

Abstract
Down syndrome (DS) is one of the most common causes of intellectual disability. Although DS accounts for only 15% of all individuals with intellectual disabilities, adults with DS account for approximately 60% of individuals with intellectual disabilities and Alzheimer’s disease. This is thought to be because of overproduction of the β-amyloid (Aβ) protein due to trisomy for the Aβ precursor protein gene on chromosome 21. Pittsburgh compound B (PiB) is a noninvasive in vivo positron emission tomography tracer used to image amyloid deposition in living humans. Studies using PiB have shown an age-dependent asymptomatic amyloid deposition in more than 20% of the cognitively normal elderly population. Presymptomatic carriers of presenilin (PS-1) and Aβ precursor protein gene mutations who are destined to develop Alzheimer’s disease also show preclinical amyloid deposition. This report describes a pilot study involving the use of PiB in seven adults with DS (age: 20–44 years). Compared with objective cutoffs for amyloid positivity in older non-DS cognitively normal control subjects, only two of the seven DS subjects (age: 38 and 44 years) showed increased PiB retention. The remaining five subjects aged between 20 and 35 years showed no detectable increase in PiB retention. Interestingly, the two subjects who showed elevated PiB retention showed a striatal-predominant pattern similar to that previously reported for PS-1 mutation carriers. These results demonstrate the feasibility of conducting PiB positron emission tomography scanning in this special population, and suggest a link between Aβ overproduction and early striatal deposition of fibrillar Aβ.
Keywords: Down syndrome, Intellectual disability, Alzheimer’s disease, Pittsburgh compound B, Amyloid deposition
1. Introduction
Down syndrome (DS) is one of the most common causes of intellectual disability, accounting for approximately 15% of all individuals with intellectual disabilities. The incidence of DS is 1:800 live births, with an estimated 7000 babies born with DS annually [1]. Although the incidence of Alzheimer’s disease (AD) among individuals with intellectual disabilities has been found to be no different than among those in the neurotypical population [2], adults with DS comprise up to 60% of individuals with intellectual disabilities who show signs of AD [3,4]. Evidence suggests that individuals with DS experience premature aging, perhaps as much as 20 years earlier than would be expected in normal aging. It is also known that individuals with DS develop AD at an early age and progress rapidly [5].
Although the average life expectancy of individuals with DS remains lower than that of neurotypical adults, the number of older DS adults has been increasing. The current mean life expectancy exceeds 50 years, with 20% or more of the DS population now aged >55 years [6,7]. The fact that more than a third of DS adults aged >50 years and more than one half of DS adults aged >60 years have been diagnosed with AD presents a significant public health problem [8]. Additionally, it has been recently demonstrated that DS subjects aged >45 years show significant cortical Pittsburgh compound B (PiB) uptake, regardless of dementia diagnosis [9]. It is important to note that age-associated decline among nondemented individuals with DS through the fifth and even sixth decade of life is not inevitable, although is does occur in many individuals [10]. Consequently, changes in cognitive functioning in this population are likely to be indicative of early stages of AD, after other potential causal factors, such as hypothyroidism or depression, are ruled out.
Postmortem studies of brain tissue in individuals with DS have shown neuropathological changes similar to those observed in individuals diagnosed with AD, characterized by the presence of neurofibrillary tangles and neuritic plaques in the brains of almost all DS individuals by 40 years of age [11,12]. In one study, the mean age at onset of dementia in patients with DS was 56 years, and prevalence increased from 11% between the ages of 40 and 49 years to 77% between the ages of 60 and 69 years. All subjects aged ≥70 years had dementia [5]. In contrast, the prevalence of dementia in the non-DS population from all causes among those aged ≤65 years was found to be <5% and it increased to 13% for those aged >65 years [13]. It is believed that this high incidence of dementia in the DS population is due to the extra copy of chromosome 21, which codes for the β-amyloid (Aβ) precursor protein (APP) gene. Studies by Hyman and colleagues have shown that the level of amyloid deposition in the brains of individuals with DS is higher than in individuals with AD [11,14]. This may be particularly true in individuals with the trisomy 21 variant, the most common cause of DS.
