transient cerebral ischemia (BR Hu, unpublished observation).
Stress Granules after Brain Ischemia and Reperfusion
Although there are important unanswered questions regarding expression of the UPR after I/R, the downstream consequences of PERK-mediated eIF2α phosphorylation have implications beyond preventing formation of ternary complexes. As detailed above, eIF2α phosphorylation leads to inhibition of polysome formation, and is the main physiologic trigger for SG formation (Anderson and Kedersha, 2002). eIF2α serine 51-to-alanine mutants are incapable of forming SGs (McEwen et al, 2005), and transfection of cells with ser to asp 51 eIF2α induces SGs, even in the absence of exogenous stress (Kedersha et al, 2002). Since eIF2α phosphorylation does not distinguish selectively vulnerable brain regions (e.g., Figure 1), it was possible that a downstream effect of eIF2α phosphorylation did distinguish vulnerable and resistant neurons. Hence, studies of SGs were undertaken. At present, there have been three studies of SGs which suggest there are substantial differences in the behavior of SGs between ischemic vulnerable and resistant brain regions.
Direct visualization of SGs used IF microscopy to view punctate cytoplasmic localization of TIA-1 in hippocampal neurons after cardiac arrest-induced global brain ischemia. Kayali et al (2005) used small ribosomal protein S6 colocalization with cyto-plasmic TIA-1 as a marker for SGs, and DeGracia et al (2006) used colocalization of cytoplasmic TIA-1 and eIF4G. Both studies provided quantitative and qualitative evidence that SGs in vulnerable CA1 neurons were different than SGs in neurons of resistant regions.
Estimates of the number of SGs per cell were determined because the number of SGs should parallel levels of eIF2α phosphorylation, as per the equilibrium depicted in Figure 4. Such a correlation was observed in resistant hippocampal areas, where the number of SGs peaked at 10 mins reperfusion, when eIF2α phosphorylation was maximum (e.g., Figure 1), and declined back to baseline at 4 h reperfusion, when eIF2α phosphorylation had decreased 80% from its peak at 10 mins reperfusion. However, in CA1, the total number of S6-containing SGs did not change with reperfusion (Kayali et al, 2005). Stress granules containing eIF4G in CA1 showed a trend towards decreasing at 10 mins reperfusion, an increase at 90 mins reperfusion, and return to control levels at 4 h reperfusion, suggesting a dynamic flux of eIF4G out of and back into SGs within the first 4 h reperfusion (DeGracia et al, 2006). However, eIF2α(P) is known to increase in CA1 pyramidal neurons to the same degree as in other hippocampal regions (DeGracia et al, 1997), so these results suggest a defect in SG formation exclusively in CA1. As discussed above, formation of SGs is a protective response during stress induced translation arrest; hence, their increase in hippocampal regions is likely neuroprotective, and the lack of increase in CA1 is expected to be detrimental.
Ischemia and reperfusion-induced changes in eIF4G provide a model to account for the differences in SGs between CA1 and resistant regions. Proteolysis of eIF4G is greater in CA1 than in DG/CA3 (Martin de la Vega et al, 2001), which may, in part account for the trend towards decreasing eIF4G in SGs of CA1 neurons at 10 mins reperfusion. Similarly, there is a corresponding decrease in eIF4F in CA1 compared with CA3 (Martin de la Vega et al, 2001). Since eIF4F catalyzes formation of the 48S preinitiation complex, one would expect a decrease in the 48S preinitiation complex specifically in CA1. Since it is the 48S preinitiation complex, or a modified form of it (Anderson and Kedersha, 2002), that is sequestered in SGs, then a decrease in 48S preinitiation complexes in CA1 would preclude an increase in the total number of SGs in CA1, as seen in the Kayali et al (2005) study. Additionally, aggregates of eIF4G were seen to form at very high levels in CA1 compared with CA3 after 90 mins reperfusion, and it is tempting to speculate that these may represent eIF4G (or its fragments) accumulating in the ubi-protein complexes discussed above. Hence, these are additional mechanisms whereby eIF4G proteolysis may contribute to translation dysfunction in vulnerable reperfused neurons.
With regard to S6-containing SGs, two major qualitative differences were observed in Kayali et al (2005). First, software reconstruction microscopy techniques allowed visualization of SGs in three dimensions. This led to the novel discovery that SGs had height and possessed an irregular columnar appearance in profile. The height profile of SGs differed between resistant and vulnerable neurons. Although providing only a course-grained indicator, these profiles suggest differences in SGs between CA1 and CA3 neurons. In resistant neurons at 4h reperfusion, the profile of the SGs consisted of a core structure in which S6 and TIA-1 were colocalized, surrounded by a flanking area in which free S6 was present, suggesting perhaps a dissolution of the SGs in the resistant neurons. In contrast, in CA1, only a spherical core of colocalized TIA-1 and S6 was visible, with no surrounding area, suggesting a tight association of S6 and TIA-1.
The second major qualitative observation was that, at 4 h reperfusion, all S6 was exclusively localized in SGs only in CA1 pyramidal neurons (Figure 6). Free cytoplasmic S6 was not observed in CA1 pyramidal neurons, but was present in all other hippocampal cell types, including CA1 interneurons. This is a significant finding as it can completely account for translation arrest exclusively in CA1 pyramidal neurons at 4 h reperfusion, when the levels of eIF2α(P) are only approximately three-fold over baseline (as compared with 12-fold over baseline at 10 mins reperfusion). The basis for this finding is not presently known and represents an important area for continuing investigation. Western blots of S6 in unfractionated whole hippocampal homogenates indicated a 50% decrease in S6 signal at 4 h reperfusion (Kayali et al, 2005). This would suggest that there is either a loss of S6 (and hence of 40S subunits), in CA1 pyramidal neurons, or that the 40S subunits that are not sequestered in SGs undergo some type of change that masks their antigenicity (e.g., by becoming bound in protein aggregates).
Figure 6.
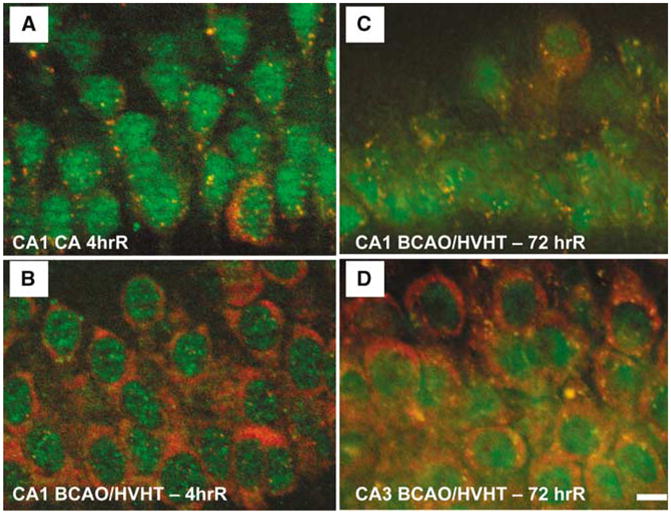
Stress granules in ischemic and reperfused CA1. (A) At 4 h reperfusion after a 10 mins cardiac arrest (CA), small ribosomal protein S6 (red) is exclusively localized in SGs, as marked by cytoplasmic colocalization with TIA-1 (green). (B) After 4 h reperfusion after 10 mins forebrain ischemia induced by bilateral carotid artery occlusion and hypovolemic hypotension (BCAO/HVHT) to 40mm Hg, CA1 pyramidal neurons resemble controls with robust free S6 staining, and a few cytoplasmic SGs. (C) At 3 days reperfusion after 10 mins BCAO/HVHT, CA1 pyramidal neurons show S6 to be exclusively localized to SGs, similar to the 4 h cardiac arrest animals. (D) However, at 3 days reperfusion after 10 mins BCAO/HVHT, pyramidal neurons of CA3 resemble controls, with robust free S6 staining, and a few cytoplasmic SGs. In (A) and (C), pyramidal layer interneurons in CA1, identified by their large cytoplasm, show staining patterns like controls, indicating that the alteration of SGs is exclusively localized to pyramidal neurons and not interneurons in CA1, consistent with known patterns of protein synthesis inhibition and cell death. Immunohistochemistry was performed as described in Kayali et al (2005). Scale bar in (D) is 10 μm and applies to all four panels.
