

Abstract
Despite the frequency of seizure disorders in the human population, the genetic and physiological basis for these defects has been difficult to resolve. Although many genetic contributions to seizure susceptibility have been identified, these involve disparate biological processes, many of which are not neural specific. The large number and heterogeneous nature of the genes involved makes it difficult to understand the complex factors underlying the etiology of seizure disorders. Examining the effect known genetic mutations have on seizure susceptibility is one approach that may prove fruitful. This approach may be helpful in both understanding how different physiological processes affect seizure susceptibility and identifying novel therapeutic treatments. We review here factors contributing to seizure susceptibility in Drosophila, a genetically tractable system that provides a model for human seizure disorders. Seizure-like neuronal activities and behaviors in the fruit fly are described, as well as a set of mutations that exhibit features resembling some human epilepsies and render the fly sensitive to seizures. Especially interesting are descriptions of a novel class of mutations that are second-site mutations that act as seizure suppressors. These mutations revert epilepsy phenotypes back to the wild-type range of seizure susceptibility. The genes responsible for seizure suppression are cloned with the goal of identifying targets for lead compounds that may be developed into new antiepileptic drugs.
I. Introduction
A. Studying Human Seizure Disorders with Drosophila
Using Drosophila to model human disease is an especially attractive idea. The goal is to leverage the sophisticated genetic, molecular genetic, and transgenic experimental capabilities available in the fly to uncover fundamental biological principles underlying the causes and cures of human pathology, in much the same way as was done previously for classical genetics, developmental biology, and neurobiology. Also compelling is the prospect that novel therapeutics might be identified by studying disease-causing genes followed by targeted drug development, or by establishing platforms for high-throughput drug screening. In this chapter, we discuss the progress made by us, and others, in the development of a Drosophila model for studying human epilepsy. The model is based mainly on a “forward genetics” approach for studying seizure disorders in the fly with electrophysiological and behavioral assays for evoking seizures, and identification of mutations that affect susceptibility to seizure: seizure-sensitive, seizure-resistant, and seizure-suppressor mutations.
The idea that fruit flies can have seizures and serve as a model for human epilepsy is somewhat surprising, yet there are important similarities at the cellular and subcellular levels between fly and mammalian nervous systems, particularly in excitable membrane components. Voltage-gated and ligand-gated signaling molecules such as Na+, K+, and Ca2+ channels, and acetylcholine, glutamate, and gamma-aminobutyric acid (GABA) transmitter receptors all have highly conserved homologues in the fruit fly. There are some features of the mammalian central nervous system that are thought to contribute to the severity of some epilepsies that are not present in the fly. For example, in flies the central nervous system is organized as a ganglionic structure with synaptic neuropilar regions, whereas the mammalian cortex is organized into synaptic layers. Nevertheless, in the fly, electrical shock of sufficient intensity delivered to the brain of adult Drosophila elicits neuronal spiking activity that is seizure-like in appearance (Kuebler and Tanouye, 2000; Kuebler et al., 2001; Lee and Wu, 2002; Pavlidis and Tanouye, 1995; Song and Tanouye, 2006), similar to other animals with complex nervous systems, including humans.
B. Human Idiopathic Epilepsies and Channelopathies
Human seizure disorders are a serious health concern because of the large number of people affected and the inadequacy of available treatments. Seizures are abnormal, synchronous, and rhythmic firing of neurons in the central nervous system. About 10% of the population will experience a seizure during their lifetime, and about 1% of people suffer from the persistent, spontaneous seizures that define epilepsy (Shneker and Fountain, 2003). A further difficulty is intractable epilepsy: about 15 million people worldwide cannot adequately control their seizures with any available medication. Thus, overall, seizure disorders represent a pervasive class of neurological disease with unsatisfactory treatment options.