Definitive diagnosis of AD relies on the demonstration of amyloid plaques and neurofibrillary tangles at autopsy [15,16]. The time course of amyloid deposition in AD has not been definitively elucidated, but evidence gained through postmortem studies of individuals with DS suggests that amyloid deposition begins over a decade before the clinical symptoms of dementia. Studies in carriers of PS-1 mutations have shown clear evidence that Aβ deposition predates dementia by at least 10 years [17]. Unexpectedly, PiB retention in some presymptomatic PS-1 mutation carriers appears to begin in the striatum [17], an area affected later in the course of late-onset AD [18].
This report describes a pilot study using PiB positron emission tomography (PET) in nondemented DS subjects. The objectives of this study were threefold. First, we sought to demonstrate the feasibility of conducting PiB PET studies in this special population. Second, we sought to assess the pathophysiological process of fibrillar Aβ deposition in the brains of DS subjects of increasing age. Finally, PET data from these subjects were compared with historical PiB PET data obtained from normal control subjects between the ages of 35 and 80 years.
2. Methods
2.1. Participant characterization
Subjects were recruited over a 22-month period from our university’s adult DS center and psychiatric clinic for adults with developmental disabilities. In addition, letters were sent out to families with DS patients in western Pennsylvania (using the DS center’s database). Interested families were contacted by telephone and underwent a thorough screening to determine study eligibility (i.e., subjects with a history of claustrophobia, with metal in their bodies, or with a current AD diagnosis were excluded). As a result, all subjects who were invited to participate in the study met study inclusion criteria. All subjects and their caregivers (when appropriate) provided informed consent for both clinical examination and the PET imaging protocol. This study was approved by the Human Use Subcommittee of the Radioactive Drug Research Committees and the Institutional Review Board of the University of Pittsburgh. Subjects had to be at least 20 years of age, have an intelligence quotient of ≥40, and have documented evidence of trisomy 21. Subjects could have no evidence of significant cognitive decline over the previous 1 year using a “Stability/Decline Scale” designed specifically for this study (see later in the text). Other exclusion criteria included any significant disease or unstable medical condition that could affect neuropsychological testing and conditions for which magnetic resonance imaging (MRI) was contraindicated.
2.2. Initial screening
Participants initially provided consent and were subsequently assessed to obtain baseline measures of cognitive and adaptive functioning using the measures mentioned in the following text.
2.2.1. Stanford–Binet Abbreviated Battery IQ
All subjects were given the Stanford–Binet Abbreviated Battery IQ [19] to obtain an updated estimate of cognitive functioning. The Stanford–Binet Abbreviated Battery IQ includes both a verbal and nonverbal subscale and produces an abbreviated intelligence quotient score (mean: 100; SD: 15) as well as a mental age.
2.2.2. Severe Impairment Battery
The Severe Impairment Battery [20] is composed of 40 simple one-step command items that are scored on a 3-point scale. Overall, scores can range from 0 to 100, with lower scores indicating greater levels of impairment.
2.2.3. Stability/Decline Scale for DS
The Stability/Decline Scale for DS was developed specifically for the current study. Caregivers who had consistent and substantial (≥4 days/week) contact with the subject for at least 1 year were asked to rate the subject’s functioning in areas of language, understanding, memory, self-care, and work/social functioning using a 3-point scale (as being better, unchanged, or worse). Any report of worsening function in any of the five areas was followed up with unstructured questions to determine whether the change was clinically significant. Any subject with a verified decrement in functioning was excluded from the study.
2.3. Image acquisition and analysis
Subjects who met inclusion criteria at the conclusion of the initial assessment were scheduled for imaging studies. PET and MRI were performed within 5 weeks of cognitive assessments.