To test longer durations of reperfusion, the bilateral carotid artery occlusion plus hypovolemic hypotension (BCAO/HVHT) model of forebrain ischemia (Smith et al, 1984) was used to assess SGs, via S6 and TIA-1 antibodies, at 72 h reperfusion (DJ DeGracia, unpublished results). CA1 neurons qualitatively resemble controls at 4 h reperfusion with this model (Figure 6B). At 72h reperfusion, after 10 mins BCAO/HVHT, CA1 neurons showed prominent SGs and a dearth of cytoplasmic S6 staining (Figure 6C). In contrast, at 72 h reper-fusion, CA3 pyramidal neurons resembled controls (Figure 6D). Since CA1 neurons are dead by 72 h reperfusion, accumulation of SGs may be a result of and not a cause of cell death. Ongoing work is addressing this issue. However, the resemblance between CA1 neurons at 4 h after cardiac arrest and 72 h after BCAO/HVHT suggests that similar processes are operating, though at a slower rate after BCAO/HVHT.
The third study to indirectly evaluate SGs showed that TIA-1 is present in highly insoluble ultracentrifugation gradient fractions of detergent-washed 10,000g pellets from reperfused animals after focal ischemia (BR Hu, unpublished observations). In controls, TIA-1 was not observed in this fraction. The level of TIA-1 in these gradient fractions increased with longer reperfusion durations. This result is particularly intriguing as others have shown that, after standard homogenization procedures, SGs enter the soluble fraction on differential centrifugation (Nover et al, 1983, 1989), and TIA-1 runs in the lightest (least dense) fractions after density gradient ultracentrifugation (Kedersha et al, 1999). The detection of TIA-1 in insoluble protein aggregate-containing fractions of reperfused brain suggests that SGs may be ‘trapped’ in insoluble protein aggregates at later reperfusion durations (Zhang et al, 2006). Thus, the trapping of SGs in protein aggregates in CA1 may account for the qualitative differences in SGs described above, and additionally indicate that a convergence of these mechanisms further contributes to irreversible translation arrest in ischemic vulnerable neurons.
Kayali et al (2005) presented the unexpected observation of a basal level of SGs in control brains, which occurred whether the anesthetic agent used was Ketamine, halothane or CO2 (DJ DeGracia, unpublished observation). These were observed in neurons of all forebrain regions including the cerebral cortex, thalamus, amygdala, and basal ganglia. A basal level of SGs in controls is consistent with the low levels of eIF2α(P) in control brains (~1% of total eIF2α; DeGracia et al, 1997). Additionally, studies of SGs in cell culture show some degree of cytoplasmic localization of TIA-1 in controls, which has led Kedersha et al (1999) to suggest that accumulation of SGs after cell stress represents an ‘extreme case’ of SG formation. The physiological function of SGs in control neurons is currently under investigation.
Stress Granule Ultrastructure
The ‘dark substance’ of Kirino et al (1984) and the ‘dark material’ of Deshpande et al (1992) described above are now known to be a proteinaceous substance (misfolded protein aggregates) containing high levels of ubi-proteins, molecular chaperones, and folding enzymes, as well as components of protein translational machinery (Hu et al, 2000, 2001, 2004; Hu, 2006; Liu et al, 2004, 2005a, b; Liu and Hu, 2004; Zhang et al, 2006). Here, we would like to comment on the potential relationship of these EM-identified structures to SGs, in the context of ultrastructural correlates of SGs.
Kayali et al (2005) described electron dense particles of 100 to 200 nm constitutively present in hippocampal control neurons. Based on the size and morphological similarity to immunogold-EM-identified SGs described in cultured cells in Gilks et al (2004), these particles were taken to be the ultra-structural correlates of SGs. Similar to the IF identified SGs, these particles increased in number in CA3 but not CA1 at 10 mins reperfusion. The inconsistency between the size of the EM particles and SGs as detected by IF (~1 to 5 μm) was explained by the tendency of several of the 100 to 200 nm particles to be in close proximity over micron sized areas, and the suggestion that IF thus detected clusters of the EM particles. Definitive identification of the 100 to 200 nm particles as SGs (via immunogold labeling) has not yet been performed.
We note that Kayali et al (2005) is not the first EM description of such structures in neurons. Electron microscopic descriptions of such particles in neurons date back several decades (Shimizu and Ishii, 1965; Gambetti et al, 1968). Older studies described these particles (Shimizu and Ishii, 1965) or noted conditions, such as puromycin administration (Gambetti et al, 1968), which enhanced their formation. This latter point is significant as puromycin enhances formation of SGs, even in the absence of exogenous stress (Kedersha et al, 1999). Salient descriptions indicated the particles were: (1) cytoplasmic, (2) not membrane enclosed, (3) surrounded by, or in contact with ribosomes, and (4) were sometimes in association with the outer face of the nuclear membrane or ER membranous sacs.
Stress Granules and Protein Aggregates
What is the relationship, if any, between the particles identified by the Hu and DeGracia labs by both IF and EM? Kayali et al (2005) showed that TIA-1/S6 immunoreactive SGs were present in control neurons, increased in resistant CA3, DG and hilar neurons, but were unchanged in CA1 during the early period of reperfusion (Kayali et al, 2005). In contrast, ubi-protein immunoclusters viewed by confocal microscopy were not present in control cells, highly accumulated in all ischemic neurons including CA1, CA3, DG, and neocortical neurons during the early periods of reperfusion (Hu et al, 2000, 2001; Liu et al, 2005a, b). These differences follow from the respective compositions of the particles: ubi-protein clusters are composed of misfolded or damaged proteins that are conjugated by ubiquitin, whereas SGs are composed of the poly-A mRNAs (Nover et al, 1989; Kedersha et al, 1999), 40S ribosomal subunits and several initiation factors (Kedersha et al, 2002; Kimball et al, 2003), but not 60S ribosomal subunits (Kedersha et al, 2002). During 4 to 24 h reperfusion, ubi-protein immuno-clusters changed further between resistant and vulnerable neurons, abating in most resistant neurons but persisting with different shape and distribution in CA1 neurons (Hu et al, 2000, 2001; Liu et al, 2005b). The number of SGs returned to control levels in all hippocampal neurons by 4 h of reperfusion, with, however, indications that the compositions of the SGs was substantially altered exclusively in CA1 pyramidal neurons as compared with controls (Kayali et al, 2005; DeGracia et al, 2006).
