

Abstract
This Minireview provides an overview on the development of silyl ketene imines and their recent applications in catalytic, enantioselective reactions. The unique structure of the ketene imine allows a diverse range of reactivity patterns and provides solutions to existing challenges in the enantioselective construction of quaternary stereogenic carbon centers and cross-benzoin adducts. A variety of reactions for which silyl ketene imines have been applied are presented with an overall goal of inspiring new uses for these underutilized nucleophiles.
Keywords: asymmetric catalysis, cyanides, silanes, synthetic methods, umpolung
1. Introduction
Silyl ketene imines (SKIs) are a class of silylated nucleophiles prepared by the selective N silylation of nitrile anions with electrophilic silylating reagents (1; Scheme 1A). SKIs belong to a broader class of compounds known as cumulenes, which are molecules possessing at least two or more cumulative double bonds, and include functional groups such as allenes and ketenes.[1] SKIs are more generally related to enoxysilane nucleophiles which are prepared by the selective O silylation of enolates.[2] Enoxysilanes have found extensive use as nucleophiles in catalytic, enantioselective reactions such as Mukaiyama-type Michael[3] and carbonyl addition reactions.[4]
Scheme 1.
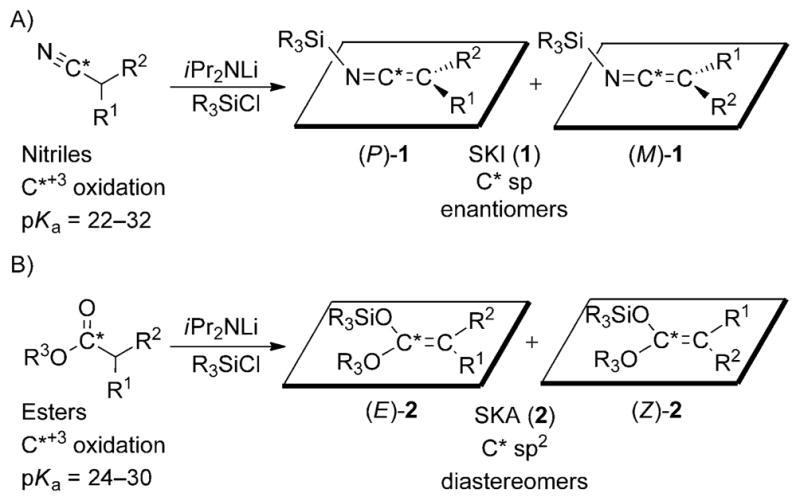
A) Synthesis of SKIs from nitriles yielding P and M enantiomers, where R1 >R2. B) Comparison to the synthesis of SKAs from esters yielding E and Z diastereomers.
From an elementary viewpoint, SKIs can be seen as the nitrile analogues of ester- or amide-derived enoxysilanes (2; Scheme 1B). For example, esters and nitriles both have a carbon atom in the +3 oxidation state and exhibit similar pKa values for deprotonation of the α-hydrogen atom. Furthermore, the synthesis of SKIs and silyl ketene acetals (SKAs) follow the exact same procedures. Although many similarities exist between these two classes of nucleophiles, structurally, SKIs and enoxysilanes are very different. The characteristic feature of SKIs is the pair of orthogonal substituent planes that impart an axis of chirality whenever R1 and R2 are dissimilar, and results in the formation of a racemic mixture of P and M enantiomers. Conversely, SKAs are planar, achiral compounds which are obtained as a mixture of E and Z diastereomers.
The unique structure of SKIs has the potential to offer significant advantages compared to enoxysilanes, especially in the context of setting quaternary stereogenic carbon atoms. One of the key issues in constructing these centers is overcoming the additional steric repulsion associated with forming a fully substituted tetrahedral carbon atom. Developing reliable catalytic, enantioselective methods that meet this challenge continues to inspire synthetic chemists.[5]
Although a number of elegant solutions have been developed, a useful class of reactions that has remained refractory for the synthesis of quaternary stereogenic carbon atoms is the addition of disubstituted enoxysilanes to carbonyl electrophiles. Overcoming the steric requirements in the addition of these nucleophiles is exacerbated by their planar structure, which places a significant amount of the steric bulk within the plane of the reacting trigonal carbon atom. Examination of potential open transition structures for Lewis acid catalyzed carbonyl additions of disubstituted silyl ketene imines and enoxysilanes clearly shows the advantages that SKIs offer (Scheme 2).
Scheme 2.
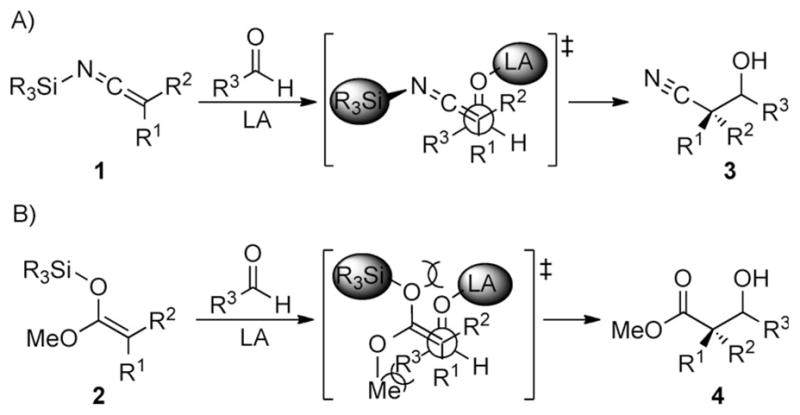
A) Open transition state for a synclinal approach of an SKI to a Lewis acid activated aldehyde. B) Comparison to open transition state for synclinal approach of an SKA. LA = Lewis acid
The presence of an sp-hybridized carbon atom in the SKI allows the sterically bulky silyl group to be placed in the plane perpendicular to and distal from the reacting carbon center (Scheme 2A). This arrangement avoids many of the unfavorable steric interactions that are encountered in the more congested approach of an enoxysilane (Scheme 2B), and could significantly reduce the energy of the transition state leading to C–C bond formation.
A second impediment to developing diastereo- and enantioselective additions of disubstituted enoxysilanes is the inability to prepare geometrically defined α,α-disubstituted enolate or enolate equivalents.[6] The addition of enoxysilanes to activated carbonyl groups typically proceed through open transition structures, and the lack of a stereo-defined enolate can often be reflected in poor anti/syn diastereoselectivity in the product. SKIs also offer a potential solution to this issue by virtue of their structure. Because SKIs contain one sp2-hybidized and one sp-hybridized carbon atom, the problems associated with selectively preparing a single geometrical isomer are completely avoided. However, unsymmetrically substituted SKIs are chiral and their chirality raises additional concerns for developing catalytic, enantioselective processes because each enantiomer could react at a different rate in the catalytic cycle, or worse, lead to different stereoselectivities. However, these issues would be mitigated if the SKI enantiomers had a low-energy barrier to racemization (see Section 2.3).
In this Minireview, recent advances in SKI chemistry are presented with a focus on how their unique structure can provide significant advantages over more commonly employed enoxysilanes.[7] The first part of the review (Sections 2 and 3) provides introductory material on the synthesis, structure, and early uses of SKIs in non-stereoselective transformations. The second half focuses on more recent applications of SKIs in catalytic, enantioselective reactions. The emphasis in Section 4 is on setting quaternary stereogenic centers, and Section 5 describes a new class of heteroatom-substituted SKIs which can serve as acyl anion equivalents in enantioselective addition reactions.
2. Synthesis, Structure, and Properties of SKIs
The deprotonation of nitriles generates ambident anions which can undergo reactions with electrophiles at either the carbon or nitrogen atom. The alkylation of nitrile anions with alkyl halides is a well-established method and has been widely used for the preparation of substituted nitriles.[8] In general, the reaction of nitrile anions with carbon-based electrophiles occurs with very high site selectivity for C versus N alkylation, although some exceptions with extremely hindered disubstituted nitriles reacting with secondary alkyl halides are known.[9] Beginning in the late 1950s researchers became interested in studying the reactions of nitrile anions with silicon-based electrophiles.
2.1. Initial Isolation and Characterization
The first synthesis of an SKI was reported by Prober in 1956.[10] Upon exposure of acetonitrile to metallic sodium and chlorotrimethylsilane (TMSCl) a new species was formed that had a characteristic absorbance in the IR region at ν̃ = 2030 cm−1. The assignment of this new compound as an SKI was based on the similarity of the intense IR band to previously reported N-alkyl ketene imine resonances.[11] Later, a more detailed synthesis and characterization of an SKI was reported by Krüger and Rochow in 1963 (Scheme 3).[12] These authors examined the reaction of acetonitrile with excess sodium hexamethyldisilazide (NaHMDS), and it resulted in the isolation of an ether-insoluble white solid. Treatment of the solid with excess TMSCl produced a yellow liquid in low yield with spectroscopic data consistent with tris(trimethylsilyl)ketene imine (5). A more-detailed investigation by West and Gornowicz on the same reaction identified the isolated white solid as the monosodium salt of acetonitrile (NaCH2CN).[13] Formation of SKI 5 was then proposed to occur by initial silylation of the monoanion and subsequent deprotonation/silylation cycles, aided by the acidifying effect of silicon on the α proton. Neither of the authors of these initial disclosures reported any applications or further study of 5.
Scheme 3.
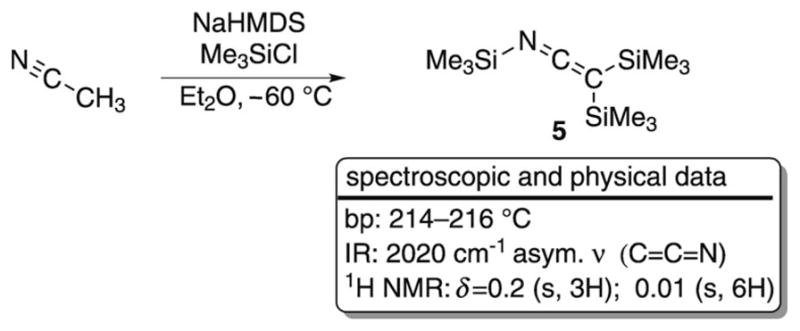
Synthesis and spectroscopic data for SKI 5.