Seizures can result from a variety of brain insults including head trauma, fever, illness, and electroconvulsive shock, but a main source of seizure susceptibility appears to be genetic predisposition. More than 70 genes have been linked to epilepsies from work done on inherited disorders in humans, mice, and flies (Noebels, 2003). These genes encode a variety of products ranging from ion channel proteins to tRNAs. The number of disparate genes involved in epilepto-genesis is further confounded by there frequently being no obvious functional relationships between mutation and seizure susceptibility, thereby complicating our ability to understand epilepsy on a mechanistic level (Jacobs et al., 2001). The clearest cases appear to be the epilepsies that are channelopathies, that is, due to mutations within ion channel genes (Catterall et al., 2008; Reid et al., 2009).
An example of channelopathies is epilepsies caused by mutation of the voltage-gated Na+ channel gene SCN1A (Meisler and Kearney, 2005; Mahoney et al., 2009; Mulley et al., 2005). In humans, such mutations are associated with three forms of epilepsy: GEFS+ (generalized epilepsy with febrile seizures plus), characterized by febrile seizures that persist beyond the age of 6 years; SMEI (severe myoclonic epilepsy in infancy), an intractable epilepsy frequently resulting in convulsive status epilepticus; and ICEGTC (intractable childhood epilepsy with generalized tonic–clonic), an atypical SMEI that does not cause myoclonic seizures. In flies, severe seizure-like behaviors and electrical abnormalities are caused by the parabss mutations that have been recently mapped to the Drosophila Na+ channel gene para; however, all other para mutations cause hypoexcitability and are generally seizure-resistant mutations that can act as seizure suppressors.
An interesting example of an epilepsy that is not a channelopathy is a familial myoclonus-ataxia epilepsy caused by mutation of the human PRICKLE1 gene (Bassuk et al., 2008; Tao et al., 2011). Mutant homologues of human PRICKLE1 in several model organisms, Drosophila, mouse, and zebrafish, also caused seizure sensitivity when mutated (Tao et al., 2011). The PRICKLE1 gene product is a LIM domain protein that modulates Wnt/Fz signaling. The fly pricklesple(pksple) mutant has been shown to be critical for planar polarity in imaginal discs, but had not been previously known to cause neurological phenotypes (Gubb et al., 1999).
II. Seizure Studies in Drosophila
A. Seizure-Like Electrical Activity in Adult Flies
Electrophysiology methods were developed in flies to evoke seizures and record subsequent seizure-like electrical activity (Kuebler and Tanouye, 2000; Kuebler et al., 2001; Lee and Wu, 2002; Pavlidis and Tanouye, 1995; Pavlidis et al., 1994). Especially valuable is the capability of quantifying seizure susceptibility that allows comparisons between wild-type and mutant flies, and with other genetic and chemical manipulations (Kuebler and Tanouye, 2000; Kuebler et al., 2001; Song and Tanouye, 2006). Seizure-like electrical activity is evoked by short wave trains of high-frequency electrical stimuli (HFS, 0.4 ms pulses at 200 Hz for 300 ms) delivered to the brain at sufficient intensity and recorded from thoracic flight muscles. This seizure-like activity reflects motoneuron action potentials on a one-to-one basis. Seizures are extensive, spreading to at least 14 different muscles, containing 60 muscle fibers innervated by 58 motoneurons. Seizures are evoked in an all-or-nothing manner: HFS stimuli below a threshold voltage never elicit seizures, whereas just above threshold, seizures are always elicited. Seizure threshold is similar for all individuals of a given genotype. Thus, each genotype possesses a characteristic seizure susceptibility that may be quantified by its threshold voltage. Normal seizure susceptibility, the wild-type range, is about 30–40 V HFS (Canton Special = 30.1, Oregon R = 39.3 V HFS; Kuebler et al., 2001).