Therefore, the respective particles identified in the Hu and DeGracia studies display distinct characteristics and behaviors, although both show specific differential changes between vulnerable and resistant neurons. Thus, co-translational protein aggregates and SGs are two separate and different phenomena contributing to alterations in ribosomes in postischemic neurons. Co-translational protein aggregation is an irreversible process, caused by misfolded proteins in which all components of translational complexes, including both 40S and 60S ribosomal subunits are present (Liu et al, 2005a, b; Zhang et al, 2006; Hu, 2006). Stress granules contain the 48S preinitiation complex, TIA-1, and a number of other RNA-interacting proteins (Anderson and Kedersha, 2006). After reperfusion, SG formation is reversible in resistant neurons, and their composition at later reperfusion requires investigation. Because SGs are increased in ischemic resistant neurons during early reperfusion, in conjunction with the observation of TIA-1 in insoluble ultracentrifuge fractions, it remains possible that SG components such as TIA-1 are trapped into protein aggregates in ischemic vulnerable neurons during the late period of reperfusion after brain ischemia (Zhang et al, 2006).
The above results indicate clearly that ischemic vulnerable neurons show major differences from resistant neurons with regard to the persistence of the stress stimulus in the form of co-translational protein aggregates, and with factors related to the maintenance of translation arrest in the form of alterations of SGs. To summarize, both resistant and vulnerable areas initially show evidence of ubi-protein complexes during early reperfusion, but these clear from resistant areas and persist in vulnerable areas. Ethanolic phosphotungstic acid-reactive and ubiquitin-immunogold positive protein aggregates are visible by EM only from 4 h of reperfusion onward exclusively in CA1 neurons, and progressively increased until cell death (Hu et al, 2000, 2001; Liu et al, 2004, 2005a, b). Changes in SGs, between vulnerable and resistant neurons, in terms of quantitative behavior and composition, occur rapidly with reperfusion. In both cases, persistence of protein aggregates and alterations in SGs, these strictly correlate with ischemic vulnerable cell populations. Thus, it is not unreasonable to state that these factors contribute to irreversible translation arrest. Discerning why these differences exist is expected to provide a causal link between irreversible translation arrest and post-ischemic DND.
Closing Comments
In closing we would like to consider possible mechanisms by which irreversible inhibition of protein syntheses contributes to delayed neuronal demise after a brief episode of brain ischemia, and discuss some possible future directions in the area. Protein metabolism and turnover are fundamental processes for cell survival, and expectedly, irreversible deprivation of these processes will ultimately lead to cell death.
There are at least two means by which irreversible aggregation of protein synthesis machinery may contribute to DND: (1) irreversible protein aggregates clump with the ER, mitochondrial, nuclear and cell membranes, thus damaging these subcellular structures (Hu et al, 2000), and (2) accumulation in the plasma membranes of large quantities of denatured or damaged proteins, without appropriate turnover because of irreversible inhibition of protein synthesis, may serve as a stimulant attracting microglia or macrophages to remove them (Liu et al, 2005a; Zhang et al, 2006; Hu, 2006). The latter explanation is consistent with the fact that DND after a brief episode of brain ischemia is not associated with cell membrane rupture as seen in classical necrosis, but rather by cell shrinkage to form dark (dead) neurons (Smith et al, 1984). This is also consistent with the fact that microglia and macrophages migrate into the CA1 pyramidal layer during post-ischemic DND, and eventually remove these dark neurons (Zheng and Yenari, 2004).
Pertaining to irreversible dysfunction of SGs, it is plausible to hypothesize that proteolytic alterations of eIF4G underlie SG changes. Reperfused CA3/DG show less eIF4G degradation and higher levels of eIF4F than CA1 after lethal I/R (Martin de la Vega et al, 2001). Further, IPC decreases the amount of eIF4G degradation during subsequent lethal ischemia, as compared with unconditioned animals (Garcia et al, 2004). Preconditioned, reperfused CA1 neurons do not display persistent translation arrest, but do display transient eIF2α(P) (Garcia et al, 2004). As eIF4G is known to be degraded by calpain I in the ischemic brain (Neumar et al, 1998), this suggests calpain-mediated proteolysis of eIF4G contributes, at least in part, to SG dysfunction specifically in CA1 by preventing delivery of mRNA-bound 40S subunits to SGs. This mechanism could be tested in vivo by administering calpain inhibitors and assessing SG behavior.
With regard to in vitro model systems of potential utility for further investigating these mechanisms, the above postulated model of SG dysfunction is based on the presence of two exogenous stresses: first, ER stress triggers eIF2α phosphorylation (and subsequent SG formation) and second, excessive Ca2+ entry triggers pathologic calpain 1 activation. Hence, in vitro systems that simulate multiple simultaneous damage mechanisms may provide models for the phenomenology we have identified here. For example, Paschen et al (2001) showed that sequential treatment of primary neurons with hydrogen peroxide (an oxidative stress) and thapsigargin (an ER stressor) blunted the ER stress response of the cells, producing a phenomenology in the cells that more resembles the case after in vivo brain I/R, where production of UPR transcripts is substantially below expected (e.g., as in Figure 5). It is worth investigating whether similar multidamage models can recreate the irreversible aggregation of proteins or alterations of SGs that occur in the in vivo reperfused brain.
To conclude, many investigators have clearly recognized the correlation between irreversible translation arrest and the death of selectively vulnerable neurons after brain I/R. The converse of this insight is as equally important: postischemic neurons that recover protein synthesis survive. These correlations suggest that understanding why vulnerable neurons display irreversible translation arrest will allow opportunities to recover translation through intervention, and thereby salvage vulnerable neuron populations. These insights have been the underlying impetus to discover the mechanisms of translation arrest in the reperfused brain.
Here, we have developed the notion that translation arrest is an integrated component of cellular stress responses. This allows recognition that postischemic translation arrest is a specific example of processes generally expressed in stressed eukaryotic cells. Recognition of the various phases of translation arrest (e.g., stress stimulus, followed by induction, maintenance, and termination of translation arrest) allows us to organize the present body of data. We have discussed two lines of recent evidence suggesting that irreversible translational arrest may be caused by: (1) co-translational protein misfolding and aggregation; and (2) dysfunction of SGs, even after the main inducer, eIF2α phosphorylation, abates. These results provide confidence that knowledge of the relationship between irreversible translation arrest and cell death will offer new inroads for the development of therapies to halt DND after brain I/R.
Acknowledgments
This work was supported by the National Institute of Neurological Disorders and Stroke (NS044100 to DJD; NS40407 and NS36386 to BRH).