2.2. Synthesis from Alkyl-Substituted Nitriles
The first useful procedure for preparing SKIs was reported by Watt.[14] Previous studies by Watt had shown that carbon-based electrophiles, such as alkyl halides, react with ambident nitrile anions at the carbon terminus to afford C-alkylation products in high selectivity.[14,15] Following up on this report, Watt examined the product distributions for reactions of various disubstituted nitrile anions with trialkyl-silyl chlorides. Interestingly, the opposite site selectivity was observed in these reactions, thus yielding N-silyl ketene imines in excellent yields (Scheme 4).
Scheme 4.

Synthesis of SKIs from disubstituted nitriles. LDA =lithium diisopropylamide, TBS =tert-butyldimethylsilyl, THF = tetrahydrofuran.
This study further demonstrated the importance of disubstitution on the nitrile for obtaining high selectivities and yields of SKIs. Monosubstituted nitriles yielded SKIs only after initial in situ Cα silylation (Scheme 5) and the reaction conditions had to be reoptimized to favor this isomer by increasing the amounts of LDA and TBSCl. Combining monosubstituted nitriles with equimolar amounts of LDA and TBSCl did not result in isolation of monosubstituted silyl ketene imines, but rather just reduced yields of the α-silylated SKIs 12.
Scheme 5.
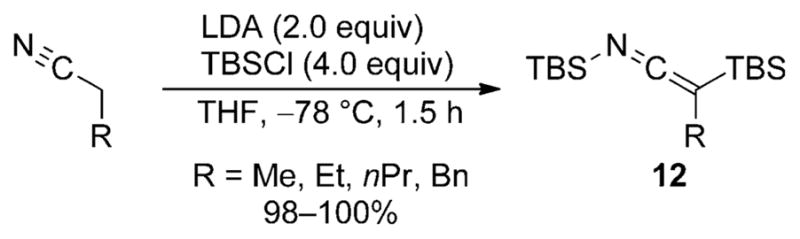
Competitive C silylation of monosubstituted nitriles.
2.3. Factors Influencing C versus N Silylation
The size of the alkyl substituent in the chlorosilane can also have an impact on the site of silylation (N vs C) for nitrile anions, an effect also studied by Watt[15] and others.[16] For example, lithiation of 2-phenylpropionitrile with LDA at low temperature and subsequent trapping of the anion with the less sterically encumbered TMSCl affords the C-silyl nitrile 13 in excellent yield and selectivity (Scheme 6).[17]
Scheme 6.
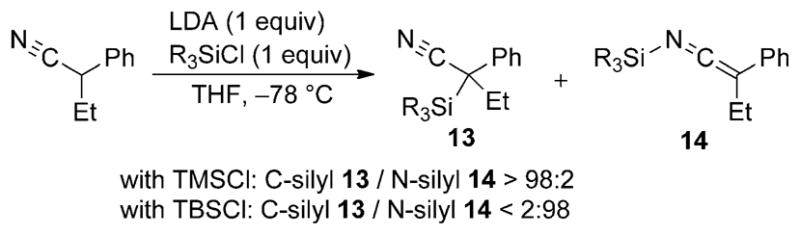
Effect of chlorosilane on C versus N silylation of nitrile anions.
This outcome contrasts the results obtained with the sterically bulky TBSCl, which affords SKI 14 exclusively and in high yield. In some cases, SKIs are known to isomerize to the C-silylated isomers upon heating. These observations are consistent with the C-silyl isomer being the more thermodynamically favored isomer. Sterically hindered silylating agents likely favor reaction at the nitrogen atom, because the C=N subunit in the nitrile anion is relatively unencumbered.
2.4. Stereoisomerization of SKIs
The perpendicular substituent planes present in SKIs impart an axis of chirality whenever the substituents are dissimilar. The configurational stability of the SKIs will play an important role in developing catalytic, enantioselective processes, because each enantiomer could react differently in the presence of a chiral catalyst. Unfortunately, SKIs are not reported to manifest their intrinsic chirality on the 1H or 13C NMR timescale, thus making determination of their configurational stabilities extremely difficult. However, related studies on the configurational stability of other ketene imines have been performed and can be used for comparison.
The stereoisomerization of both N-alkyl and N-aryl ketene imines has been studied by observing the NMR coalescence temperature of the signals for diastereotopic methyl and/or benzylic protons in the ketene imines by Jochims et al.[18] The results demonstrate that both N-aryl and N-alkyl ketene imines racemize quickly in solution at room temperature with experimentally determined energy barriers ranging from 30–65 kJmol−1. Computational studies, also reported by Jochims, suggest a mechanism that involves the intermediate 15, which contains a linear (C–C≡N–R)+ fragment (Scheme 7). A similar mechanism for topomerization of imines has been suggested with experimentally determined values of 57–64 kJmol−1.[19]
Scheme 7.

Stereoisomerization of N-aryl and N-alkyl ketene imines.
N-Alkyl ketene imines are typically on the higher end of the racemization barriers and the authors have noted that, in general, the energies are lowered by electron-withdrawing substituents at either the N or C terminus.[18] Therefore, N-silyl ketene imines likely have inversion barriers closer to those observed for N-alkyl ketene imines, which are in the range of 40–60 kJmol−1.
3. SKIs in Uncatalyzed Reactions
Despite the early reports on the synthesis and characterization of SKIs by Krüger, and the later improvements by Watt, very few examples of SKIs as nucleophiles have appeared. Surprisingly, the related enoxysilane nucleophiles derived from ketones, esters, and amides were concurrently being developed for uses in a wide variety of synthetic transformations.[2a–c]
3.1. Aldol and Michael-Type Reactions
An early report from Frainnet and co-workers describes the carbonyl addition reactions of SKIs.[20] These authors established that SKIs are competent carbon nucleophiles for the addition to a range of different carbonyl electrophiles including aromatic aldehydes, α,β-unsaturated aldehydes, ketones, and acid halides. For example, the combination of N-(trimethylsilyl)diphenylketene imine (10) with benzaldehyde, in the absence of solvent leads to an exothermic reaction from which the β-silyloxy nitrile 16 could be isolated in high yield (Scheme 8).[20] An electron-poor aromatic aldehyde, 4-chlorobenzaldehyde, also undergoes addition in high yield, but no further studies on the scope of this reaction with respect to the nucleophile or other aldehydes were reported.
Scheme 8.
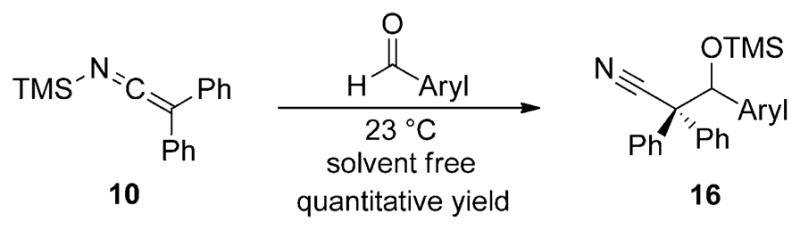
Aldol addition of SKI 10 to aromatic aldehydes.
Interestingly, a competitive 1,4-addition pathway is observed with olefinic aldehydes under identical reaction conditions (Scheme 9).[20] In the addition of 10 to cinnamaldehyde, a 63:37 ratio of 1,4- to 1,2-addition products (17 and 18, respectively) is observed. However, with crotonaldehyde as the electrophile, the addition proceeds exclusively through the 1,4-addition pathway to afford a saturated aldehyde product in high yield after hydrolysis of the trimethylsilyl enol ether.
Scheme 9.

Competitive 1,4- and 1,2-addition of SKI 10 with enals.
The reaction of 10 with various ketone electrophiles was also examined. Unlike the additions to aldehydes, which proceed in the absence of any promoters, ketones require substoichiometric amounts of HgI2 to engage in reaction (Scheme 10). The undesired enolization of the ketone is a competitive process for many of the substrates studied, and limits the generality of this process.
Scheme 10.

Addition of SKI 10 to ketones.
3.2. Acylation Reactions
The C acylation of 10 with acyl chlorides was also briefly examined by Frainnet and co-workers.[21] The reactions are performed in the absence of a promoter or solvent to afford the β-keto nitrile products 19 in high yields (Scheme 11). Both aromatic and aliphatic acyl chlorides participate in the acylation reaction to generate ketone products in similar yields.
Scheme 11.
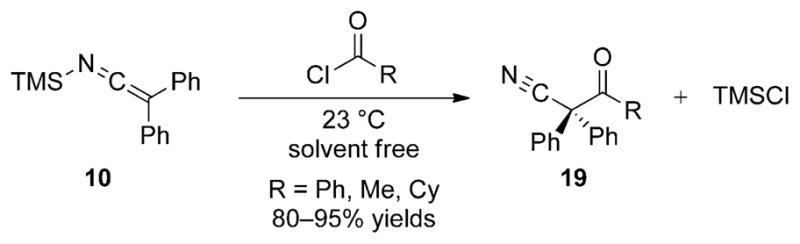
Uncatalyzed acylation of SKI 10 with acid halides.
A more detailed investigation on the acylation of SKIs was reported by Meier and Würthwein after the initial disclosure by Frainnet.[22] Remarkably, these authors observed competitive N acylation with extremely hindered SKIs. For example, the reaction of di-tert-butyl silyl ketene imine 20 with benzoyl chloride yields selectively the N-benzoyl ketene imine 21 in 52% yield (Scheme 12).
Scheme 12.