2.4. Magnetic resonance imaging
Before the PET imaging sessions, a spoiled gradient recalled MR scan (1.5 T) was obtained for each subject for MR–PET image co-registration and anatomical region-of-interest (ROI) definition, as described previously [21,22]. The MR-based partial volume correction corresponded to a two-component approach that corrected the PET PiB and fluorodeoxyglucose measures for the dilutional effect of expanded cerebrospinal fluid accompanying normal aging or disease-related cerebral atrophy [23]. ROIs were separately hand-drawn on the co-registered MR image and included the following: frontal cortex; anterior cingulate/medial frontal cortex; lateral temporal, mesial temporal, occipital, parietal, precuneus/posterior cingulate, and sensorimotor cortices; anterior–ventral striatum; pons; and subcortical white matter. A cerebellar ROI was used as the reference region. These regions have been previously described in detail [24].
2.5. PiB PET imaging
High specific activity of [11C]PiB was produced as described previously [22,25]. The subjects were then injected with 15 mCi of [11C]PiB and were allowed to relax quietly in a chair for approximately 25 minutes, after which they were placed on the scanner table. A 10-minute transmission scan was followed by a 20-minute PiB PET study (4 ×300-second frames) from 40 to 60 minutes after injection, acquired as participants rested with eyes open in a quiet, dimly lit room. Analysis of the PiB PET data used the standardized uptake value ratio (SUVR) at 40 to 60 minutes postinjection. To determine the SUVR, the SUV was first determined by normalizing the regional tissue radioactivity concentration to injected dose and body mass (unitless measure, assuming 1 g/cm3 tissue density), and each regional SUV was then divided by the SUV value of the cerebellar reference region that is representative of free and nonspecific radiotracer retention. These SUVR values obtained at 40 to 60 minutes postinjection have been shown to compare favorably with measures of PiB retention, based on arterial data [21,26].
3. Results
Eight subjects consented to participate in the study and completed the initial screening assessment. Seven subjects completed the scanning component of the protocol (the eighth subject completed the MRI scan but was unable to complete the PiB PET scan). Table 1 provides demographic information for the eight subjects. Subjects ranged in age from 20 to 44 years, and the sample included six males and two females. All subjects functioned within the moderate range of intellectual disability, with mental ages ranging from 4 years 3 months to 6 years 4 months. All of these subjects were cognitively stable and considered nondemented when enrolled.
Table 1.
Subject demographics
| Subject number | Age | Sex | Mental age | SIB score | Follow-up |
|---|---|---|---|---|---|
| 1 | 44 | M | 4 years 11 months | 83 | Alzheimer’s |
| 2 | 34 | M | 5 years 1 month | 87 | |
| 3* | 26 | M | 4 years 3 months | 89 | No change |
| 4 | 38 | M | 5 years 5 months | 100 | No change |
| 5 | 22 | M | 4 years 9 months | 87 | |
| 6 | 20 | F | 6 years 4 months | 96 | |
| 7 | 35 | F | 5 years 2 months | 98 | No change |
| 8 | 20 | M | 4 years 3 months | 88 | No change |
Abbreviation: SIB, Severe Impairment Battery.
Could not complete scan.
The PiB images of the two oldest DS subjects (subjects 1 and 4) are shown in Figure 1. The pattern of PiB retention in subject 1 was very similar to that seen previously in late-onset AD patients [18], except for relatively increased signal in the striatum. This striatal predominance was even more marked for subject 4, who had PiB retention almost restricted to the striatum. This pattern was very reminiscent of PiB retention in pre-symptomatic PS-1 mutation carriers [17]. The other five younger DS subjects showed no evidence of specific PiB retention in neocortex (Figure 2) and appeared very similar to PiB-negative cognitively normal control subjects [18,27–29]. Further, all SUVR data were analyzed using pons as a reference region, and no differences were observed when compared with SUVR cerebellar data.
Fig. 1.