References
- Althausen S, Mengesdorf T, Mies G, Olah L, Nairn AC, Proud CG, Paschen W. Changes in the phosphorylation of initiation factor eIF-2alpha, elongation factor eEF-2 and p70 S6 kinase after transient focal cerebral ischaemia in mice. J Neurochem. 2001;78:779–87. doi: 10.1046/j.1471-4159.2001.00462.x. [DOI] [PubMed] [Google Scholar]
- Anderson P, Kedersha N. Visibly stressed: the role of eIF2, TIA-1, and stress granules in protein translation. Cell Stress Chaperones. 2002;7:213–21. doi: 10.1379/1466-1268(2002)007<0213:vstroe>2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson P, Kedersha N. RNA granules. J Cell Biol. 2006;13172:803–8. doi: 10.1083/jcb.200512082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angelidis CE, Lazaridis I, Pagoulatos GN. Aggregation of hsp70 and hsc70 in vivo is distinct and temperature-dependent and their chaperone function is directly related to non-aggregated forms. Eur J Biochem. 1999;259:505–12. doi: 10.1046/j.1432-1327.1999.00078.x. [DOI] [PubMed] [Google Scholar]
- Araki T, Kato H, Inoue T, Kogure K. Regional impairment of protein synthesis following brief cerebral ischemia in the gerbil. Acta Neuropathol (Berl) 1990;199079:501–5. doi: 10.1007/BF00296109. [DOI] [PubMed] [Google Scholar]
- Beere HM. Death versus survival: functional interaction between the apoptotic and stress-inducible heat shock protein pathways. J Clin Invest. 2005;115:2633–9. doi: 10.1172/JCI26471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodsch W, Takahashi K, Barbier A, Ophoff BG, Hossmann KA. Cerebral protein synthesis and ischemia. Prog Brain Res. 1985;63:197–210. doi: 10.1016/S0079-6123(08)61984-6. [DOI] [PubMed] [Google Scholar]
- Brostrom CO, Brostrom MA. Regulation of translational initiation during cellular responses to stress. Prog Nucleic Acid Res Mol Biol. 1998;58:79–125. doi: 10.1016/s0079-6603(08)60034-3. [DOI] [PubMed] [Google Scholar]
- Bukau B, Hesterkamp T, Luirink J. Growing up in a dangerous environment: a network of multiple targeting and folding pathways for nascent polypeptides in the cytosol. Trends Cell Biol. 1996;6:480–6. doi: 10.1016/0962-8924(96)84946-4. [DOI] [PubMed] [Google Scholar]
- Burda J, Chavko M. Mechanism of protein synthesis inhibition in CNS during postischaemic reperfusion. Physiol Res. 1991;40:395–402. [PubMed] [Google Scholar]
- Burda J, Martin ME, Garcia A, Alcazar A, Fando JL, Salinas M. Phosphorylation of the alpha subunit of initiation factor 2 correlates with the inhibition of translation following transient cerebral ischaemia in the rat. Biochem J. 1994;302:335–8. doi: 10.1042/bj3020335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burry RW, Lasher RS. A quantitative electron microscopic study of synapse formation in dispersed cell cultures of rat cerebellum stained either by Os-UL or by EPTA. Brain Res. 1978;147:1–15. doi: 10.1016/0006-8993(78)90768-0. [DOI] [PubMed] [Google Scholar]
- Calfon M, Zeng H, Urano F, Till JH, Hubbard SR, Harding HP, Clark SG, Ron D. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature. 2002;3415:92–6. doi: 10.1038/415092a. [DOI] [PubMed] [Google Scholar]
- Chavko M, Burda J, Danielisova V, Marsala J. Molecular mechanisms of ischemic damage of spinal cord. Gerontology. 1987;33:220–6. doi: 10.1159/000212881. [DOI] [PubMed] [Google Scholar]
- Clemens MJ. Initiation factor eIF2 alpha phosphorylation in stress responses and apoptosis. Prog Mol Subcell Biol. 2001;27:57–89. doi: 10.1007/978-3-662-09889-9_3. [DOI] [PubMed] [Google Scholar]
- Cooper HK, Zalewska T, Kawakami S, Hossmann KA, Kleihues P. Delayed inhibition of protein synthesis during recirculation after compression ischemia of the rat brain. Acta Neurol Scand Suppl. 1977;64:130–1. [PubMed] [Google Scholar]
- DeGracia DJ. Acute and persistent protein synthesis inhibition following cerebral reperfusion. J Neurosci Res. 2004;77:771–6. doi: 10.1002/jnr.20225. [DOI] [PubMed] [Google Scholar]
- DeGracia DJ, Kumar R, Owen CR, Krause GS, White BC. Molecular pathways of protein synthesis inhibition during brain reperfusion: implications for neuronal survival or death. J Cereb Blood Flow Metab. 2002;22:127–41. doi: 10.1097/00004647-200202000-00001. [DOI] [PubMed] [Google Scholar]
- DeGracia DJ, Montie HL. Cerebral ischemia and the unfolded protein response. J Neurochem. 2004;91:1–8. doi: 10.1111/j.1471-4159.2004.02703.x. [DOI] [PubMed] [Google Scholar]
- DeGracia DJ, Neumar RW, White BC, Krause GS. Global brain ischemia and reperfusion: modifications in eukaryotic initiation factors associated with inhibition of translation initiation. J Neurochem. 1996;67:2005–12. doi: 10.1046/j.1471-4159.1996.67052005.x. [DOI] [PubMed] [Google Scholar]
- DeGracia DJ, Rafols JA, Morley SJ, Kayali F. Immunohistochemical mapping of total and phosphorylated eukaryotic initiation factor 4G in rat hippocampus following global brain ischemia and reperfusion. Neuroscience. 2006;139:1235–48. doi: 10.1016/j.neuroscience.2006.01.038. [DOI] [PubMed] [Google Scholar]
- DeGracia DJ, Sullivan JM, Neumar RW, Alousi SS, Hikade KR, Pittman JE, White BC, Rafols JA, Krause GS. Effect of brain ischemia and reperfusion on the localization of phosphorylated eukaryotic initiation factor 2 alpha. J Cereb Blood Flow Metab. 1997;17:1291–302. doi: 10.1097/00004647-199712000-00004. [DOI] [PubMed] [Google Scholar]
- Dember LM, Kim ND, Liu KQ, Anderson P. Individual RNA recognition motifs of TIA-1 and TIAR have different RNA binding specificities. J Biol Chem. 1996;2271:2783–8. doi: 10.1074/jbc.271.5.2783. [DOI] [PubMed] [Google Scholar]
- Deshpande J, Bergstedt K, Linden T, Kalimo H, Wieloch T. Ultrastructural changes in the hippocampal CA1 region following transient cerebral ischemia: evidence against programmed cell death. Exp Brain Res. 1992;88:91–105. doi: 10.1007/BF02259131. [DOI] [PubMed] [Google Scholar]
- Dewar D, Graham DI, Teasdale GM, McCulloch J. Cerebral ischemia induces alterations in tau and ubiquitin proteins. Dementia. 1994;5:168–73. doi: 10.1159/000106716. [DOI] [PubMed] [Google Scholar]
- Dienel GA, Pulsinelli WA, Duffy TE. Regional protein synthesis in rat brain following acute hemispheric ischemia. J Neurochem. 1980;35:1216–26. doi: 10.1111/j.1471-4159.1980.tb07878.x. [DOI] [PubMed] [Google Scholar]
- Doutheil J, Althausen S, Gissel C, Paschen W. Activation of MYD116 (gadd34) expression following transient forebrain ischemia of rat: implications for a role of disturbances of endoplasmic reticulum calcium homeostasis. Brain Res Mol Brain Res. 1999;863:225–32. doi: 10.1016/s0169-328x(98)00276-9. [DOI] [PubMed] [Google Scholar]
