

Abstract
Scheuthauer-Marie-Sainton syndrome also known as Cleidocranial dysplasia (CCD) is an autosomal dominant disorder characteristically presented with multiple supernumerary teeth; partial or complete absence of the clavicles; and open sagittal sutures and fontanelles. This condition was first reported by Meckel in 1760. There is also evidence that it existed in the prehistoric man. More than 1,000 cases have been reported in the medical literature regarding this syndrome. A case of a 35-year-male of CCD with multiple supernumerary teeth is being reported. The diagnostic and management aspects of this syndrome are discussed.
Keywords: Autosomal dominant, cleidocranial dysplasia, hypoplastic clavicle, retained teeth, scheuthauer-marie-sainton syndrome, supernumerary teeth
Introduction
Cleidocranial dysplasia (CCD), also known as Marie and Sainton's disease or Scheuthauer-MarieSainton syndrome or Mutational Dysostosis. It is a congenital disorder of autosomal dominant inheritance affecting both sexes equally.[1] Mutations in the core binding factor Alpha-1 gene located on chromosome 6p21 have been suggested to be the cause of CCD.[2–4] Rearrangement of long arm of chromosome numbers 6 and 8 has also been suggested in favor of CCD. It is also considered to be an autosomal dominant skeletal dysplasia caused by mutations in the bone/cartilage specific osteoblast transcription factor RUNX2 gene.
The most striking features of this syndrome are partial or complete absence of clavicles causing unusual mobility of the shoulders (seen in about 10% of cases) and late closure of fontanelles resulting in frontal bossing. Prolonged retention of deciduous teeth with delay in eruption of succedaneous teeth is the characteristic oral finding. Involvement of facial bones, altered eruption patterns and presence of multiple supernumerary teeth makes it significant for a dentist.
Case Report
A 35-year-old male patient reported with complaint of absence of teeth in upper and lower arches. His medical history was non-contributory. A detailed history ruled out any familial inheritance. His facial feature was characteristic with a sunken nasal bridge and frontal bossing. Brachycephalic head and hypertelorism were other salient features noticed on examination. He was able to adduct his shoulders when asked to bring his shoulders forward to the midline. This confirmed an anomaly in clavicle formation [Figure 1]; oral examination showed prolonged retention of deciduous teeth.
Figure 1.

Adduction of shoulders
The patient was subjected to radiographic examinations of chest, skull, hand wrist, cephalometric, and panoramic radiographs. Panoramic radiographs revealed abnormally retained primary teeth, nine impacted permanent and supernumerary teeth (excluding second and third molars) [Figure 2]. Skull AP view showed open sutures with large wormian bones [Figure 3]. Lateral cephalogram showed shortening of cranial base anteriorly; in the posterior region The physiological mandibular angle was replaced by a rounded outer contour of the mandible. Supernumerary teeth were also seen [Figure 4]. Radiograph chest PA showed narrowing of thorax and ribs. Clavicles were absent [Figure 5]. Impacted teeth were removed and prosthetic rehabilitation was done.
Figure 2.
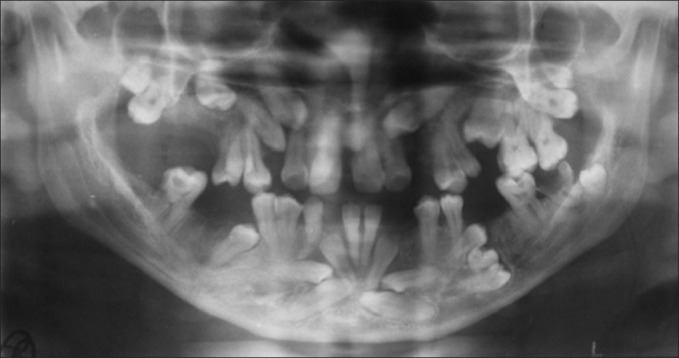
Panoramic radiograph
Figure 3.