B. Seizure Disorders in Flies: Seizure Sensitivity in the Bang-Sensitive Paralytic Mutant Class
An obvious attraction of working with Drosophila is to take advantage of valuable mutant collections that may be screened electrophysiologically for alterations in seizure susceptibility. All of the mutants in one behavioral class, called the bang-sensitive (BS) paralytic class, were consistently found to be sensitive to seizure (Kuebler et al., 2001; Pavlidis and Tanouye, 1995). The BS class contains 14 mutant alleles representing 12 genes and a variety of gene products (Table I). Three BS mutants have been used as experimental representatives of the class. (1) The slamdance (sda) mutant displays a weak seizure sensitivity that is fully penetrant, but is the easiest to suppress by drug or suppressor mutant. (2) The easily shocked (eas) mutant shows moderate seizure sensitivity. (3) The bang senseless1 allele of the paralyzed gene (parabss1) is a severe BS mutation: electrophysiologically and behaviorally the most sensitive to seizure with a prominent tonic–clonic-like phenotype. It is the most difficult BS mutant to suppress genetically and pharmacologically, and was presented to model human intractable epilepsy (Parker et al., 2011).
Table I.
Seizure-Sensitive Mutants and Their Gene Products.
| Seizure-Sensitive Mutant | Seizure Threshold (V HFS) | Gene Product |
|---|---|---|
| paralyzed (parabss1, parabss2) | 3.2, 3.7 | Na+ channel (Parker et al., 2011) |
| easily shocked (eas) | 3.4 | Ethanolamine kinase (Pavlidis et al., 1994) |
| slamdance (sda) | 6.7 | Aminopeptidase N (Zhang et al., 2002) |
| bang sensitive (bas1, bas2) | 7.6, 3.8 | Unknown |
| technical knockout (tko) | 9.9 | ribosomal protein S12 (Royden et al., 1987) |
| jitterbug ( jbug ) | 10.5 | Unknown |
| couch potato (cpo) | 11.1 | RNA-binding protein (Glasscock and Tanouye, 2005) |
| kazachoc (kcc) | 17.0 | K+ Cl− cotransporter (Hekmat-Scafe et al., 2006) |
| knockdown (kdn) | 20.2 | Citrate synthase (Fergestad et al., 2006) |
| prickle (pksple) | Unknown | LIM domain protein (Tao et al., 2011) |
| stress-sensitive (sesB) | Unknown | Adenine nucleotide translocase (Zhang et al., 1999) |
| rock-n-roll (rnr) | Unknown | Unknown |
Seizure threshold is voltage of high-frequency stimulation (V HFS). For comparison the seizure threshold is 30.1 V HFS for Canton-Special wild-type flies (Kuebler et al., 2001).
The parabss1 mutation (formerly named bss1) is presented as an example of the BS mutant class. Like most other BS mutants, parabss1 flies display unexceptional behaviors under normal conditions. Feeding, grooming mating behaviors, and phototaxis and geotaxis responses all appear normal. Overall activity levels are unaltered; flies are neither hyperactive nor sluggish. Abnormal behavior is induced in parabss1 following a mechanical shock, a tap of the culture vial or brief vortex mixing (a “bang”). The resulting behavioral phenotype is complex with six distinguishable phases (Fig. 1A).
Fig. 1. Drosophila parabss1mutant behavior.

(A) Cartoon depicting stereotypic behavioral phenotype of parabss1 flies subjected to a mechanical shock (10 s vortex = BANG): initial seizure-like behavior, followed by complete paralysis, then a tonic–clonic-like period that is unique to parabss1 and not evident in other BS mutant genotypes. In the figure, one clonus-like event is depicted, but the number can vary, as can the duration of the period. The tonic–clonic-like period is followed by a recovery seizure and the fly then recovers. Not depicted is a quiescent period of variable duration often observed between the recovery seizure and recovery, as well as the refractory period during which flies are resistant to further seizures that occurs immediately following recovery. (B) For parabss1/Y hemizygous males, recovery time from behavioral paralysis is substantially longer than for parabss1/+ heterozygous females or for another BS mutant, easPC80/Y.