- Frydman J. Folding of newly translated proteins in vivo: the role of molecular chaperones. Annu Rev Biochem. 2001;70:603–47. doi: 10.1146/annurev.biochem.70.1.603. [DOI] [PubMed] [Google Scholar]
- Gale M, Jr, Tan SL, Katze MG. Translational control of viral gene expression in eukaryotes. Microbiol Mol Biol Rev. 2000;64:239–80. doi: 10.1128/mmbr.64.2.239-280.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gambetti P, Gonatas NK, Flexner LB. The fine structure of puromycin-induced changes in mouse entorhinal cortex. J Cell Biol. 1968;36:379–90. doi: 10.1083/jcb.36.2.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia L, Burda J, Hrehorovska M, Burda R, Martin ME, Salinas M. Ischaemic preconditioning in the rat brain: effect on the activity of several initiation factors, Akt and extracellular signal-regulated protein kinase phosphorylation, and GRP78 and GADD34 expression. J Neurochem. 2004;88:136–47. doi: 10.1111/j.1471-4159.2004.02188.x. [DOI] [PubMed] [Google Scholar]
- Giffard RG, Yenari MA. Many mechanisms for hsp70 protection from cerebral ischemia. J Neurosurg Anesthesiol. 2004;16:53–61. doi: 10.1097/00008506-200401000-00010. [DOI] [PubMed] [Google Scholar]
- Gilks N, Kedersha N, Ayodele M, Shen L, Stoecklin G, Dember LM, Anderson P. Stress granule assembly is mediated by prion-like aggregation of TIA-1. Mol Biol Cell. 2004;15:5383–98. doi: 10.1091/mbc.E04-08-0715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gingras AC, Raught B, Sonenberg N. eIF4 initiation factors: effectors of mRNA recruitment to ribosomes and regulators of translation. Annu Rev Biochem. 1999;68:913–63. doi: 10.1146/annurev.biochem.68.1.913. [DOI] [PubMed] [Google Scholar]
- Gueydan C, Droogmans L, Chalon P, Huez G, Caput D, Kruys V. Identification of TIAR as a protein binding to the translational regulatory AU-rich element of tumor necrosis factor alpha mRNA. J Biol Chem. 1999;22274:2322–6. doi: 10.1074/jbc.274.4.2322. [DOI] [PubMed] [Google Scholar]
- Halford NG, Hey S, Jhurreea D, Laurie S, McKibbin RS, Zhang Y, Paul MJ. Highly conserved protein kinases involved in the regulation of carbon and amino acid metabolism. J Exp Bot. 2004;55:35–42. doi: 10.1093/jxb/erh019. [DOI] [PubMed] [Google Scholar]
- Hardesty B, Tsalkova T, Kramer G. Co-translational folding. Curr Opin Struct Biol. 1999;9:111–4. doi: 10.1016/s0959-440x(99)80014-1. [DOI] [PubMed] [Google Scholar]
- Harding HP, Calfon M, Urano F, Novoa I, Ron D. Transcriptional and translational control in the mammalian unfolded protein response. Annu Rev Cell Dev Biol. 2002;18:575–99. doi: 10.1146/annurev.cellbio.18.011402.160624. [DOI] [PubMed] [Google Scholar]
- Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M, Ron D. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol Cell. 2000;6:1099–108. doi: 10.1016/s1097-2765(00)00108-8. [DOI] [PubMed] [Google Scholar]
- Harding HP, Zhang Y, Ron D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature. 1999;21397:271–4. doi: 10.1038/16729. [DOI] [PubMed] [Google Scholar]
- Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M, Sadri N, Yun C, Popko B, Paules R, Stojdl DF, Bell JC, Hettmann T, Leiden JM, Ron D. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell. 2003;11:619–33. doi: 10.1016/s1097-2765(03)00105-9. [DOI] [PubMed] [Google Scholar]
- Hartl FU, Hayer-Hartl M. Molecular chaperones in the cytosol: from nascent chain to folded protein. Science. 2002;295:1852–8. doi: 10.1126/science.1068408. [DOI] [PubMed] [Google Scholar]
- Hayashi T, Saito A, Okuno S, Ferrand-Drake M, Dodd RL, Chan PH. Damage to the endoplasmic reticulum and activation of apoptotic machinery by oxidative stress in ischemic neurons. J Cereb Blood Flow Metab. 2005;25:41–53. doi: 10.1038/sj.jcbfm.9600005. [DOI] [PubMed] [Google Scholar]
- Hayashi T, Saito A, Okuno S, Ferrand-Drake M, Dodd RL, Nishi T, Maier CM, Kinouchi H, Chan PH. Oxidative damage to the endoplasmic reticulum is implicated in ischemic neuronal cell death. J Cereb Blood Flow Metab. 2003;23:1117–28. doi: 10.1097/01.WCB.0000089600.87125.AD. [DOI] [PubMed] [Google Scholar]
- Hayashi T, Tanaka J, Kamikubo T, Takada K, Matsuda M. Increase in ubiquitin conjugates dependent on ischemic damage. Brain Res. 1993;620:171–3. doi: 10.1016/0006-8993(93)90288-x. [DOI] [PubMed] [Google Scholar]
- Haze K, Yoshida H, Yanagi H, Yura T, Mori K. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol Biol Cell. 1999;10:3787–99. doi: 10.1091/mbc.10.11.3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hossmann KA. Disturbances of cerebral protein synthesis and ischemic cell death. Prog Brain Res. 1993;96:167–77. doi: 10.1016/s0079-6123(08)63265-3. [DOI] [PubMed] [Google Scholar]
- Hu BR. Co-translational protein folding and aggregation after brain ischemia. In: Abel Lajtha., editor. Handbook of neurochemistry and molecular neurobiology. 3rd. Vol. 23. Berlin Heidelberg New York: 2006. acute ischemic injury and repair in the nervous system (Vol Editor: Pak Chan) in press. [Google Scholar]
- Hu BR, Janelidze S, Ginsberg MD, Busto R, Perez-Pinzon M, Sick TJ, Siesjo BK, Liu CL. Protein aggregation after focal brain ischemia and reperfusion. J Cereb Blood Flow Metab. 2001;21:865–75. doi: 10.1097/00004647-200107000-00012. [DOI] [PubMed] [Google Scholar]
- Hu BR, Martone ME, Liu CL. Protein aggregation, unfolded protein response and delayed neuronal death after brain ischemia. In: Buchan VA, Ito U, editors. Maturation phenomenon in cerebral ischemia. Berlin Heidelberg: Springer-Verlag; 2004. pp. 225–37. [Google Scholar]
- Hu BR, Martone ME, Jones YZ, Liu CL. Protein aggregation after transient cerebral ischemia. J Neurosci. 2000;20:3191–9. doi: 10.1523/JNEUROSCI.20-09-03191.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu BR, Park M, Martone ME, Fischer WH, Ellisman MH, Zivin JA. Assembly of proteins to postsynaptic densities after transient cerebral ischemia. J Neurosci. 1998;18:625–33. doi: 10.1523/JNEUROSCI.18-02-00625.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu BR, Wieloch T. Stress-induced inhibition of protein synthesis initiation: Modulation of initiation factor 2 and guanine nucleotide exchange factor activity following transient cerebral ischemia in the rat. J Neurosci. 1993;13:1830–8. doi: 10.1523/JNEUROSCI.13-05-01830.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai H, Harland J, McCulloch J, Graham DI, Brown SM, Macrae IM. Specific expression of the cell cycle regulation proteins, GADD34 and PCNA, in the peri-infarct zone after focal cerebral ischaemia in the rat. Eur J Neurosci. 2002;15:1929–36. doi: 10.1046/j.1460-9568.2002.02025.x. [DOI] [PubMed] [Google Scholar]