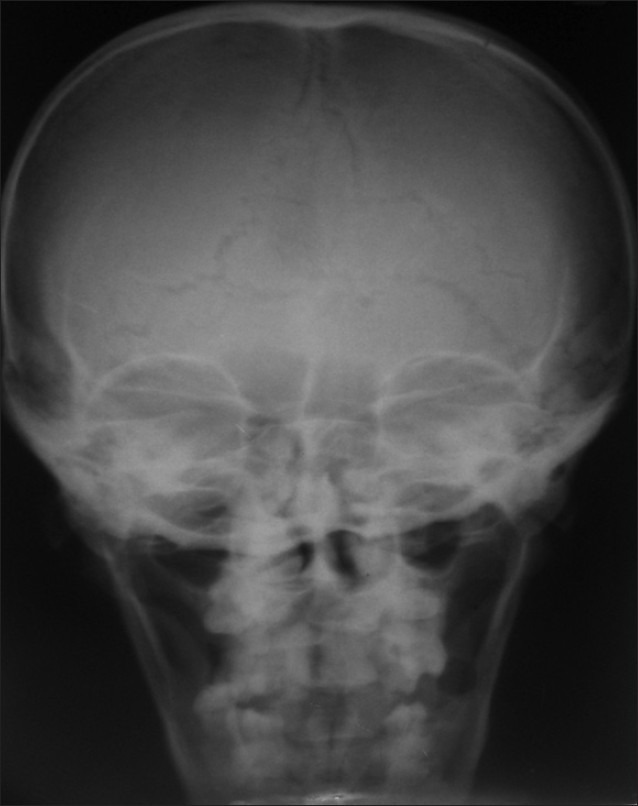
Skull AP view
Figure 4.
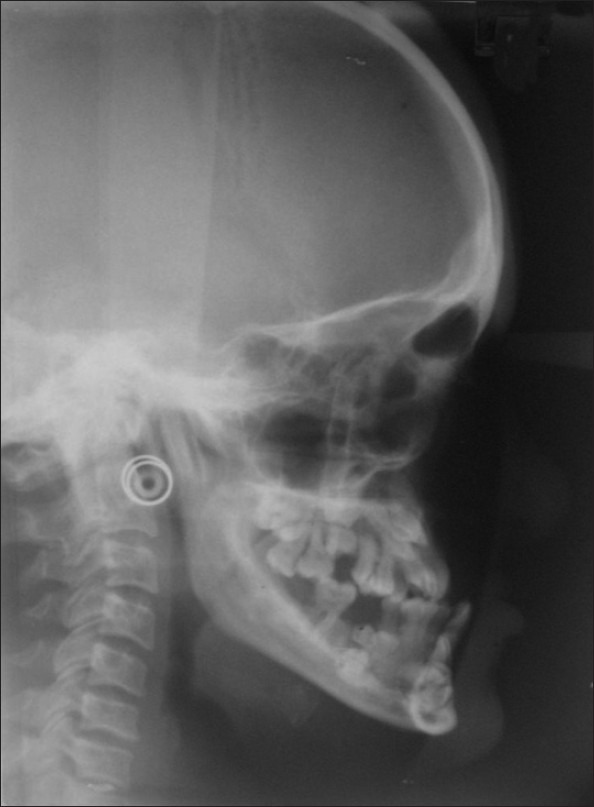
Lateral cephalogram
Figure 5.
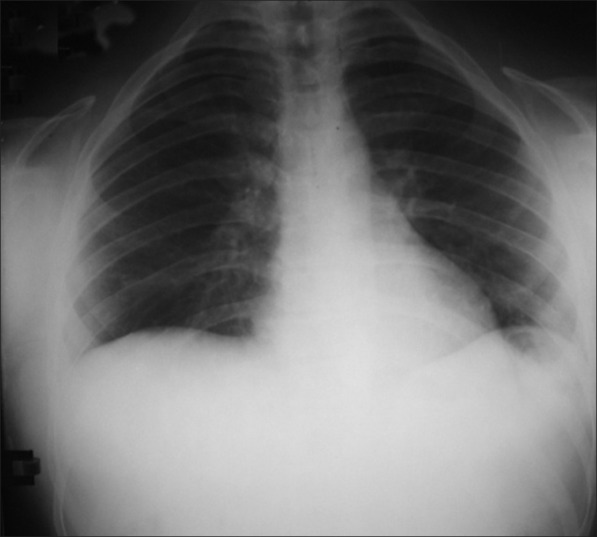
Chest PA view
Discussion
The clinical findings of Scheuthauer-Marie - Sainton syndrome, although present at birth, are often either missed or diagnosed at a much later date. Initial diagnosis is based on clinical appearance. The radiographic evaluation of patients is the most important and reliable means to confirm the diagnosis.
The affected individuals are of short stature, with a broad head and frontoparietal bossing. Depressed nasal bridge and hypertelorism are additional features of this disease. Intraorally they exhibit a high narrow arched palate with enlarged mandibles.[1,2]
One of the characteristic oral finding is prolonged retention of the deciduous teeth and a delay in eruption of the permanent teeth. Various views regarding the etiology of noneruption, such as lack of cellular cementum, defectiveness in postcementum formation, presence of thick connective tissue between oral epithelium and dental follicle, delayed tooth formation and maturation are suggested.[1]
It is characteristic for numerous unerupted supernumerary teeth to be found by radiographic examination. Crypt formation around impacted teeth and ectopic teeth also have been reported.[1] These are mainly seen in mandibular premolar and incisor areas. The reason for the formation of multiple supernumerary teeth is still unknown. A short lower facial height, anterior inclination of mandible and mandibular prognathism has also been reported.