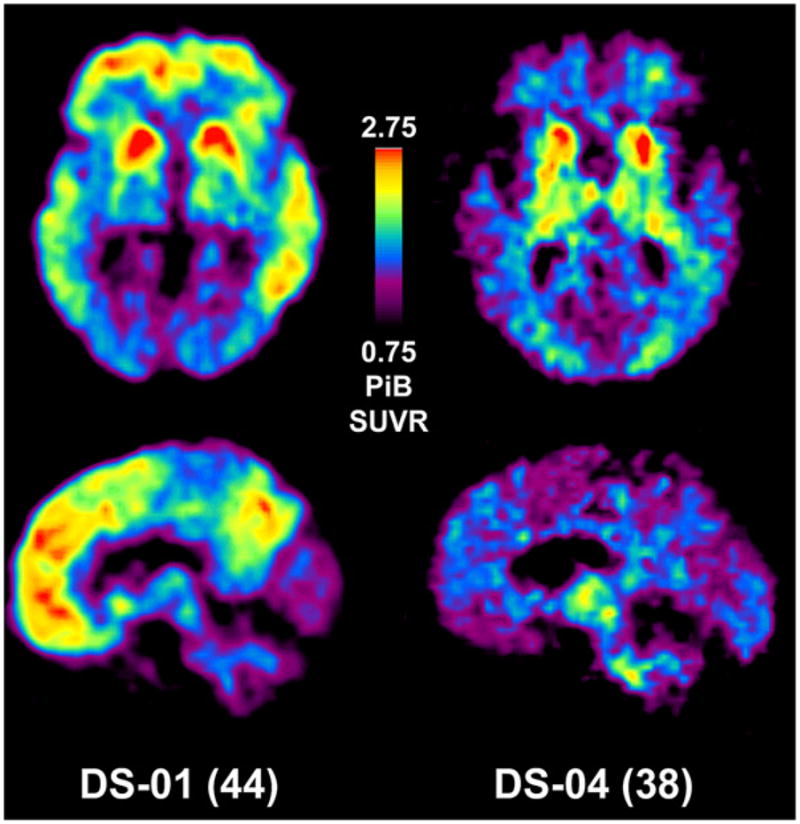
Pittsburgh compound B (PiB) standardized uptake value ratio images of two PiB-positive subjects with Down syndrome (DS). Axial images are shown at the top and sagittal images at the bottom. Note that the scan of subject DS-1 is very similar to those seen in late-onset Alzheimer’s disease (AD) (17), but with a predominant striatal signal. High PiB retention in subject DS-4 is mainly limited to the anterior striatum, similar to presenilin-1 mutation carriers (16).
Fig. 2.
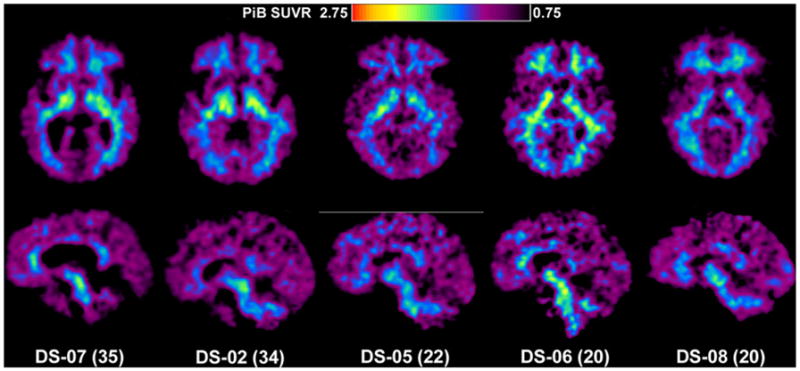
PiB standardized uptake value ratio images of five PiB-negative subjects with DS. The axial images are shown at the top and the sagittal images at the bottom. Note that all of these scans are typical of normal control subjects and show only nonspecific PiB retention in white matter.
Quantitative SUVR data demonstrate that subject 1 had PiB retention in the lower end of the AD range in frontal cortex, anterior cingulate cortex, precuneus/posterior cingulate cortex, anterior–ventral striatum, and lateral temporal cortex (Figure 3). This subject also had more modest elevations in parietal and sensorimotor cortices relative to control subjects and the other DS subjects (Figure 3). No elevation in PiB retention was observed in mesial temporal or occipital cortices, or in the pons and subcortical white matter areas of subject 1. As the images would predict, subject 4 showed a quantitative increase in PiB retention only in the striatum (Figure 3). This subject tended to be at the top of the range of the younger DS subjects in all of the other cortical areas in which PiB shows large increases in AD, but these values were still well within the PiB-negative control range.