- Ito U, Spatz M, Walker JT, Jr, Klatzo I. Experimental cerebral ischemia in mongolian gerbils I Light microscopic observations. Acta Neuropathol (Berl) 1975;32:209–23. doi: 10.1007/BF00696570. [DOI] [PubMed] [Google Scholar]
- Kaufman RJ. Stress signaling from the lumen of the endoplasmic reticulum: coordination of gene transcriptional and translational controls. Genes Dev. 1999;1513:1211–33. doi: 10.1101/gad.13.10.1211. [DOI] [PubMed] [Google Scholar]
- Kawakami A, Tian Q, Duan X, Streuli M, Schlossman SF, Anderson P. Identification and functional characterization of a TIA-1-related nucleolysin. Proc Natl Acad Sci USA. 1992;1589:8681–5. doi: 10.1073/pnas.89.18.8681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayali F, Montie HL, Rafols JA, DeGracia DJ. Prolonged translation arrest in reperfused hippocampal cornu Ammonis 1 is mediated by stress granules. Neuroscience. 2005;134:1223–45. doi: 10.1016/j.neuroscience.2005.05.047. [DOI] [PubMed] [Google Scholar]
- Kedersha N, Anderson P. Stress granules: sites of mRNA triage that regulate mRNA stability and translatability. Biochem Soc Trans. 2002;30:963–9. doi: 10.1042/bst0300963. [DOI] [PubMed] [Google Scholar]
- Kedersha N, Chen S, Gilks N, Li W, Miller IJ, Stahl J, Anderson P. Evidence that ternary complex (eIF2–GTP–tRNA(i)(Met))-deficient preinitiation complexes are core constituents of mammalian stress granules. Mol Biol Cell. 2002;13:195–210. doi: 10.1091/mbc.01-05-0221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedersha N, Cho MR, Li W, Yacono PW, Chen S, Gilks N, Golan DE, Anderson P. Dynamic shuttling of TIA-1 accompanies the recruitment of mRNA to mammalian stress granules. J Cell Biol. 2000;11151:1257–68. doi: 10.1083/jcb.151.6.1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedersha NL, Gupta M, Li W, Miller I, Anderson P. RNA-binding proteins TIA-1 and TIAR link the phosphorylation of eIF-2alpha to the assembly of mammalian stress granules. J Cell Biol. 1999;27147:1431–42. doi: 10.1083/jcb.147.7.1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YK, Jang SK. Continuous heat shock enhances translational initiation directed by internal ribosomal entry site. Biochem Biophys Res Commun. 2002;20297:224–31. doi: 10.1016/s0006-291x(02)02154-x. [DOI] [PubMed] [Google Scholar]
- Kimball SR, Horetsky RL, Ron D, Jefferson LS, Harding HP. Mammalian stress granules represent sites of accumulation of stalled translation initiation complexes. Am J Physiol Cell Physiol. 2003;284:C273–84. doi: 10.1152/ajpcell.00314.2002. [DOI] [PubMed] [Google Scholar]
- Kirino T. Delayed neuronal death in the gerbil hippocampus following ischemia. Brain Res. 1982;6239:57–69. doi: 10.1016/0006-8993(82)90833-2. [DOI] [PubMed] [Google Scholar]
- Kirino T. Ischemic tolerance. J Cereb Blood Flow Metab. 2002;22:1283–96. doi: 10.1097/01.WCB.0000040942.89393.88. [DOI] [PubMed] [Google Scholar]
- Kirino T, Sano K. Fine structural nature of delayed neuronal death following ischemia in the gerbil hippocampus. Acta Neuropathol (Berl) 1984;62:209–18. doi: 10.1007/BF00691854. [DOI] [PubMed] [Google Scholar]
- Kirino T, Tamura A, Sano K. Delayed neuronal death in the rat hippocampus following transient forebrain ischemia. Acta Neuropathol (Berl) 1984;64:139–47. doi: 10.1007/BF00695577. [DOI] [PubMed] [Google Scholar]
- Kleihues P, Hossmann KA. Protein synthesis in the cat brain after prolonged cerebral ischemia. Brain Res. 1971;2435:409–18. doi: 10.1016/0006-8993(71)90484-7. [DOI] [PubMed] [Google Scholar]
- Kumar R, Azam S, Sullivan JM, Owen C, Cavener DR, Zhang P, Ron D, Harding HP, Chen JJ, Han A, White BC, Krause GS, DeGracia DJ. Brain ischemia and reperfusion activates the eukaryotic initiation factor 2alpha kinase, PERK. J Neurochem. 2001;77:1418–21. doi: 10.1046/j.1471-4159.2001.00387.x. [DOI] [PubMed] [Google Scholar]
- Kumar R, Krause GS, Yoshida H, Mori K, DeGracia DJ. Dysfunction of the unfolded protein response during global brain ischemia and reperfusion. J Cereb Blood Flow Metab. 2003;23:462–71. doi: 10.1097/01.WCB.0000056064.25434.CA. [DOI] [PubMed] [Google Scholar]
- Kusmierczyk AR, Martin J. Chaperonins-keeping a lid on folding proteins. FEBS Lett. 2001;505:343–57. doi: 10.1016/s0014-5793(01)02838-1. [DOI] [PubMed] [Google Scholar]
- Latchman DS. Protective effect of heat shock proteins in the nervous system. Curr Neurovasc Res. 2004;1:21–7. doi: 10.2174/1567202043480206. [DOI] [PubMed] [Google Scholar]
- Le Guiner C, Lejeune F, Galiana D, Kister L, Breathnach R, Stevenin J, Del Gatto-Konczak F. TIA-1 and TIAR activate splicing of alternative exons with weak 5′ splice sites followed by a U-rich stretch on their own pre-mRNAs. J Biol Chem. 2001;2276:40638–46. doi: 10.1074/jbc.M105642200. [DOI] [PubMed] [Google Scholar]
- Liu CL, Chen S, Kamme F, Hu BR. Ischemic preconditioning prevents protein aggregation after transient cerebral ischemia. Neuroscience. 2005a;134:69–80. doi: 10.1016/j.neuroscience.2005.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu CL, Ge P, Zhang F, Hu BR. Co-translational protein aggregation after transient cerebral ischemia. Neuroscience. 2005b;134:1273–84. doi: 10.1016/j.neuroscience.2005.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu CL, Hu BR. Alterations of N-ethylmaleimide-sensitive atpase following transient cerebral ischemia. Neuroscience. 2004;128:767–74. doi: 10.1016/j.neuroscience.2004.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu CL, Martone ME, Hu BR. Protein ubiquitination in postsynaptic densities after transient cerebral ischemia. J Cereb Blood Flow Metab. 2004;24:1219–25. doi: 10.1097/01.WCB.0000136706.77918.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnusson K, Wieloch T. Impairment of protein ubiquitination cause delayed neuronal death. Neurosci Lett. 1989;96:264–70. doi: 10.1016/0304-3940(89)90389-3. [DOI] [PubMed] [Google Scholar]
- Martin de la Vega C, Burda J, Nemethova M, Quevedo C, Alcazar A, Martin ME, Danielisova V, Fando JL, Salinas M. Possible mechanisms involved in the down-regulation of translation during transient global ischaemia in the rat brain. Biochem J. 2001;1357:819–26. doi: 10.1042/0264-6021:3570819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martone ME, Jones YZ, Young SJ, Ellisman MH, Zivin JA, Hu BR. Modification of postsynaptic densities after transient cerebral ischemia: a quantitative and three-dimensional ultrastructural study. Neuroscience. 1999;19:1988–97. doi: 10.1523/JNEUROSCI.19-06-01988.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCaig D, Imai H, Gallagher L, Graham DI, Harland J, Moira Brown S, Mhairi Macrae I. Evolution of GADD34 expression after focal cerebral ischaemia. Brain Res. 2005;91034:51–61. doi: 10.1016/j.brainres.2004.11.058. [DOI] [PubMed] [Google Scholar]