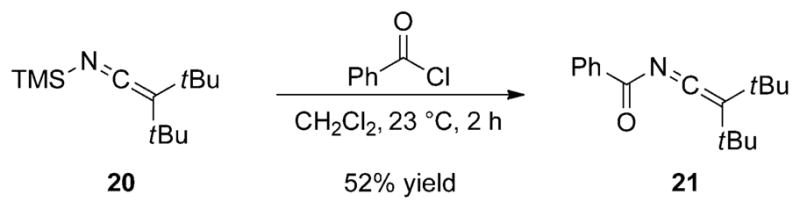
Competitive N acylation of sterically hindered SKI 21.
3.3. Aldol-Type Addition and Subsequent Peterson Elimination
The addition of bis(trimethylsilyl)ketene imine 22 to aldehydes in the presence of Lewis acids has been investigated by Matsuda and co-workers (Scheme 13).[23] The direct aldolization product of this reaction is β-silylcarbinol 23, which undergoes a Peterson-type elimination in the presence of boron trifluoride to afford the 2-alkenenitriles 24 in moderate to good yields. Aldehydes containing a linear aliphatic chain produced Z alkenes whereas α-branched aldehydes led to the isomeric E alkenes. Because of the stereospecificity of the Peterson elimination,[24] this change in selectivity can be rationalized in terms of the diastereoselectivity of the addition of 22 to α-branched versus unhindered aldehydes. Ketones also undergo the addition/elimination reaction, but with reduced yields and selectivities.[25]
Scheme 13.
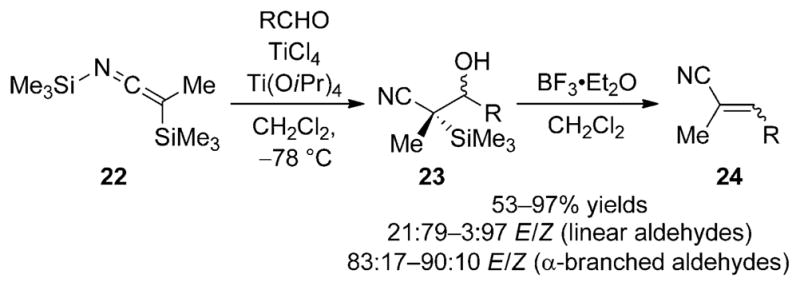
Aldol addition of SKI 22 and subsequent Peterson elimination.
3.4. Oxidative Decyanations
A procedure for converting secondary nitriles into ketones through the intermediacy of an SKI was developed by Watt and co-workers.[15,26] This process renders alkyl nitriles acyl anion equivalents, thus greatly extending the utility of the cyano functional group. To achieve the umpolung, secondary nitriles are converted into SKIs which can be iodinated at the α-carbon atom with iodine in THF (Scheme 14). The resulting α-iodo nitriles 25 undergo oxidative decyanation in the presence of silver oxide to produce the ketone products 26 in moderate to good overall yields.
Scheme 14.
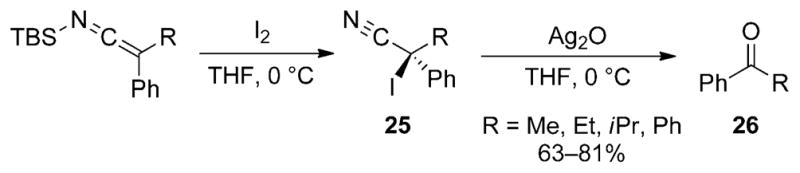
Oxidative decyanation of SKIs to afford ketone products.
3.5. Preparation and Reactions of Silyl Vinylketene Imines
The preparation of SKIs from enolizable, unsaturated nitriles offers new opportunities in reactivity by extension of the nucleophilic character to a remote γ-carbon atom. However, preparation of these unsaturated nucleophiles can be complicated by competing Cα or Cγ silylation. Although a number of reports have detailed the synthesis of SKIs from alkyl nitriles, very little research has been conducted on the preparation of N-silyl vinylketene imines derived from unsaturated nitriles. A single report on the conversion of allyl cyanide into an N-silyl vinylketene imine 27 was disclosed by Ghosez and co-workers (Scheme 15).[27] The synthesis involves the double lithiation of allyl cyanide with an excess of LDA and subsequent trapping of the dianion with an excess of triisopropylsilyl chloride (TIPSCl) to afford 27 in good yield and high selectivity. The sensitivity of allyl nitrile toward additional Cα silylation is noted even with the use of bulky silylating agents such as TBSCl.
Scheme 15.
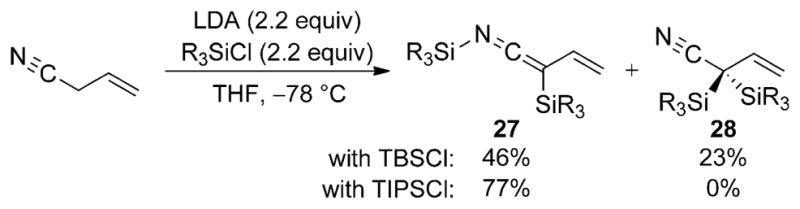
Synthesis of N-silyl vinylketene imine from allyl cyanide.
In its first synthetic application, the N-silyl vinylketene imine 29 served as a diene in [4+2] cycloaddition reactions with acetylenic esters (Scheme 16). Typically the neat ketene imine is heated to 150°C in the presence of an equimolar amount of alkynyl dienophile to yield the substituted anilines 30 after desilylation with KF in refluxing MeOH.[27]
Scheme 16.
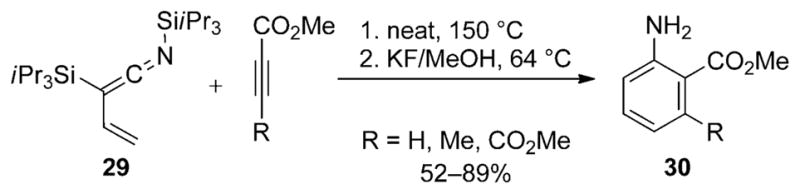
[4+2] cycloaddition of N-silyl vinylketene imine 29.
4. Catalytic, Enantioselective Reactions of SKIs
4.1. Acylation Reaction
In spite of the fact that SKIs were known to be competent nucleophiles in additions to carbonyl electrophiles, it was not until 2005 that the first catalytic, asymmetric reaction was reported.[17] In previous studies, the planar chiral pyridine derivative 31 was shown to catalyze the enantioselective acylation of SKAs.[28] To extend this work toward the synthesis of nitriles containing a quaternary stereogenic center, Fu and Mermerian studied the acylation of SKIs under similar reaction conditions (Scheme 17).[17] In the presence of 5 mol% of the Lewis base catalyst 31, SKIs undergo acylations with propionic anhydride in good yields and moderate to good enantioselectivities. The reaction exhibits excellent substrate scope for additions of aryl-substituted SKIs, however dialkyl-substituted nucleophiles are unreactive under the optimized reaction conditions. Other electrophilic reagents such as cyanoformates and chloroformates also participate in the reaction but with inferior enantioselectivities.
Scheme 17.

Catalytic, enantioselective acylation of SKIs.
A mechanistic hypothesis suggests that the acylation proceeds through a dual activation pathway which involves first, a reaction of the Lewis base catalyst with an acid anhydride to produce a reactive acylpyridinium cation. The propionate counterion formed during this activation step can then activate the SKI by coordination to the silyl group, thus potentially leading to an anionic nitrile intermediate. Experimental evidence in support of a dual activation pathway is provided by the observation that an α-trimethylsilyl nitrile also participates in the acylation reaction with similar yield, enantioselectivity, and the same sense of asymmetric induction as is observed with SKIs. This hypothesis is also consistent with the low reactivity observed with dialkyl-substituted SKIs, which lack an anion stabilizing group.
4.2. Aldol Reaction
The aldol reaction represents a powerful and predictable method for the stereoselective construction of carbon–carbon bonds, and a number of useful catalyst systems for setting both secondary and tertiary stereogenic centers have been realized.[4d,29] However, a significant, unsolved problem in aldol methodology still remains, namely the synthesis of quaternary stereogenic carbon centers, which in large measure has been hampered by the inability to control the geometry of disubstituted enolates.[6] To address this problem and empower the aldol reaction for the synthesis of quaternary stereogenic carbon centers, studies in these laboratories have focused on the Lewis base catalyzed additions of SKIs to aldehydes (Scheme 18).[30]
Scheme 18.

Catalytic, enantioselective aldol reaction of SKIs.
Preceding reports showed that the combination of SiCl4 and Lewis base catalyst (R,R)-39 provides an effective catalyst system for the addition of silyl enol ethers and silyl ketene acetals to aldehydes.[31] Accordingly, the aldol addition of a variety of aryl- and alkyl-substituted SKIs with aromatic aldehydes was examined under similar reaction conditions. The resulting β-hydroxy nitriles containing a quaternary stereogenic center (40–49) are isolated in good yield and with excellent levels of diastereo- and enantioselectivity. Broad substrate scope in both the SKI and aromatic aldehyde is observed. However, aliphatic aldehydes are unreactive under the optimized reaction conditions.
The reaction rates, measured by in situ IR monitoring, for the additions of analogously substituted SKIs and SKAs are vastly different.[30b] For example, SKIs typically undergo addition to aromatic aldehydes in less than 15 minutes, whereas disubstituted SKAs are mostly unreactive under the same reaction conditions. The disparity in rates for these two nucleophiles suggests a higher energy barrier for the addition of disubstituted SKAs, potentially because of the added nonbonded repulsion encountered in the transition structure (see, Scheme 2B).