Fig. 3.

PiB retention in clinically unimpaired control subjects (red triangles), DS subjects (circles), and AD patients (blue squares). The cutoff between PiB-positive and PiB-negative for each brain region is shown by a white bar. Black diamonds demonstrate the mean ±SD for the control and AD groups. Subject DS-1 is marked with an “x” inside the circle, and subject DS-4 is marked with a filled blackcircle. Brain area abbreviations: FRC, frontal; ACG, anterior cingulate gyrus; PRC, precuneus; AVS, anterior ventral striatum; LTC, lateral temporal; PAR, parietal; MTC, mesial temporal; OCC, occipital (includes calcarine); SMC, sensorimotor; PON, pons; SWM, subcortical white matter.
Charts of five subjects who continue to be followed were reviewed to determine whether any changes in status had been noted. As indicated in Table 1, four of the five subjects had no evidence of symptoms suggestive of AD at follow-up after 3 to 5 years. However, subject 1, whose pattern was very reminiscent of PiB retention in late-onset AD subjects, was diagnosed with AD approximately 1 year after participation in the current study.
4. Discussion
This article describes a pilot study using PiB PET in adults with DS. The study results demonstrate the feasibility of conducting PiB PET scanning in this special population. Seven of eight subjects successfully completed the MRI/PET scanning protocol. Although all subjects were deemed to be free of dementia (and did not display any recent changes in adaptive functioning), one subject showed evidence of a pattern of PiB retention that was similar to that seen in late-onset AD patients, and another subject displayed focal PiB retention in the striatum, a pattern similar to that observed in presymptomatic PS-1 mutation carriers [17]. Additionally, these data also support the recent findings of the pilot study by Landt et al. [9], demonstrating an age-related increase in PiB binding in DS subjects with and without dementia. PiB retention in this small DS cohort appeared to be related to age; the single subject aged >40 years was the only subject who displayed a pattern of PiB retention suggestive of late-onset AD and who subsequently converted to AD at follow-up.
It is important to note that although only two subjects displayed PiB retention, this does not preclude the presence of some type of Aβ deposition in the brains of the younger DS subjects. It has been shown that one subject who displayed no evidence of PiB retention at the time of the PET scan, showed pathological evidence of diffuse neocortical Aβ plaques at autopsy 3 years later [30]. Indeed, it has been reported that extensive oligomeric Aβ deposits can exist in subjects with DS [31]. Additionally, it has been reported that amyloid is primarily found in the form of diffuse neocortical plaques in the brains of younger individuals with DS (<30 years) [32]. It has been shown that PiB stains compact/cored plaques more prominently than diffuse plaques [33].
It is of interest to note the similarity between the striatal-predominant pattern of Aβ deposition observed in the two nondemented DS patients aged >38 years and the striatal-predominant pattern of Aβ deposition previously reported in nondemented carriers of mutations in PS-1, APP, and, perhaps, PS-2 genes [17,34–37]. The apparent commencement of PiB-detectable Aβ deposition in the striatum of these two groups is distinct from that in late-onset AD, where the first sites of Aβ deposition appear to be in the midline orbital–frontal region and the precuneus, although the striatum is later involved in essentially all AD cases [18,27,28]. The cause of this early, focal striatal Aβ deposition in autosomal dominant early-onset AD is not known. Detection of a similar phenomenon in nondemented DS patients suggests that a common thread may be either overproduction of the longer, more aggregation-prone Aβ-42 peptide or regional metabolic or clearance differences that are displayed with Aβ overproduction in these two younger cohorts. Interestingly, it has been reported that in an animal model of DS, overexpression of APP in the striatum appears to peak earlier than in neocortical areas [38].