- McEwen E, Kedersha N, Song B, Scheuner D, Gilks N, Han A, Chen JJ, Anderson P, Kaufman RJ. Heme-regulated inhibitor kinase-mediated phosphorylation of eukaryotic translation initiation factor 2 inhibits translation, induces stress granule formation, and mediates survival upon arsenite exposure. J Biol Chem. 2005;29280:16925–33. doi: 10.1074/jbc.M412882200. [DOI] [PubMed] [Google Scholar]
- Meijer HA, Thomas AA. Control of eukaryotic protein synthesis by upstream open reading frames in the 5′-untranslated region of an mRNA. Biochem J. 2002;1367:1–11. doi: 10.1042/BJ20011706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mengesdorf T, Althausen S, Oberndorfer I, Paschen W. Response of neurons to an irreversible inhibition of endoplasmic reticulum Ca(2+)-ATPase: relationship between global protein synthesis and expression and translation of individual genes. Biochem J. 2001;15356:805–12. doi: 10.1042/0264-6021:3560805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mengesdorf T, Proud CG, Mies G, Paschen W. Mechanisms underlying suppression of protein synthesis induced by transient focal cerebral ischemia in mouse brain. Exp Neurol. 2002;177:538–46. doi: 10.1006/exnr.2002.8002. [DOI] [PubMed] [Google Scholar]
- Meriin AB, Sherman MY. Role of molecular chaperones in neurodegenerative disorders. Int J Hyperthermia. 2005;21:403–19. doi: 10.1080/02656730500041871. [DOI] [PubMed] [Google Scholar]
- Mies G, Ishimaru S, Xie Y, Seo K, Hossmann KA. Ischemic thresholds of cerebral protein synthesis and energy state following middle cerebral artery occlusion in rat. J Cereb Blood Flow Metab. 1991;11:753–61. doi: 10.1038/jcbfm.1991.132. [DOI] [PubMed] [Google Scholar]
- Morimoto T, Ide T, Ihara Y, Tamura A, Kirino T. Transient ischemia depletes free ubiquitin in the gerbil hippocampal CA1 neurons. Am J Pathol. 1996;148:249–57. [PMC free article] [PubMed] [Google Scholar]
- Morris DR, Geballe AP. Upstream open reading frames as regulators of mRNA translation. Mol Cell Biol. 2000;20:8635–42. doi: 10.1128/mcb.20.23.8635-8642.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagomi T, Kirino T, Kanemitsu H, Tsujita Y, Tamura A. Early recovery of protein synthesis following ischemia in hippocampal neurons with induced tolerance in the gerbil. Acta Neuropathol (Berl) 1993;86:10–5. doi: 10.1007/BF00454892. [DOI] [PubMed] [Google Scholar]
- Neumar RW, DeGracia DJ, Konkoly LL, Khoury JI, White BC, Krause GS. Calpain mediates eukaryotic initiation factor 4G degradation during global brain ischemia. J Cereb Blood Flow Metab. 1998;18:876–81. doi: 10.1097/00004647-199808000-00007. [DOI] [PubMed] [Google Scholar]
- Nevins TA, Harder ZM, Korneluk RG, Holcik M. Distinct regulation of internal ribosome entry site-mediated translation following cellular stress is mediated by apoptotic fragments of eIF4G translation initiation factor family members eIF4GI and p97/DAP5/NAT1. J Biol Chem. 2003;7278:3572–9. doi: 10.1074/jbc.M206781200. [DOI] [PubMed] [Google Scholar]
- Nover L, Scharf KD, Neumann D. Formation of cytoplasmic heat shock granules in tomato cell cultures and leaves. Mol Cell Biol. 1983;3:1648–55. doi: 10.1128/mcb.3.9.1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nover L, Scharf KD, Neumann D. Cytoplasmic heat shock granules are formed from precursor particles and are associated with a specific set of mRNAs. Mol Cell Biol. 1989;9:1298–308. doi: 10.1128/mcb.9.3.1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novoa I, Zeng H, Harding HP, Ron D. Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2alpha. J Cell Biol. 2001;28153:1011–22. doi: 10.1083/jcb.153.5.1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowak TS., Jr Synthesis of a stress protein following transient ischemia in the gerbil. J Neurochem. 1985;45:1635–41. doi: 10.1111/j.1471-4159.1985.tb07236.x. [DOI] [PubMed] [Google Scholar]
- Nowak TS., Jr Protein synthesis and the heart shock/stress response after ischemia. Cerebrovasc Brain Metab Rev. 1990;2:345–66. [PubMed] [Google Scholar]
- Nowak TS, Jr, Jacewicz M. The heat shock/stress response in focal cerebral ischemia. Brain Pathol. 1994;4:67–76. doi: 10.1111/j.1750-3639.1994.tb00812.x. [DOI] [PubMed] [Google Scholar]
- Ouyang YB, Hu BR. Protein ubiquitination in rat brain following hypoglycemic coma. Neurosci Lett. 2001;298:159–62. doi: 10.1016/s0304-3940(00)01721-3. [DOI] [PubMed] [Google Scholar]
- Owen CR, Kumar R, Zhang P, McGrath BC, Cavener DR, Krause GS. PERK is responsible for the increased phosphorylation of eIF2alpha and the severe inhibition of protein synthesis after transient global brain ischemia. J Neurochem. 2005;94:1235–42. doi: 10.1111/j.1471-4159.2005.03276.x. [DOI] [PubMed] [Google Scholar]
- Oyadomari S, Mori M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2004;11:381–9. doi: 10.1038/sj.cdd.4401373. [DOI] [PubMed] [Google Scholar]
- Panniers R. Translational control during heat shock. Biochimie. 1994;76:737–47. doi: 10.1016/0300-9084(94)90078-7. [DOI] [PubMed] [Google Scholar]
- Paschen W. Disturbances of calcium homeostasis within the endoplasmic reticulum contribute to the development of ischemic-cell damage. Med Hypotheses. 1996;47:283–8. doi: 10.1016/s0306-9877(96)90068-7. [DOI] [PubMed] [Google Scholar]
- Paschen W. Shutdown of translation: lethal or protective? Unfolded protein response versus apoptosis. J Cereb Blood Flow Metab. 2003;23:773–9. doi: 10.1097/01.WCB.0000075009.47474.F9. [DOI] [PubMed] [Google Scholar]
- Paschen W, Aufenberg C, Hotop S, Mengesdorf T. Transient cerebral ischemia activates processing of xbp1 messenger RNA indicative of endoplasmic reticulum stress. J Cereb Blood Flow Metab. 2003;23:449–61. doi: 10.1097/01.WCB.0000054216.21675.AC. [DOI] [PubMed] [Google Scholar]
- Paschen W, Hayashi T, Saito A, Chan PH. GADD34 protein levels increase after transient ischemia in the cortex butnot in the CA1 subfield: implications for post-ischemic recovery of protein synthesis in ischemia-resistant cells. J Neurochem. 2004;90:694–701. doi: 10.1111/j.1471-4159.2004.02555.x. [DOI] [PubMed] [Google Scholar]
- Paschen W, Mengesdorf T, Althausen S, Hotop S. Peroxidative stress selectively down-regulates the neuronal stress response activated under conditions of endoplasmic reticulum dysfunction. J Neurochem. 2001;76:1916–24. doi: 10.1046/j.1471-4159.2001.00206.x. [DOI] [PubMed] [Google Scholar]