An initial seizure is typical for BS mutants, lasting several seconds, and characterized by leg shaking, abdominal muscle contractions, wing flapping and scissoring, and proboscis extensions.
An initial paralytic period is also typical of BS mutants; parabss1 flies are immobile and unresponsive to mechanical stimulus, as described previously (Ganetzky and Wu, 1982).
Unlike other BS mutants, initial paralysis in parabss1 is followed by an extended period of tonic–clonic-like activity. During this period, the fly is mainly quiescent, resembling a tonic phase. The quiescence is broken up by multiple bouts of clonus-like activity.
Recovery seizure is also typical of all BS mutants including parabss1 flies; this seizure resembles the initial seizure and clonus-like activity.
A refractory period is observed in parabss1, as in all other BS mutants during which behaviorally normal flies cannot be induced to have further seizures.
Finally, there is complete recovery and parabss1 flies regain bang sensitivity.
The total paralytic recovery time for parabss1 (Fig. 1B) is longer than that for other BS mutants, mainly because of the tonic–clonic activity; mean recovery time for parabss1 is ~240 s compared with sda or eas, 38 and 81 s, respectively.
C. Seizure Sensitivity in parabss1 and Electrophysiology of Tonic–Clonic-Like Spontaneous Firing
The electrophysiology phenotype of parabss1 is generally similar to that seen in other BS mutants, even though more extreme in some aspects. As was the case for behavior, electrophysiology is normally unexceptional in parabss1 mutants as determined using the adult giant fiber (GF) system neural circuit as proxy (Tanouye and Wyman, 1980). Thus, for example, single-pulse stimulation of the GF produced evoked muscle potential responses in the dorsal longitudinal muscles that were normal in appearance, threshold (2.04 ± 0.48 V), and latency (2.07 ms). Also normal was the ability to follow high-frequency stimulation and respond to short interpulse twin pulse intervals.
Seizure-like electrical activity in parabss1 mutants is evoked by HFS stimuli at exceptionally low stimulation voltages (4.4 V HFS; Fig. 2), substantially lower than for wild type and slightly lower than for eas and sda BS mutant males (5.13 and 6.2 V HFS, respectively). Thus, using the criterion of HFS threshold, parabss1 mutants are about seven times more seizure-sensitive than Canton-Special wild-type flies (Kuebler and Tanouye, 2000; Lee and Wu, 2002; Parker et al., 2011). Electrophysiological features of evoked parabss1 seizures are complex (Fig. 3).
Fig. 2. The BS mutantsdais more susceptible to seizures than wild type, and therefore has a lower seizure threshold.
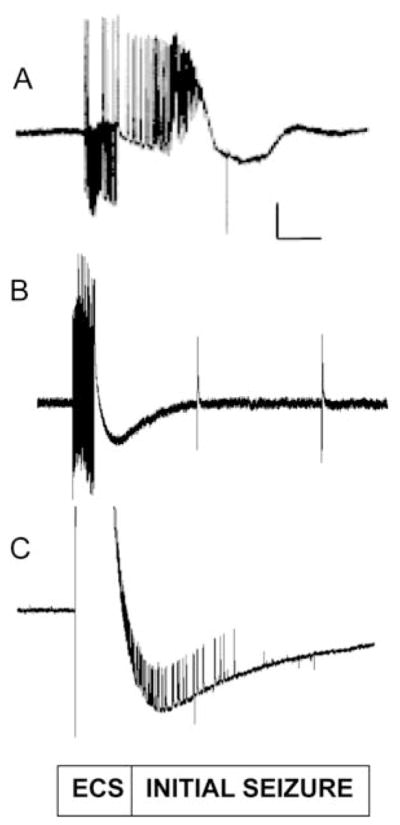
(A) seizure-like activity (initial seizure) is elicited in a parabss1 fly by a high-frequency stimulus of low strength (4 V) and displayed at a high sweep speed. (B) A low-voltage high-frequency (HF) stimulus of 8 V fails to elicit a seizure in a wild-type Canton-Special (CS) fly because the stimulus is below the seizure threshold. Following the HF stimulus artifact, there is no seizure activity observed in this recording displayed at a high sweep speed. Note also that there is no period of synaptic failure and single-pulse stimulation of the giant fiber (GF) (0.5 Hz) continues to evoke dorsal longitudinal muscle (DLM) potentials. Two such effective single-pulse stimuli are depicted in this trace; each was effective in evoking a DLM potential. (C) Seizure-like activity is elicited in a wild-type CS fly by a high-voltage HF stimulus (32 V), which is above the threshold for seizure. The seizure in this recording begins within the large stimulus artifact and is displayed at a high sweep speed. Vertical calibration bar is 20 mV. Horizontal calibration bar is 300 ms. Adapted from Kuebler et al., 2001.