4.3. Mannich Reaction
The addition of enolates or enolate equivalents to imines has been of great interest owing to the usefulness of the β-amino carbonyl products and their application to the preparation of β-amino acids.[32] Compared to aldehydes, imines represent a more difficult class of acceptors in view of the reduced electrophilicity of the imine carbon atom.[33] However, the electrophilicity of imines can be augmented by introduction of an electron-withdrawing N substituent, which greatly increases the utility of imines in a number of different reactions.[34] Despite these advances, preparing quaternary stereogenic centers by the enantioselective addition of disubstituted enolate or enolate equivalents to imines is exceedingly rare.[35] This paucity of methods could again arise from the additional strain energy encountered in the formation of quaternary carbon centers, and becomes especially difficult to surmount with the reduced electrophilicity associated with an imine acceptor.
4.3.1. Mannich Reaction of Copper Ketene Imines
An innovative strategy for setting quaternary stereogenic centers in Mannich reactions has been developed by Shibasaki and co-workers.[36] Decarboxylation of α-cyanocarboxylates in the presence of copper(I) acetate and the chiral phosphine ligand (R)-50 generates the N-metalloketene imines 51 (Scheme 19).
Scheme 19.

In situ generation of copper ketene imines and subsequent Mannich reaction.
Enantioselective addition of these reactive nucleophiles to N-diphenylphosphinoyl-activated imines produces β-amino cyano compounds containing a quaternary stereogenic center in good yield and stereoselectivity. Aromatic and aliphatic imines react with similar yields and enantioselectivities, but a significant reduction in the diastereoselectivity is observed for addition to aliphatic imines (compare 52 to 57). In addition, the best results are observed when phenyl-substituted α-cyanocarboxylic acids are employed. The authors do not provide spectroscopic evidence for the structure of the metalloid ketene imine, so it is difficult to rule out the possibility of a C-bound copper intermediate as the active nucleophile.
4.3.2. Mannich Reaction of SKIs
Enantioselective additions of SKIs to acyl hydrazones catalyzed by stoichiometric quantities of a chiral silicon Lewis acid have recently been reported by Leighton and co-workers.[37] The Mannich reaction allows access to the β-hydrazido nitriles 62, which contain a quaternary stereogenic center in moderate to good yields and with good diastereo- and enantioselectivities (Scheme 20).
Scheme 20.
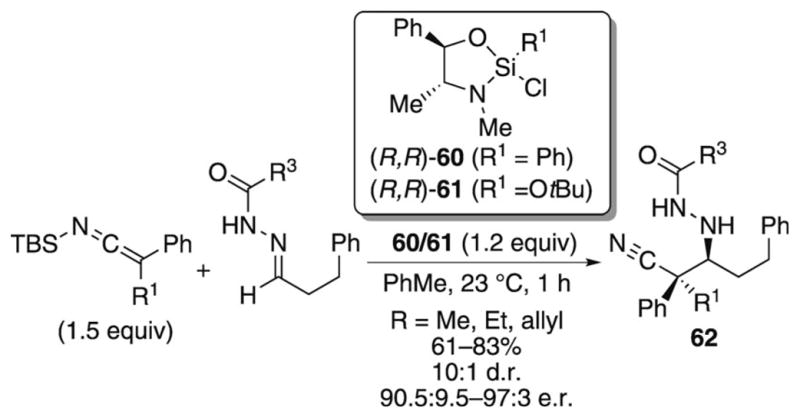
Enantioselective Mannich reaction of SKIs.
Hydrazones derived from aliphatic and aromatic aldehydes participate in the reaction, but consistently lower yields are observed with aliphatic hydrazones. The large difference in reactivity between SKIs and SKAs observed in the aldol addition is seen here as well; the authors report that disubstituted SKAs do not react to any appreciable extent with these hydrazone electrophiles.
Application of this method for the synthesis of pyrrolidines was reported by Leighton and Vu (Scheme 21).[38] The Mannich reaction of the allyl-substituted SKIs 63 with hydrazones results in the formation of the bishomoallyic hydrazides 64. These substrates participate in a thermal hydroamination reaction[39] to afford the substituted pyrrolidines 65 in good yields and stereoselectivities.
Scheme 21.
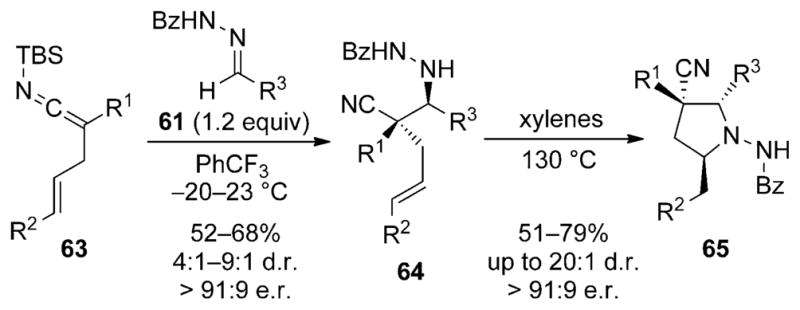
Synthesis of substituted pyrrolidines using SKIs. Bz =benzoyl.
4.4. Conjugate Addition
The conjugate addition of nucleophiles to α,β-unsaturated acceptors leading to 1,5-dicarbonyl compounds has been a productive area for the stereoselective synthesis of compounds containing quaternary carbon centers.[5e] The majority of examples rely on the addition of stabilized nucleophiles such as β-keto esters, β-diketones, and α-cyano ketones reacting with cyclic and acyclic enones. The conjugate addition of simple disubstituted enolates derived from esters, nitriles, or ketones is largely an unsolved problem.[40]
The seminal work of Frainnet and co-workers demonstrated the ability of SKIs to undergo site-selective 1,4-additions with α,β-unsaturated aldehydes and ketones.[20] In continuation of this early precedent, studies in our laboratories focused on the Lewis base catalyzed conjugate addition of SKIs to unsaturated electrophiles (Scheme 22).[41]
Scheme 22.
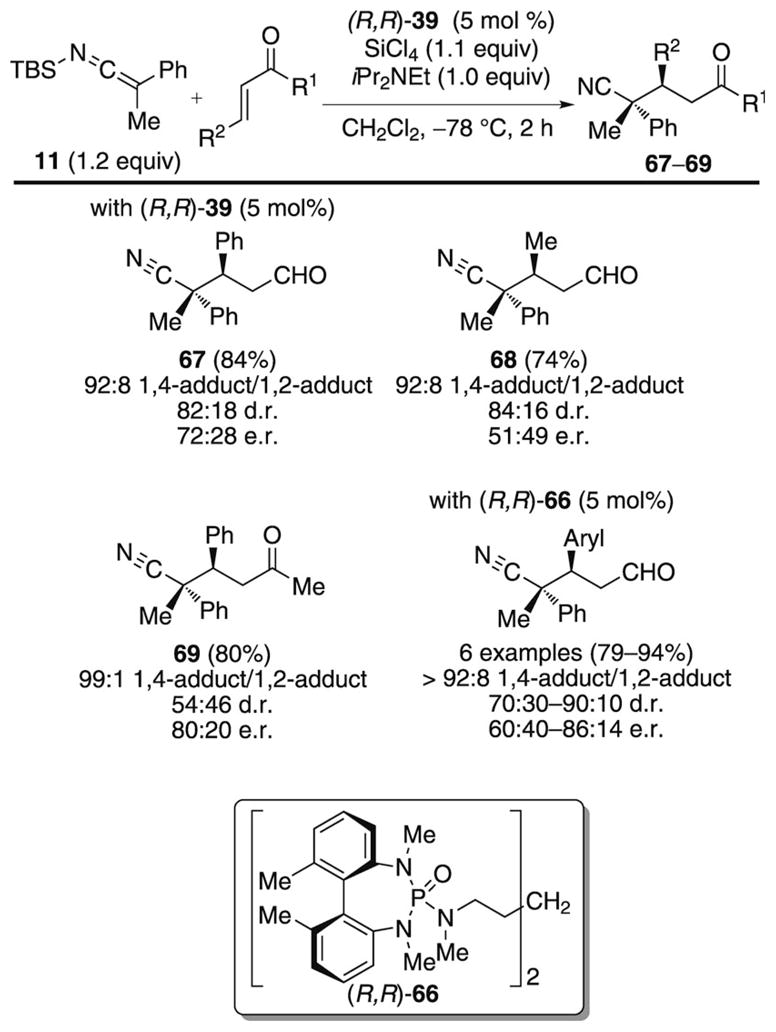
Catalytic, enantioselective conjugate addition of SKI 11.
An initial survey of substrates with SKI 11 and Lewis base catalyst (R,R)-39 identified α,β-unsaturated aldehydes and ketones as promising candidates which yielded γ-cyano carbonyl compounds in a greater than 90:10 ratio of the 1,4-to 1,2-addition products and moderate stereoselectivities. Efforts to increase the enantioselectivity by structural modification of the Lewis base proved unfruitful and only a moderate increase in the diastereoselectivity with the biphenyl-based phosphoramide catalyst (R,R)-66 could be achieved. An examination of the substrate scope for aromatic enals showed that both electron-rich and heteroaromatic enals undergo site-selective 1,4-addition in good yield and moderate enantioselectivity.
Catalyst loading studies demonstrate that the reduced enantioselectivity is not solely a result of a competitive achiral background reaction. Other possible factors contributing to the moderate enantioselectivity are: 1) the inability of the catalyst to control the s-cis versus s-trans conformation of the enal and 2) the stereocontrolling influence of the chiral Lewis base is too far removed from the site of addition.