- Perales C, Carrasco L, Ventoso I. Cleavage of eIF4G by HIV-1 protease: effects on translation. FEBS Lett. 2003;2533:89–94. doi: 10.1016/s0014-5793(02)03764-x. [DOI] [PubMed] [Google Scholar]
- Pickering BM, Willis AE. The implications of structured 5′ untranslated regions on translation and disease. Semin Cell Dev Biol. 2005;16:39–47. doi: 10.1016/j.semcdb.2004.11.006. [DOI] [PubMed] [Google Scholar]
- Proud CG. eIF2 and the control of cell physiology. Semin Cell Dev Biol. 2005;16:3–12. doi: 10.1016/j.semcdb.2004.11.004. [DOI] [PubMed] [Google Scholar]
- Pulsinelli WA, Brierley JB, Plum F. Temporal profile of neuronal damage in a model of transient forebrain ischemia. Ann Neurol. 1982;11:491–8. doi: 10.1002/ana.410110509. [DOI] [PubMed] [Google Scholar]
- Rajdev S, Hara K, Kokubo Y, Mestril R, Dillmann W, Weinstein PR, Sharp FR. Mice overexpressing rat heat shock protein 70 are protected against cerebral infarction. Ann Neurol. 2000;47:782–91. [PubMed] [Google Scholar]
- Ron D. Translational control in the endoplasmic reticulum stress response. J Clin Invest. 2002;110:1383–8. doi: 10.1172/JCI16784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ron D. Signaling cascades regulating NMDA receptor sensitivity to ethanol. Neuroscientist. 2004;10:325–36. doi: 10.1177/1073858404263516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheper GC, van Wijk R, Thomas AM. Regulation of the activity of eukaryotic initiation factors in stressed cells. Prog Mol Subcell Biol. 2001;27:40–56. [PubMed] [Google Scholar]
- Shang J, Lehrman MA. Discordance of UPR signaling by ATF6 and Ire1p-XBP1 with levels of target transcripts. Biochem Biophys Res Commun. 2004;30:390–6. doi: 10.1016/j.bbrc.2004.03.058. [DOI] [PubMed] [Google Scholar]
- Sharp FR, Massa SM, Swanson RA. Heat-shock protein protection. Trends Neurosci. 1999;22:97–9. doi: 10.1016/s0166-2236(98)01392-7. [DOI] [PubMed] [Google Scholar]
- Shen X, Ellis RE, Lee K, Liu CY, Yang K, Solomon A, Yoshida H, Morimoto R, Kurnit DM, Mori K, Kaufman RJ. Complementary signaling pathways regulate the unfolded protein response and are required for C elegans development. Cell. 2001;28107:893–903. doi: 10.1016/s0092-8674(01)00612-2. [DOI] [PubMed] [Google Scholar]
- Shimizu N, Ishii S. Electron-microscopic observations on nucleolar extrusion in nerve cells of the rat hypothalamus. Z Zellforsch Mikrosk Anat. 1965;3067:367–72. doi: 10.1007/BF00339382. [DOI] [PubMed] [Google Scholar]
- Siesjo BK. Mechanisms of ischemic brain damage. Crit Care Med. 1988;16:954–63. doi: 10.1097/00003246-198810000-00006. [DOI] [PubMed] [Google Scholar]
- Simon RP, Cho H, Gwinn R, Lowenstein DH. The temporal profile of 72-kDa heat-shock protein expression following global ischemia. J Neurosci. 1991;11:881–9. doi: 10.1523/JNEUROSCI.11-03-00881.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith ML, Bendek G, Dahlgren N, Rosen I, Wieloch T, Siesjo BK. Models for studying long-term recovery following forebrain ischemia in the rat 2 A 2-vessel occlusion model. Acta Neurol Scand. 1984;69:385–401. doi: 10.1111/j.1600-0404.1984.tb07822.x. [DOI] [PubMed] [Google Scholar]
- Tajiri S, Oyadomari S, Yano S, Morioka M, Gotoh T, Hamada JI, Ushio Y, Mori M. Ischemia-induced neuronal cell death is mediated by the endoplasmic reticulum stress pathway involving CHOP. Cell Death Differ. 2004;11:403–15. doi: 10.1038/sj.cdd.4401365. [DOI] [PubMed] [Google Scholar]
- Thilmann R, Xie Y, Kleihues P, Kiessling M. Persistent inhibition of protein synthesis precedes delayed neuronal death in postischemic gerbil hippocampus. Acta Neuropathol (Berl) 1986;71:88–93. doi: 10.1007/BF00687967. [DOI] [PubMed] [Google Scholar]
- Tian Q, Streuli M, Saito H, Schlossman SF, Anderson P. A polyadenylate binding protein localized to the granules of cytolytic lymphocytes induces DNA fragmentation in target cells. Cell. 1991;167:629–39. doi: 10.1016/0092-8674(91)90536-8. [DOI] [PubMed] [Google Scholar]
- Vass K, Welch WJ, Nowak TS., Jr Localization of 70-kDa stress protein induction in gerbil brain after ischemia. Acta Neuropathol (Berl) 1988;77:128–35. doi: 10.1007/BF00687422. [DOI] [PubMed] [Google Scholar]
- Voellmy R. On mechanisms that control heat shock transcription factor activity in metazoan cells. Cell Stress Chaperones. 2004;9:122–33. doi: 10.1379/CSC-14R.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieloch T, Bergstedt K, Hu BR. Protein phosphorylation and the regulation of mRNA translation following cerebral ischemia. Prog Brain Res. 1993;96:179–91. doi: 10.1016/s0079-6123(08)63266-5. [DOI] [PubMed] [Google Scholar]
- Wieloch T, Hu BR, Boris-Moller A, Cardell M, Kamme F, Kurihara J, Sakata K. Intracellular signal transduction in the postischemic brain. Adv Neurol. 1996;71:371–87. [PubMed] [Google Scholar]
- Yenari MA, Dumas TC, Sapolsky RM, Steinberg GK. Gene therapy for treatment of cerebral ischemia using defective herpes simplex viral vectors. Ann NY Acad Sci. 2001;939:340–57. doi: 10.1111/j.1749-6632.2001.tb03643.x. [DOI] [PubMed] [Google Scholar]
- Yoshida H, Okada T, Haze K, Yanagi H, Yura T, Negishi M, Mori K. ATF6 activated by proteolysis binds in the presence of NF-Y (CBF) directly to the cis-acting element responsible for the mammalian unfolded protein response. Mol Cell Biol. 2000;20:6755–67. doi: 10.1128/mcb.20.18.6755-6767.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimine T, Hayakawa T, Kato A, Yamada K, Matsumoto K, Ushio Y, Mogami H. Autoradiographic study of regional protein synthesis in focal cerebral ischemia with TCA wash and image subtraction techniques. J Cereb Blood Flow Metab. 1987;7:387–93. doi: 10.1038/jcbfm.1987.81. [DOI] [PubMed] [Google Scholar]
- Zhang F, Liu CL, Hu BR. Irreversible aggregation of protein synthesis machinery after focal brain ischemia. J Neurochem. 2006;98:102–12. doi: 10.1111/j.1471-4159.2006.03838.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K, Kaufman RJ. The unfolded protein response: a stress signaling pathway critical for health and disease. Neurology. 2006;2466:S102–9. doi: 10.1212/01.wnl.0000192306.98198.ec. [DOI] [PubMed] [Google Scholar]
- Zhang T, Delestienne N, Huez G, Kruys V, Gueydan C. Identification of the sequence determinants mediating the nucleo-cytoplasmic shuttling of TIAR and TIA-1 RNA-binding proteins. J Cell Sci. 2005;1118:5453–63. doi: 10.1242/jcs.02669. [DOI] [PubMed] [Google Scholar]
- Zheng Z, Yenari MA. Post-ischemic inflammation: molecular mechanisms and therapeutic implications. Neurol Res. 2004;26:884–92. doi: 10.1179/016164104X2357. [DOI] [PubMed] [Google Scholar]