Hypoplasia of the masseter muscles is another feature, which eventually leads to hyperfunction of the temporal muscles. This will make the anterior border of the mandibular ramus parallel to the posterior border, and the coronoid process will be directed upwards and backwards in such cases.[3]
Radiographic examination reveals the widely patent anterior fontanelle and sutures with wormian bones in cranium. The clavicles typically are reduced to single or double fragments on each side with middle part being deficient. The widened symphysis pubis (distance between pubic bones) results from a delay in ossification during adulthood. Other changes include hypoplasia and anterior rotation of the iliac wings and wide sacroiliac joints. The femoral epiphyses are large, the femoral necks broad. Spina bifida occulta is occasionally observed in the cervical and upper thoracic levels. Hands and feet demonstrate various anomalies including shortening and broadening of carpal, metacarpal, tarsal, metatarsal bones. There is hypoplasia of distal phalanges with massive epiphyses (marked in thumb) and shortening and premature epiphyseal closure of middle phalanges.[4,5]
Congenital pseudoarthrosis is characterized by the absence of one of the clavicles (usually the right clavicle). In majority of the cases, involvement is unilateral with a marked predominance of the right side. The cases are sporadic, there is no other bone involvement, and most cases presumably heal malformed bones. Pycnodysostosis or the Maroteaux-Lamy syndrome is characteized by osteosclerosis, delayed suture closures, dysplasia of distal phalanges, anodontias, and delayed eruption of permanent teeth osteosclerosis along with the absence of supernumerary teeth are sufficient to distinguish Scheuthauer-Marie-Sainton syndrome from pyknodysostosis.[4,5]
Genetic counselling is appropriate for prospective parents with a family history of Scheuthauer-MarieSainton syndrome, where one or both parents are affected.[4] No specific treatment exists for CCD, as the bony abnormalities cause little problem. Complications may arise during delivery in case of narrow pelvis Since Scheuthauer-Marie-Sainton syndrome patients generally have multiple impacted permanent teeth, prolonged retention of primary teeth, and multiple supernumerary teeth, they can develop masticatory problems with aging. Many approaches have been suggested for treatment of such problems, including the following: Removal of the impacted permanent, supernumerary and deciduous teeth, combined with installation of over-dentures or orthodontic traction of the impacted permanent teeth;[6,7] removal of the supernumerary teeth immediately after completion of mineralization of their crowns combined with removal of the overlying bone of the permanent teeth to facilitate their eruption. Thus, two critical factors in the success of these treatments are the correct timing of the treatment and the number of supernumerary teeth. In our case, a supernumerary tooth was extracted and prosthetic rehabilitation was given.
Footnotes
Source of Support: Nil,
Conflict of Interest: None declared
References
- 1.Shafer WG, Hine MK, Levy BM. A Text Book of Oral Pathology. 5th ed. New Delhi: Elsevier; 2006. pp. 994–7. [Google Scholar]
- 2.Neville BW, Damm DD, Allen CM, Bouquot JE. Oral and Maxillofacial Pathology. 2nd ed. Philadelphia: WB Saunders Co; 2002. pp. 537–9. [Google Scholar]
- 3.Furuuchi T, Kochi S, Sasano T, Iikubo M, Komai S, Igari K. Morphologic characteristics of masseter muscle in cleidocranial dysplasia: A report of 3 cases. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2005;99:185–90. doi: 10.1016/j.tripleo.2004.03.011. [DOI] [PubMed] [Google Scholar]
- 4.Mundlos S. Cleidocranial dysplasia: Clinical and molecular genetics. J Med Genet. 1999;36:177–82. [PMC free article] [PubMed] [Google Scholar]
- 5.Tanaka JL, Ono E, Filho EM, Castilho JC, Moraes LC, Moraes ME. Cleidocranial dysplasia: Importance of radiographic images in diagnosis of the condition. J Oral Sci. 2006;48:161–6. doi: 10.2334/josnusd.48.161. [DOI] [PubMed] [Google Scholar]
- 6.Jensen BL, Kreiborg S. Dental treatment strategies in cleidocranial dysplasia. Br Dent J. 1992;172:243–7. doi: 10.1038/sj.bdj.4807836. [DOI] [PubMed] [Google Scholar]
- 7.Davies TM, Lewis DH, Gillbe GV. The surgical and orthodontic management of unerupted teeth in cleidocranial dysostosis. Br J Orthod. 1987;14:43–7. doi: 10.1179/bjo.14.1.43. [DOI] [PubMed] [Google Scholar]