4.5. Vinylogous Additions
The preceding examples have all focused on the synthesis of quaternary stereogenic carbon centers by reaction at the α position of dialkyl-substituted SKIs. By applying the principle of vinylogy to SKIs it is possible to extend the nucleophilic character to the γ-carbon atom through introduction of a double bond.[42] Site-selective γ addition of vinylogous SKIs with carbonyl compounds would produce α,β-unsaturated nitriles containing a γ-stereogenic center. Subsequent transformation of the unsaturated nitrile would allow access to α,β-unsaturated acids, aldehydes, and amides as well as allylic amines.[43] Despite the synthetic utility of the products, enantioselective vinylogous additions of allylic nitriles are limited.[43a,44]
To expand the scope of nucleophilic allylic nitriles in asymmetric synthesis, our laboratories recently reported a method for the Lewis base catalyzed vinylogous aldol additions of the N-silyl vinylketene imine 70 derived from 2-methyl-3-butenenitrile.[45] The inherent problem associated with the synthesis of this class of SKIs is achieving N-silylation over competitive Cα or Cγ silylation (see, Section 3.5). The use of a sterically encumbered silylating agent in combination with an allylic nitrile containing an α-alkyl substituent allows the preparation of the vinyl SKI 70 in good yield and high selectivity (Scheme 23).
Scheme 23.
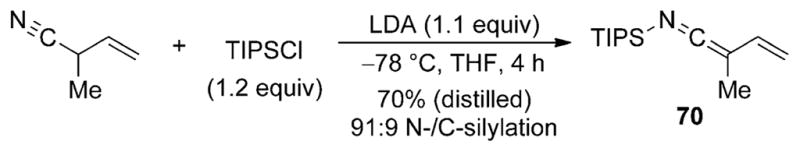
Synthesis of N-silyl vinylketene imine 70.
The N-silyl vinylketene imine 70 undergoes Lewis base catalyzed vinylogous aldol reactions with a wide range of acceptors, including aliphatic, olefinic, and aromatic aldehydes (Scheme 24). These reactions are characterized by excellent site selectivity which favors γ addition of the nucleophile to the aldehyde acceptor and yields α,β-unsaturated nitriles with high preference for formation of the E-double isomer. Importantly, aliphatic aldehydes participate in the reaction to afford nitrile products in moderate to good yields and high enantioselectivities, albeit at longer reaction times (24 h). Olefinic and aromatic aldehydes react with much faster rates (ca. 2 h), and overall afford nitriles in good yield and enantioselectivity. The lowest selectivities observed are for electron-poor and extremely hindered aromatic aldehydes. In these cases an achiral background reaction may be competitive.
Scheme 24.

Catalytic, enantioselective vinylogous addition of SKI 70.
5. α-Heteroatom-Substituted SKIs
Incorporation of a cyano group into the carbon framework of a molecule imparts novel reactivity which can be clearly distinguished from related ester or amide functionality. Similar to carbonyl groups, the cyano group inductively and mesomerically stabilizes negative charge and can be utilized for generating nucleophilic character at the α-carbon atom through deprotonation. Although this mode of reactivity is analogous to any carbonyl compound, the unique geometry of the resulting ketene imines has distinct advantages for the preparation of quaternary stereogenic carbon centers, as was illustrated in previous examples. A fundamental way in which cyano and carbonyl functions differ is manifested in the ability of the cyano group to act as a nucleofuge. This very simple difference in leaving group ability between these two groups has profound implications on the modes of reactivity available to nitrile nucleophiles.
5.1. Cyanide Catalyzed Benzoin Reaction
An instructive example of how the cyano group can impact reactivity is provided by the now classic cyanide-catalyzed benzoin reaction, first reported by Wöhler and Liebig in 1832 (Scheme 25).[46] In this process, the cyanide ion adds to an aromatic aldehyde to afford a cyanohydrin which is now activated for deprotonation of the former aldehydic hydrogen atom. Addition of the resulting ketene imine anion 81 to a second molecule of aldehyde and subsequent proton transfer generates the unstable cyanohydrin conjugate base 82. Loss of the cyanide group by collapse of the tetrahedral carbon atom in this intermediate affords the α-hydroxy carbonyl product 83.
Scheme 25.
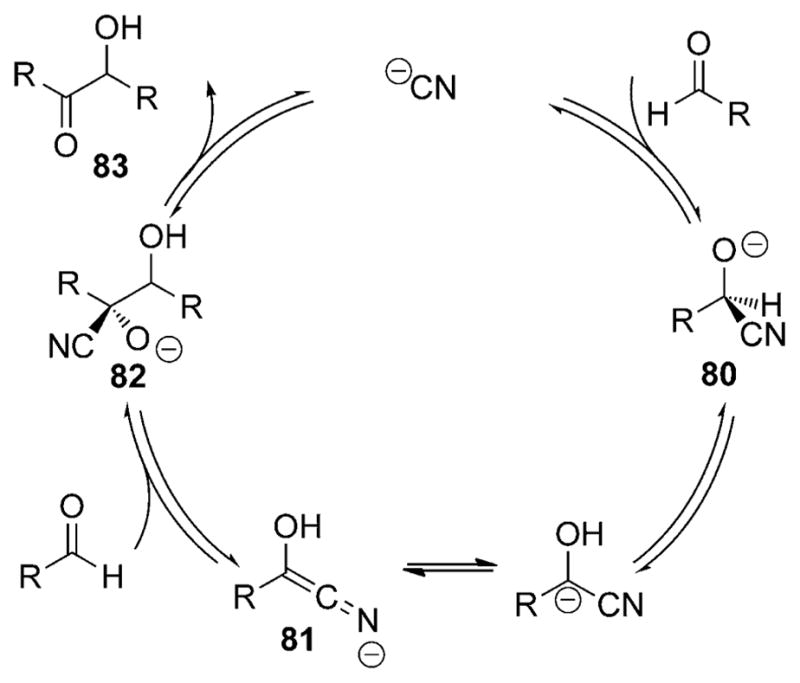
Cyanide-catalyzed benzoin reaction.
Overall, the cyanide-catalyzed coupling of aldehydes results in an even disposition of heteroatoms in the product (e.g. 1,2) and departs from the more traditional acid/base-mediated reactions of carbonyl compounds which yield only odd relationships between heteroatoms (e.g. 1,3). This fundamental switch in reaction polarity has inspired and challenged chemists to develop novel methods which allow for polarity inversion (or umpolung),[47] for example by the use of N-heterocyclic carbenes.[48] The development and application of these methods lie outside the scope of this review. However, methods for achieving polarity inversion which focus specifically on accessing ketene imine intermediates will be briefly discussed below.
5.2. Metalated Ketene Imines as Acyl Anion Equivalents
The key intermediate for accomplishing polarity inversion in the benzoin process is the ketene imine 81 (Scheme 25), which acts as an acyl anion equivalent.[47a] A number of strategies based on the generation of related ketene imine intermediates from the stoichiometric metalation of protected cyanohydrins,[49] chirally-modified cyanohydrins,[50] and chiral α-amino nitriles[51] have all been reported. Although these methods provide reliable strategies for accomplishing polarity inversion in organic reactions, they are limited by the use of reactive metalated ketene imines as well as chiral auxiliaries for achieving asymmetric induction.
An imaginative method for catalytically generating an O-silyl-protected version of metallo ketene imine intermediate 81 employs a cyanide-promoted Brook rearrangement[52] of an acyl silane, and was reported by Johnson and co-workers in 2004 (Scheme 26).[53] A successful catalytic, enantioselective acylation of the in situ generated ketene imine is achieved by employing the chiral (salen)aluminum alkoxide precatalyst 84 and benzyl cyanoformate. Interestingly, the cyanoformate plays two roles in the catalytic cycle, thus providing both the source of cyanide anion as well as an electrophilic trap for ketene imine intermediate 85. The resulting acylated cyanohydrin products are obtained in good yields, but the enantio-selectivity of the reaction reaches a maximum at 91:9 e.r.
Scheme 26.
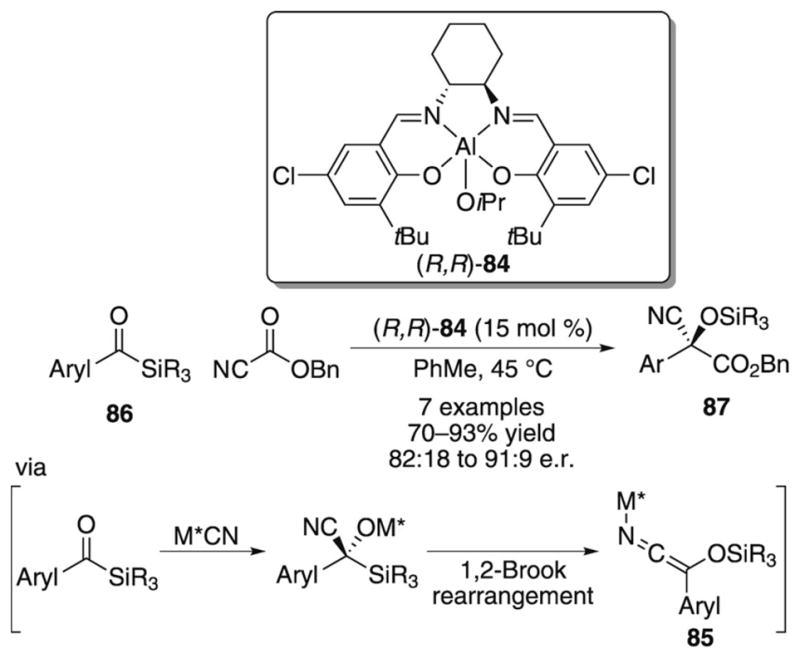
In situ generation of metallo ketene imine 85 and subsequent enantioselective acylation.