In summary, although the findings in this study may not necessarily be representative of the general adult DS population, given the small sample size, this study suggests that a larger, longitudinal study of this population is warranted and feasible. Additionally, studies that include an extended age range beyond 50 years may yield further information. It is important to determine whether the presence of amyloid deposition at baseline is a predictor of the subsequent development of dementia in this DS population, as was demonstrated in one of the subjects reported here. One of the possible challenges to conducting longitudinal scans with this population would be the possibility of limited cooperation among subjects who begin to show evidence of symptoms of dementia. In fact, a limitation of the current study was that the feasibility of conducting a PET scan in individuals with DS and dementia was not tested. Although preliminary in nature, the present study demonstrates that PiB PET imaging is possible in the DS population using current standardized scanning protocols. To our knowledge, these data also represent the largest cohort of nondemented DS subjects with PiB PET. These data further demonstrate that PiB retention is evident among the older DS subjects studied here in a pattern similar to that observed in PS-1 mutation carriers.
References
- 1.Hook EB, Cross PK, Schreinemachers DM. Chromosomal abnormality rates at amniocentesis and in live-born infants. JAMA. 1983;249:2034–8. [PubMed] [Google Scholar]
- 2.Zigman WB, Schupf N, Devenny DA, Miezejeski C, Ryan R, Urv TK, et al. Incidence and prevalence of dementia in elderly adults with mental retardation without Down syndrome. Am J Ment Retard. 2004;109:126–41. doi: 10.1352/0895-8017(2004)109<126:IAPODI>2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 3.Janicki MP, Dalton AJ. Prevalence of dementia and impact on intellectual disability services. Ment Retard. 2000;38:276–88. doi: 10.1352/0047-6765(2000)038<0276:PODAIO>2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 4.Janicki MP, Dalton AJ, Henderson CM, Davidson PW. Mortality and morbidity among older adults with intellectual disability: health services considerations. Disabil Rehab. 1999;21:284–94. doi: 10.1080/096382899297710. [DOI] [PubMed] [Google Scholar]
- 5.Visser FE, Aldenkamp AP, van Huffelen AC, Kuilman M, Overweg J, van Wijk J. Prospective study of the prevalence of Alzheimer-type dementia in institutionalized individuals with Down syndrome. Am J Ment Retard. 1997;101:400–12. [PubMed] [Google Scholar]
- 6.Malone Q. Mortality and survival of the Down’s syndrome population in Western Australia. J Ment Defic Res. 1988;32(Pt. 1):59–65. doi: 10.1111/j.1365-2788.1988.tb01388.x. [DOI] [PubMed] [Google Scholar]
- 7.Strauss D, Eyman RK. Mortality of people with mental retardation in California with and without Down syndrome, 1986–1991. Am J Ment Retard. 1996;100:643–53. [PubMed] [Google Scholar]
- 8.Prasher VP, Filer A. Behavioural disturbance in people with Down’s syndrome and dementia. J Intellect Disabil Res. 1995;39(Pt. 5):432–6. doi: 10.1111/j.1365-2788.1995.tb00547.x. [DOI] [PubMed] [Google Scholar]
- 9.Landt J, D’Abrera JC, Holland AJ, Aigbirhio FI, Fryer TD, Canales R, et al. Using positron emission tomography and carbon 11-labeled Pittsburgh compound B to image brain fibrillar {beta}-amyloid in adults with Down syndrome: safety, acceptability, and feasibility. Arch Neurol. 2011;68:890–6. doi: 10.1001/archneurol.2011.36. [DOI] [PubMed] [Google Scholar]