5.3. N-Silyl Oxyketene Imines as Acyl Anion Equivalent
All of the previous methods for achieving polarity inversion with ketene imines have focused on the use of anionic intermediates. With the exception of the work by Johnson and co-workers, this focus has greatly limited the application of these methods in catalytic, enantioselective processes because of the requirement of stoichiometrically generating reactive metalloid ketene imines. Surprisingly, very few attempts have been made at taming the reactivity of these anionic intermediates through silylation and isolation of an N-silyl ketene imine derivative. Obtaining a stable and isolable form of intermediate 81 (Scheme 25) could provide a convenient method for performing cross-benzoin reactions by sequential reaction with another aldehyde, a current challenge in acyl anion chemistry.[54]
5.3.1. Synthesis of N-Silyl Oxyketene Imines from Protected Cyano-hydrins
Only a few attempts at isolating an N-silyl derivative of a cyanohydrin anion has been disclosed. Cunico and Kuan studied the metalation of trimethylsilyl-protected cyanohydrins and the subsequent silylation of the resulting metalloid ketene imines with TMSCl (Scheme 27).[55] The ratio of C- to N-silylation products is highly responsive to the size of the alkyl substituent (R) in the starting material. Cyanohydrins containing smaller alkyl groups such as methyl, favor silylation at the carbon atom, whereas more sterically demanding isopropyl-substituted cyanohydrins react at the nitrogen atom. Intermediate results are obtained with cyanohydrins containing a 1-hexyl group, but interestingly the ratio shifts in favor of the C-silyl isomer upon standing for five days at room temperature. These results suggest that the C-silyl nitrile is the thermodynamically more stable product and that the initial product distribution is at least partially under kinetic control. The authors did not report any subsequent reactions of the N-silyl oxyketene imines.
Scheme 27.
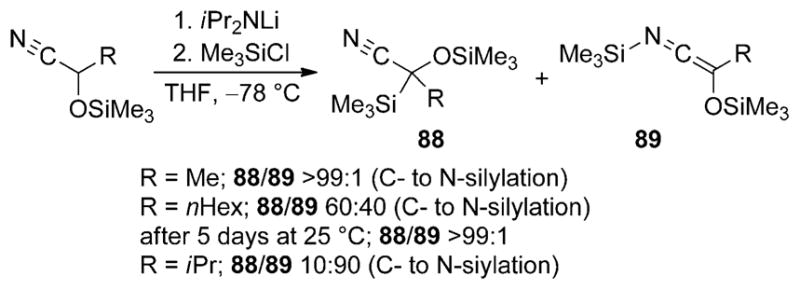
N versus C silylation of protected cyanohydrin anions.
To address the issue of N versus C silylation of protected cyanohydrin anions, efforts in our laboratories concentrated on the use of sterically demanding protecting groups and silylating agents in the reaction.[56] A variety of tert-butyl-protected cyanohydrins (90) were deprotonated with potassium hexamethyldisilazane (KHMDS) and the resulting potassio ketene imines were treated with TIPSCl to provide the N-silyl oxyketene imines 91 in high yield and with excellent selectivity for N silylation (Scheme 28). The products are obtained as yellow liquids, can be handled in air, and are stable for months when stored at 4°C. In some cases, extensive heating of the SKIs resulted in isomerization to the more stable C-silyl isomer, but the high purity observed in the crude reaction mixture obviates the need for distillation.
Scheme 28.
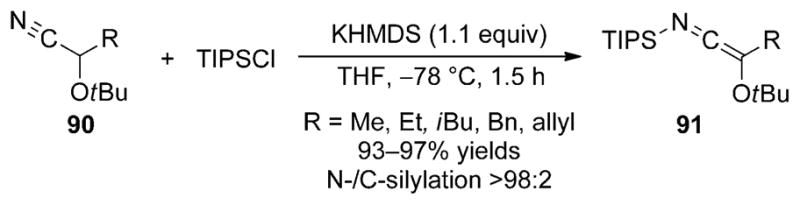
Synthesis of N-silyl oxyketene imines from tert-butyl-protected cyanohydrins.
5.3.2. Lewis Base Catalyzed Additions of N-Silyl Oxyketene Imines
N-Silyl oxyketene imines undergo highly enantioselective Lewis base catalyzed addition to aromatic aldehydes.[56] For these experiments, 2.5 mol% of the Lewis base catalyst (R,R)-39, 1.1 equivalents of SiCl4, and the aromatic aldehyde are combined prior to addition of the nucleophile to the reaction medium (Scheme 29). The direct aldol product formed in the catalytic cycle is the trichlorosilyl ether 92. The in situ generated intermediate is a result of the unique nature of this system in which the inexpensive and weak Lewis acid, SiCl4, remains covalently bound to the aldolate product while the chiral Lewis base turns over to close the catalytic cycle.[31c] Quenching the reactions under basic conditions using aqueous KF/NaHCO3 leads to the isolation of glycolate-aldol-type products containing the tertiary alcohol 92 in high yield and excellent enantioselectivity (Scheme 29). Importantly, the basic workup preserves the stereochemical integrity of the cyanocarbinol in 93, which can be formally regarded as a ketone cyanohydrin.
Scheme 29.

Catalytic, enantioselective carbonyl additions of N-silyl oxyketene imines to aldehydes leading to either aldol products or cross-benzoin adducts depending on type of reaction quench.
Alternatively, the ability of N-silyl oxyketene imines to serve as acyl anion equivalents is realized by employing an acidic quench. The trichlorosilyl group present in intermediate 92 is utilized for in situ removal of the tert-butyl ether by release of HCl from methanolysis of the labile Si–Cl bonds. Subsequent basic workup of the reaction with aqueous KF/NaHCO3 effects retro-cyanation and cross-benzoin adducts 94 are obtained directly in good yield and excellent enantio-selectivity (Scheme 29). The high level of correspondence between the enantiomeric composition of the glycolate aldol products and the corresponding cross-benzoin adducts confirms that the α-hydroxy stereogenic center is not subject to epimerization during the basic workup. Only in cases with extremely electron-deficient substrates are minor losses in the enantiomeric composition of the cross-benzoin product observed.
The versatility of the products resulting from addition of N-silyl oxyketene imines to aldehydes is illustrated by a number of manipulations of the cyano group (Scheme 30). The 4-methoxybenzyl-protected nitrile 95 (prepared on a gram scale in 95:5 d.r.) is subjected to known reaction conditions for achieving nitrile functional group conversions. The reactions allow access to useful building blocks, such as amino alcohols and α-hydroxy carbonyl compounds containing vicinally substituted tertiary and secondary stereogenic alcohols (96–99). The products are isolated in good to excellent yields and without loss of diastereomeric purity from the starting nitrile. Interestingly, in the case of methyl lithium addition, the steric hindrance imposed by the tertiary alcohol allows isolation of the direct addition product (e.g. imine 98) in high yield.[57]
Scheme 30.
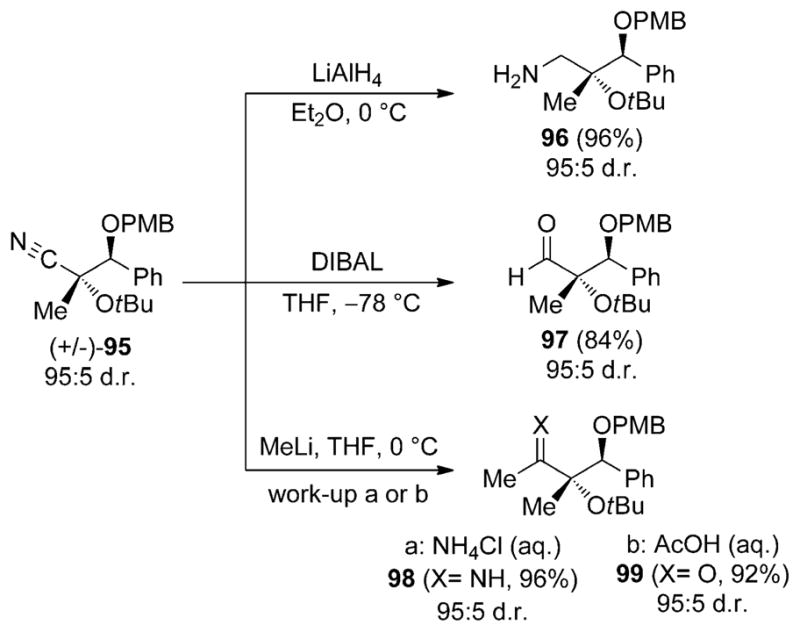
Representative transformations of protected nitrile product 95. DIBAL =diisobutylaluminium hydride, PMB = para-methoxybenzyl.
6. Summary and Outlook
Since the first reports of their synthesis nearly 60 years ago, silyl ketene imines have remained largely overlooked as nucleophilic reagents for organic transformations. Only in the past few years have researchers rediscovered these compounds and the advantages that they offer in catalytic, enantioselective transformations. The defining structural feature of SKIs is the pair of orthogonal substituent planes. This geometry allows SKIs to overcome the reactivity and selectivity issues which are often encountered in the preparation of quaternary stereogenic carbon centers. Capitalizing on the unique structure of SKIs has allowed the development of enantioselective methods for the synthesis of quaternary stereogenic carbon centers by employing classic reactions such as aldol additions, Mannich reactions, and acylations in unprecedented yields and stereoselectivities. In many cases, SKIs exhibit increased reactivity over analogously substituted SKAs and can be employed in catalytic systems previously optimized for SKAs, thus allowing facile access to quaternary stereogenic carbon centers.
Fundamentally distinct reactivity patterns are observed with SKIs derived from the selective N silylation of protected cyanohydrin anions. The resulting N-silyl oxyketene imines participate in carbonyl addition reactions, thereby yielding protected β-hydroxy cyanohydrins containing a tertiary alcohol in high yield and excellent enantioselectivity. The versatility of these products is demonstrated by the unique ability of the cyano group to either act as a leaving group, through deprotection/retrocyanation, or be functionalized through reduction or organometallic addition. The former permits access to cross-benzoin products derived from aliphatic aldehydes, in excellent enantioselectivity, and showcases the ability of N-silyl oxyketene imines to serve as acyl anion equivalents. Future variations on this umpolung-type reactivity of SKIs should focus on addressing limitations in Stetter reactions[58] and homoenolate additions.[59]
Clearly, the unique combination of structure and bonding present in SKIs offers distinct advantages over related ester and amide enolates and has allowed the preparation of challenging bond constructions, such as quaternary stereogenic centers, tertiary alcohols, and cross-benzoin adducts. Despite these advantages, SKI chemistry remains in its infancy, especially when placed in the broader context of the transformations developed for enoxysilanes. Future investigations should focus on developing metal-catalyzed processes for achieving α functionalizations of SKIs. Areas of research that may prove particularly fertile are organotransition metal catalyzed, enantioselective α arylations[60] and allylations[61] of silylated nucleophiles.
Biographies

Scott E. Denmark obtained an S.B. degree from MIT in 1975 and his D.Sc.Tech. (under the direction of Albert Eschenmoser) from the ETH Zürich in 1980. That same year he began his career at the University of Illinois and since 1991 he has been the Reynold C. Fuson Professor of Chemistry. His research interests include the invention of new synthetic reactions, exploratory organoelement chemistry and the origin of stereocontrol in fundamental carbon–carbon bond-forming processes.

Tyler W. Wilson obtained a BS in chemistry from Boise State University in 2004. He conducted graduate studies in the laboratories of Prof. Scott E. Denmark at the University of Illinois at Urbana-Champaign, earning a PhD in 2011. His doctoral research focused on the applications of silyl ketene imines in Lewis base catalyzed carbonyl additions. Currently, he is conducting post-doctoral studies with Prof. John F. Hartwig at the University of California, Berkeley.
References
- 1.a) Patai S. The Chemistry of Ketenes, Allenes and Related Compounds. Wiley; New York: 1980. [Google Scholar]; b) Schuster HF, Coppola GM. Allenes in Organic Synthesis. Wiley; New York: 1984. [Google Scholar]; c) Tidwell TT. Ketenes. Wiley; New York: 1995. [Google Scholar]
- 2.a) Brownbridge P. Synthesis. 1983:1–28. [Google Scholar]; b) Brownbridge P. Synthesis. 1983:85–104. [Google Scholar]; c) Kita Y, Tamura O, Tamura Y. J Synth Org Chem Jpn. 1986;44:1118–1133. [Google Scholar]; d) Kobayashi S, Manabe K, Ishitani H, Matsuo J-I. In: Science of Synthesis Organometallics: Silicon Compounds. Fleming I, Ley SV, editors. Vol. 4. Georg Thieme; Stuttgart: 2004. pp. 316–369. [Google Scholar]
- 3.a) Narasaka K, Soai K, Mukaiyam T. Chem Lett. 1974:1223–1224. [Google Scholar]; b) Oare DA, Heathcock CH. Top Stereochem. 1991;20:87–170. [Google Scholar]; c) Oare DA, Heathcock CH. Top Stereochem. 1989;19:227–407. [Google Scholar]
- 4.a) Mukaiyama T. Org React. 1982;28:203–331. [Google Scholar]; b) Mukaiyama T, Banno K, Narasaka K. J Am Chem Soc. 1974;96:7503–7509. [Google Scholar]; c) Mukaiyama T, Narasaka K, Banno K. Chem Lett. 1973:1011–1014. [Google Scholar]; d) Mahrwald R. Modern Aldol Reactions. Wiley-VCH; Weinheim: 2004. [Google Scholar]
- 5.a) Denissova I, Barriault L. Tetrahedron. 2003;59:10105–10146. [Google Scholar]; b) Douglas CJ, Overman LE. Proc Natl Acad Sci USA. 2004;101:5363–5367. doi: 10.1073/pnas.0307113101. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Trost BM, Jiang C. Synthesis. 2006:369–396. [Google Scholar]; d) Das JP, Marek I. Chem Commun. 2011;47:4593–4623. doi: 10.1039/c0cc05222a. [DOI] [PubMed] [Google Scholar]; e) Christoffers J, Baro A. Quaternary Stereocenters: Challenges and Solutions for Organic Synthesis. Wiley-VCH; Weinheim: 2005. [Google Scholar]
- 6.a) Manthorpe JM, Gleason JL. J Am Chem Soc. 2001;123:2091–2092. doi: 10.1021/ja0058280. [DOI] [PubMed] [Google Scholar]; b) Das JP, Chechik H, Marek I. Nat Chem. 2009;1:128–132. doi: 10.1038/nchem.131. [DOI] [PubMed] [Google Scholar]; c) Mekelburger HB, Wilcox CS. In: Comprehensive Organic Synthesis. Trost BM, editor. Vol. 2. Permagon; New York: 1991. p. 99. [Google Scholar]
- 7.For reviews on related alkyl and aryl ketene imines and ketene iminium salts, see: Snider BB. Chem Rev. 1988;88:793–811.Alajarin M, Vidal A, Tovar F. In: Targets in Heterocyclic Systems. Attanasi OA, Spinelli D, editors. Vol. 4. Royal Society of Chemistry; Cambridge: 2000. pp. 293–326.Madelaine C, Valerio V, Maulide N. Chem Asian J. 2011;6:2224–2239. doi: 10.1002/asia.201100108.Lu P, Wang Y. Chem Soc Rev. 2012;41:5687–5705. doi: 10.1039/c2cs35159e.
- 8.Arseniyadis S, Kyler KS, Watt DS. Org React. 1984;31:3–344. [Google Scholar]
- 9.Newman MS, Fukunaga T, Miwa T. J Am Chem Soc. 1960;82:873–875. [Google Scholar]
- 10.Prober M. J Am Chem Soc. 1956;78:2274–2277. [Google Scholar]
- 11.a) Stevens CL, French JC. J Am Chem Soc. 1953;75:657–660. [Google Scholar]; b) Stevens CL, French JC. J Am Chem Soc. 1954;76:4398–4402. [Google Scholar]
- 12.Krüger CR, Rochow EG. Angew Chem. 1963;75:793. [Google Scholar]
- 13.West R, Gornowicz GA. J Am Chem Soc. 1971;93:1714–1720. [Google Scholar]
- 14.Watt DS. Synth Commun. 1974;4:127–131. [Google Scholar]
- 15.Watt DS. J Org Chem. 1974;39:2799–2800. [Google Scholar]
- 16.Llonch JP, Frainnet E. C R Seances Acad Sci Ser C. 1973;276:1803. [Google Scholar]
- 17.Data in Scheme 6 is taken from the supporting information, see: Mermerian AH, Fu GC. Angew Chem. 2005;117:971–974. doi: 10.1002/anie.200461886.Angew Chem Int Ed. 2005;44:949–952. doi: 10.1002/anie.200461886.
- 18.a) Jochims JC, Lambrecht J, Burkert U, Zsolnai L, Huttner G. Tetrahedron. 1984;40:893–903. [Google Scholar]; b) Lambrecht J, Gambke B, Vonseyerl J, Huttner G, Nell GK, Herzberger S, Jochims JC. Chem Ber. 1981;114:3751–3771. [Google Scholar]
- 19.a) Knorr R, Ruhdorfer J, Mehlstaubl J, Bohrer P, Stephenson DS. Chem Ber. 1993;126:747–754. [Google Scholar]; b) Finnerty J, Mitschke U, Wentrup C. J Org Chem. 2002;67:1084–1092. doi: 10.1021/jo010398x. [DOI] [PubMed] [Google Scholar]
- 20.Cazeau P, Llonch JP, Simonin-Dabescat F, Frainnet E. J Organomet Chem. 1976;105:145–156. [Google Scholar]
- 21.Cazeau P, Llonch JP, Simonin-Dabescat F, Frainnet E. J Organomet Chem. 1976;105:157–160. [Google Scholar]
- 22.Meier S, Würthwein EU. Chem Ber. 1990;123:2339–2347. [Google Scholar]
- 23.Okada H, Matsuda I, Izumi Y. Chem Lett. 1983:97–100. [Google Scholar]
- 24.Under Lewis acidic conditions anti-elimination is strongly favored in the Peterson olefination, see: Hudrlik PF, Peterson D. J Am Chem Soc. 1975;97:1464–1468.Hudrlik PF, Peterson D, Rona RJ. J Org Chem. 1975;40:2263–2264.Ager DJ. Synthesis. 1984:384–398.
- 25.Matsuda I, Okada H, Izumi Y. Bull Chem Soc Jpn. 1983;56:528–532. [Google Scholar]
- 26.Freerksen RW, Selikson SJ, Wroble RR, Kyler KS, Watt DS. J Org Chem. 1983;48:4087–4096. [Google Scholar]
- 27.Differding E, Vandevelde O, Roekens B, Van TT, Ghosez L. Tetrahedron Lett. 1987;28:397–400. [Google Scholar]
- 28.a) Mermerian AH, Fu GC. J Am Chem Soc. 2003;125:4050–4051. doi: 10.1021/ja028554k. [DOI] [PubMed] [Google Scholar]; b) Mermerian AH, Fu GC. J Am Chem Soc. 2005;127:5604–5607. doi: 10.1021/ja043832w. [DOI] [PubMed] [Google Scholar]
- 29.Carreira EM, Fettes A, Marti C. Org React. 2006;67:1–216. [Google Scholar]
- 30.a) Denmark SE, Wilson TW, Burk MT, Heemstra JR., Jr J Am Chem Soc. 2007;129:14864–14865. doi: 10.1021/ja077134y. [DOI] [PubMed] [Google Scholar]; b) Wilson TW. PhD Thesis. University of Illinios; Urbana-Champaign: 2011. Synthesis, Study and Application of Silyl Ketene Imines in Lewis Base Catalyzed Carbonyl Addition Reactions. [Google Scholar]
- 31.Denmark SE, Wynn T, Beutner GL. J Am Chem Soc. 2002;124:13405–13407. doi: 10.1021/ja0282947.Denmark SE, Heemstra JR., Jr Org Lett. 2003;5:2303–2306. doi: 10.1021/ol034641l.Denmark SE, Beutner GL, Wynn T, Eastgate MD. J Am Chem Soc. 2005;127:3774–3789. doi: 10.1021/ja047339w.For a recent review on Lewis base catalysis, see: Denmark SE, Beutner GL. Angew Chem. 2008;120:1584–1663.Angew Chem Int Ed. 2008;47:1560–1638. doi: 10.1002/anie.200604943.