- 10.Devenny DA, Krinsky-McHale SJ, Sersen G, Silverman WP. Sequence of cognitive decline in dementia in adults with Down’s syndrome. J Intellect Disabil Res. 2000;44(Pt. 6):654–65. doi: 10.1046/j.1365-2788.2000.00305.x. [DOI] [PubMed] [Google Scholar]
- 11.Hyman BT. Down syndrome and Alzheimer disease. Progress Clin Bio Res. 1992;379:123–42. [PubMed] [Google Scholar]
- 12.Schupf N, Kapell D, Nightingale B, Rodriguez A, Tycko B, Mayeux R. Earlier onset of Alzheimer’s disease in men with Down syndrome. Neurol. 1998;50:991–5. doi: 10.1212/wnl.50.4.991. [DOI] [PubMed] [Google Scholar]
- 13.Hebert L, Scherr P, Bienias J, Bennett D, Evans D. Alzheimer’s disease in the U.S. population: prevalence estimates using the 2000 census. Arch Neurol. 2003;60:1119–22. doi: 10.1001/archneur.60.8.1119. [DOI] [PubMed] [Google Scholar]
- 14.Hyman BT, West HL, Rebeck GW, Lai F, Mann DM. Neuropathological changes in Down’s syndrome hippocampal formation. Effect of age and apolipoprotein E genotype. Arch Neurol. 1995;52:373–8. doi: 10.1001/archneur.1995.00540280059019. [DOI] [PubMed] [Google Scholar]
- 15.Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM, et al. The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD) Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology. 1991;41:479–86. doi: 10.1212/wnl.41.4.479. [DOI] [PubMed] [Google Scholar]
- 16.Workgroup. The National Institute on Aging, and Reagan Institute Working Group on Diagnostic Criteria for the Neuropathological Assessment of Alzheimer’s Disease. Consensus recommendations for the postmortem diagnosis of Alzheimer’s disease. Neurobiol Aging. 1997;18(Suppl):S1–2. [PubMed] [Google Scholar]
- 17.Klunk WE, Price JC, Mathis CA, Tsopelas ND, Lopresti BJ, Ziolko SK, et al. Amyloid deposition begins in the striatum of presenilin-1 mutation carriers from two unrelated pedigrees. J Neurosci. 2007;27:6174–84. doi: 10.1523/JNEUROSCI.0730-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Klunk WE, Engler H, Nordberg A, Wang Y, Blomqvist G, Holt DP, et al. Imaging brain amyloid in Alzheimer’s disease with Pittsburgh compound-B. Ann Neurol. 2004;55:306–19. doi: 10.1002/ana.20009. [DOI] [PubMed] [Google Scholar]
- 19.Roid GH. Stanford-Binet Intelligence Test Scales. Itasca, IL: Riverside Publishing; 2003. [Google Scholar]
- 20.Panisset M, Roudier M, Saxton J, Boller F. Severe impairment battery. A neuropsychological test for severely demented patients. Arch Neurol. 1994;51:41–5. doi: 10.1001/archneur.1994.00540130067012. [DOI] [PubMed] [Google Scholar]
- 21.Lopresti BJ, Klunk WE, Mathis CA, Hoge JA, Ziolko SK, Lu X, et al. Simplified quantification of Pittsburgh compound B amyloid imaging PET studies: a comparative analysis. J Nucl Med. 2005;46:1959–72. [PubMed] [Google Scholar]
- 22.Price JC, Klunk WE, Lopresti BJ, Lu X, Hoge JA, Ziolko SK, et al. Kinetic modeling of amyloid binding in humans using PET imaging and Pittsburgh compound-B. J Cereb Blood Flow Metab. 2005;25:1528–47. doi: 10.1038/sj.jcbfm.9600146. [DOI] [PubMed] [Google Scholar]
- 23.Meltzer C, Kinahan P, Greer P, Nichols TE, Comtat C, Cantwell MN, Lin MP, Price JC. Comparative evaluation of MR-based partial volume correction schemes for PET. J Nucl Med. 1999;40:2053–65. [PubMed] [Google Scholar]
- 24.Cohen AD, Price JC, Weissfeld LA, James J, Rosario BL, Bi W, et al. Basal cerebral metabolism may modulate the cognitive effects of Aβ in mild cognitive impairment: an example of brain reserve. J Neurosci. 2009;29:14770–8. doi: 10.1523/JNEUROSCI.3669-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wilson AA, Garcia A, Chestakova A, Kung HF, Houle S. A rapid one-step radiosynthesis of the beta-amyloid imaging radiotracer N-methyl-[C-11]2-(4′-methylaminophenyl)-6-hydroxybenzothiazole ([C-11]-6-OH-BTA-1) J Label Compd Radiopharm. 2004;47:679–82. [Google Scholar]
- 26.McNamee RL, Yee SH, Price JC, Klunk WE, Rosario B, Weissfeld L, et al. Consideration of optimal time window for Pittsburgh compound B PET summed uptake measurements. J Nucl Med. 2009;50:348–55. doi: 10.2967/jnumed.108.057612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Aizenstein HJ, Nebes RD, Saxton JA, Price JC, Mathis CA, Tsopelas ND, et al. Frequent amyloid deposition without significant cognitive impairment among the elderly. Arch Neurol. 2008;65:1509–17. doi: 10.1001/archneur.65.11.1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mintun MA, Larossa GN, Sheline YI, Dence CS, Lee SY, Mach RH, et al. [11C]PIB in a nondemented population: potential antecedent marker of Alzheimer disease. Neurology. 2006;67:446–52. doi: 10.1212/01.wnl.0000228230.26044.a4. [DOI] [PubMed] [Google Scholar]
- 29.Rowe CC, Ng S, Ackermann U, Gong SJ, Pike K, Savage G, et al. Imaging beta-amyloid burden in aging and dementia. Neurology. 2007;68:1718–25. doi: 10.1212/01.wnl.0000261919.22630.ea. [DOI] [PubMed] [Google Scholar]
- 30.Cairns NJ, Ikonomovic MD, Benzinger T, Storandt M, Fagan AM, Shah AR, et al. Absence of Pittsburgh compound B detection of cerebral amyloid {beta} in a patient with clinical, cognitive, and cerebrospinal fluid markers of Alzheimer disease: a case report. Arch Neurol. 2009;66:1557–62. doi: 10.1001/archneurol.2009.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lott IT, Head E. Alzheimer disease and Down syndrome: factors in pathogenesis. Neurobiol Aging. 2005;26:383–9. doi: 10.1016/j.neurobiolaging.2004.08.005. [DOI] [PubMed] [Google Scholar]
- 32.Iwatsubo T, Mann DM, Odaka A, Suzuki N, Ihara Y. Amyloid beta protein (A beta) deposition: a beta 42(43) precedes A beta 40 in Down syndrome [see comments] Ann Neurol. 1995;37:294–9. doi: 10.1002/ana.410370305. [DOI] [PubMed] [Google Scholar]
- 33.Ikonomovic MD, Klunk WE, Abrahamson EE, Mathis CA, Price JC, Tsopelas ND, et al. Post-mortem correlates of in vivo PiB-PET amyloid imaging in a typical case of Alzheimer’s disease. Brain. 2008;131(Pt. 6):1630–45. doi: 10.1093/brain/awn016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jayadev S, Leverenz JB, Steinbart E, Stahl J, Klunk W, Yu CE, Bird TD. Alzheimer’s disease phenotypes and genotypes associated with mutations in presenilin 2. Brain. 2010;133(Pt. 4):1143–54. doi: 10.1093/brain/awq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Koivunen J, Verkkoniemi A, Aalto S, Paetau A, Ahonen JP, Viitanen M, et al. PET amyloid ligand [11C]PIB uptake shows predominantly striatal increase in variant Alzheimer’s disease. Brain. 2008;131(Pt. 7):1845–53. doi: 10.1093/brain/awn107. [DOI] [PubMed] [Google Scholar]
- 36.Remes AM, Laru L, Tuominen H, Aalto S, Kemppainen N, Mononen H, et al. Carbon 11-labeled Pittsburgh compound B positron emission tomographic amyloid imaging in patients with APP locus duplication. Arch Neurol. 2008;65:540–4. doi: 10.1001/archneur.65.4.540. [DOI] [PubMed] [Google Scholar]
- 37.Villemagne VL, Ataka S, Mizuno T, Brooks WS, Wada Y, Kondo M, et al. High striatal amyloid beta-peptide deposition across different autosomal Alzheimer disease mutation types. Arch Neurol. 2009;66:1537–44. doi: 10.1001/archneurol.2009.285. [DOI] [PubMed] [Google Scholar]
- 38.Lack AK, Gill KE, Porrino LJ. Local cerebral glucose utilization in rats exposed to an enriched environment: a comparison to impoverishment. Pharmacol Biochem Behav. 2010;96:521–5. doi: 10.1016/j.pbb.2010.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]