- 32.a) Kobayashi S, Ishitani H. Chem Rev. 1999;99:1069–1094. doi: 10.1021/cr980414z. [DOI] [PubMed] [Google Scholar]; b) Liu M, Sibi MP. Tetrahedron. 2002;58:7991–8035. [Google Scholar]; c) Verkade JMM, van Hemert LJC, Quaedflieg P, Rutjes F. Chem Soc Rev. 2008;37:29–41. doi: 10.1039/b713885g. [DOI] [PubMed] [Google Scholar]; d) Enders D, Reinhold U. Tetrahedron: Asymmetry. 1997;8:1895–1946. [Google Scholar]
- 33.Appel R, Mayr H. J Am Chem Soc. 2011;133:8240–8251. doi: 10.1021/ja200820m. [DOI] [PubMed] [Google Scholar]
- 34.a) Petrini M, Torregiani E. Synthesis. 2007:159–186. [Google Scholar]; b) Ellman JA, Owens TD, Tang TP. Acc Chem Res. 2002;35:984–995. doi: 10.1021/ar020066u. [DOI] [PubMed] [Google Scholar]
- 35.Tiong EA, Gleason JL. Org Lett. 2009;11:1725–1728. doi: 10.1021/ol802643k. [DOI] [PubMed] [Google Scholar]
- 36.Yin L, Kanai M, Shibasaki M. J Am Chem Soc. 2009;131:9610–9611. doi: 10.1021/ja9036675. [DOI] [PubMed] [Google Scholar]
- 37.Notte GT, Vu JMB, Leighton JL. Org Lett. 2011;13:816–818. doi: 10.1021/ol103096u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vu JMB, Leighton JL. Org Lett. 2011;13:4056–4059. doi: 10.1021/ol201566u. [DOI] [PubMed] [Google Scholar]
- 39.Roveda JG, Clavette C, Hunt AD, Gorelsky SI, Whipp CJ, Beauchemin AM. J Am Chem Soc. 2009;131:8740–8741. doi: 10.1021/ja902558j. [DOI] [PubMed] [Google Scholar]
- 40.Takenaka N, Abell JP, Yamamoto H. J Am Chem Soc. 2007;129:742–743. doi: 10.1021/ja0668320. [DOI] [PubMed] [Google Scholar]
- 41.Denmark SE, Wilson TW. Synlett. 2010:1723–1728. [Google Scholar]
- 42.a) Fuson RC. Chem Rev. 1935;16:1–27. [Google Scholar]; b) Krishnamurthy S. J Chem Educ. 1982;59:543–547. [Google Scholar]; c) Denmark SE, Heemstra JR, Beutner GL. Angew Chem. 2005;117:4760–4777. doi: 10.1002/anie.200462338. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2005;44:4682–4698. doi: 10.1002/anie.200462338. [DOI] [PubMed] [Google Scholar]; d) Casiraghi G, Battistini L, Curti C, Rassu G, Zanardi F. Chem Rev. 2011;111:3076–3154. doi: 10.1021/cr100304n. [DOI] [PubMed] [Google Scholar]
- 43.a) Yazaki R, Kumagai N, Shibasaki M. J Am Chem Soc. 2010;132:5522–5531. doi: 10.1021/ja101687p. [DOI] [PubMed] [Google Scholar]; b) Verdegem PJE, Monnee MCF, Lugtenburg J. J Org Chem. 2001;66:1269–1282. doi: 10.1021/jo0009595. [DOI] [PubMed] [Google Scholar]
- 44.a) Yazaki R, Kumagai N, Shibasaki M. J Am Chem Soc. 2009;131:3195–3197. doi: 10.1021/ja900001u. [DOI] [PubMed] [Google Scholar]; b) Yazaki R, Nitabaru T, Kumagai N, Shibasaki M. J Am Chem Soc. 2008;130:14477–14779. doi: 10.1021/ja806572b. [DOI] [PubMed] [Google Scholar]
- 45.Denmark SE, Wilson TW. Angew Chem. 2012;124:3290–3293. [Google Scholar]; Angew Chem Int Ed. 2012;51:3236–3239. doi: 10.1002/anie.201108795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.a) Lapworth A. J Chem Soc. 1903;83:995–1005. [Google Scholar]; b) Wöhler F, Liebig J. Justus Liebigs Ann Chem. 1832;3:276. [Google Scholar]; c) Wöhler F, Liebig J. Ann Pharm. 1832;3:249. [Google Scholar]
- 47.a) Seebach D. Angew Chem. 1979;91:259–278. [Google Scholar]; Angew Chem Int Ed Engl. 1979;18:239–258. [Google Scholar]; b) Hase TA. Umpoled Synthons. Wiley-Interscience; New York: 1987. [Google Scholar]
- 48.a) Zeitler K. Angew Chem. 2005;117:7674–7678. [Google Scholar]; Angew Chem Int Ed. 2005;44:7506–7510. doi: 10.1002/anie.200502617. [DOI] [PubMed] [Google Scholar]; b) Marion N, Diez-Gonzalez S, Nolan IP. Angew Chem. 2007;119:3046–3058. doi: 10.1002/anie.200603380. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2007;46:2988–3000. doi: 10.1002/anie.200603380. [DOI] [PubMed] [Google Scholar]; c) Enders D, Niemeier O, Henseler A. Chem Rev. 2007;107:5606–5655. doi: 10.1021/cr068372z. [DOI] [PubMed] [Google Scholar]; d) Moore JL, Rovis T. In: Asymmetric Organocatalysis. List B, editor. Vol. 291. Springer; Berlin: 2010. pp. 77–144. [Google Scholar]
- 49.a) Stork G, Maldonado L. J Am Chem Soc. 1971;93:5286–5287. [Google Scholar]; b) Stork G, Maldonado L. J Am Chem Soc. 1974;96:5272–5274. [Google Scholar]; c) Deuchert K, Hertenst U, Hünig S. Synthesis. 1973:777–778. [Google Scholar]; d) Hünig S, Wehner G. Synthesis. 1975:391–392. [Google Scholar]; e) Hünig S, Wehner G. Synthesis. 1975:180–182. [Google Scholar]
- 50.Schrader T. Chem Eur J. 1997;3:1273–1282. [Google Scholar]
- 51.Enders D, Shilvock JP. Chem Soc Rev. 2000;29:359–373. [Google Scholar]
- 52.Brook AG. Acc Chem Res. 1974;7:77–84. [Google Scholar]
- 53.a) Nicewicz DA, Yates CM, Johnson JS. Angew Chem. 2004;116:2706–2709. doi: 10.1002/anie.200353354. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2004;43:2652–2655. doi: 10.1002/anie.200353354. [DOI] [PubMed] [Google Scholar]; b) Nicewicz DA, Yates CM, Johnson JS. J Org Chem. 2004;69:6548–6555. doi: 10.1021/jo049164e. [DOI] [PubMed] [Google Scholar]
- 54.Johnson JS. Curr Opin Drug Discovery Dev. 2007;10:691–703. [PubMed] [Google Scholar]
- 55.Cunico RF, Kuan CP. J Org Chem. 1992;57:1202–1205. [Google Scholar]
- 56.Denmark SE, Wilson TW. Nat Chem. 2010;2:937–943. doi: 10.1038/nchem.857. [DOI] [PubMed] [Google Scholar]
- 57.Manetto A, Georganakis D, Leondiadis L, Gimisis T, Mayer P, Carell T, Chatgilialoglu C. J Org Chem. 2007;72:3659–3666. doi: 10.1021/jo062518c. [DOI] [PubMed] [Google Scholar]
- 58.a) Stetter H, Kuhlmann H. Org React. 1991;40:407. [Google Scholar]; b) deAlaniz JR, Rovis T. Synlett. 2009:1189–1207. doi: 10.1055/s-0029-1216654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.a) Ahlbrecht H, Beyer U. Synthesis. 1999:365–390. [Google Scholar]; b) Nair V, Vellalath S, Babu BP. Chem Soc Rev. 2008;37:2691–2698. doi: 10.1039/b719083m. [DOI] [PubMed] [Google Scholar]; c) Nair V, Menon RS, Biju AT, Sinu CR, Paul RR, Jose A, Sreekumar V. Chem Soc Rev. 2011;40:5336–5346. doi: 10.1039/c1cs15139h. [DOI] [PubMed] [Google Scholar]
- 60.a) Johansson CCC, Colacot TJ. Angew Chem. 2010;122:686–718. [Google Scholar]; Angew Chem Int Ed. 2010;49:676–707. doi: 10.1002/anie.200903424. [DOI] [PubMed] [Google Scholar]; b) Bigot A, Williamson AE, Gaunt MJ. J Am Chem Soc. 2011;133:13778–13781. doi: 10.1021/ja206047h. [DOI] [PubMed] [Google Scholar]; c) Allen AE, MacMillan DWC. J Am Chem Soc. 2011;133:4260–4263. doi: 10.1021/ja2008906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.a) Trost BM, VanVranken DL. Chem Rev. 1996;96:395–422. doi: 10.1021/cr9409804. [DOI] [PubMed] [Google Scholar]; b) Trost BM. J Org Chem. 2004;69:5813–5837. doi: 10.1021/jo0491004. [DOI] [PubMed] [Google Scholar]; c) Mohr JT, Stoltz BM. Chem Asian J. 2007;2:1476–1491. doi: 10.1002/asia.200700183. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Hartwig JF, Stanley LM. Acc Chem Res. 2010;43:1461–1475. doi: 10.1021/ar100047x. [DOI] [PMC free article] [PubMed] [Google Scholar]