Fig. 3. Complex seizure-like electrophysiology phenotypes in parabss1 mutants.

(A) Electrical recording of seizure-like neuronal activity and synaptic failure in a parabss1/Y animal subjected to a 4-V high-frequency electrical stimulus (HFS), as well as single-pulse stimuli to trigger the giant fiber circuit, allowing assessment of synaptic function (giant fiber response, GFR). Following an initial seizure (IS) is a period of synaptic failure within which GF stimulation fails to evoke a response (SF1–5), unlike that seen prior to the seizure. As depicted, during the period of synaptic failure, spontaneous secondary seizures are observed (SS1–4). Although in this trace four secondary seizures are observed, the number is variable. A final recovery seizure (RS) is observed, and shortly thereafter, GF system transmission is restored (response recovery, RR). (B) Initial seizure and secondary seizures at higher sweep speed. (C) Recovery seizure at higher sweep speed. Calibration bar is 20 mV, 10 s in (A), 20 mV, 1.5 s in (B) and (C).
There is an initial “seizure” following delivery of a suprathreshold HFS stimulus consisting of aberrant high-frequency firing (>100 Hz) lasting for ~1 s, present in the dorsal longitudinal muscle and all other muscle fibers and motoneurons examined (Fig. 3A, B). This parabss1 seizure-like phenotype is qualitatively similar to that seen for other BS genotypes and normal flies, differing only in the HFS voltage required for eliciting it (i.e., seizure threshold), but otherwise indistinguishable in subsequent electrical activity.
After the initial seizure, the next aspect of the electrophysiology phenotype for parabss1 mutants is the sudden failure of the GF neural circuit to drive muscle potentials in the dorsal longitudinal muscle. This is termed a “synaptic failure” period as it has been shown to be due to transmission failure at many central synapses (Fig. 3A; Pavlidis and Tanouye, 1995); it is likely the cause of behavioral paralysis in parabss1 mutants and for other BS genotypes. However, although the period of synaptic failure is short in most BS mutants, for example about 38 s in sda, for parabss1, it is longer and more complex, apparently reflecting the complexity observed behaviorally as tonic–clonic-like activity.
For the purpose of describing the electrophysiology of the parabss1 tonic–clonic-like period, it is convenient to consider three seizure types: (1) initial seizure, occurring immediately after HFS; (2) secondary seizures, occurring within the tonic–clonic-like interval; and (3) recovery seizure, occurring immediately prior to long-term recovery of the GF system neural circuit response (Fig. 3). Initial and secondary seizures resemble each other in waveform and duration; recovery seizures are somewhat different in waveform and longer in duration, although the significance of this is unclear (Fig. 3C). In young parabss1 flies (1 day post-eclosion), the tonic–clonic-like period is fairly short, about 45 s, and contains just one secondary seizure. In older parabss1 flies (7 days), the tonic–clonic-like period is longer, about 75 s and typically contains three to four secondary seizures, although up to eight have been observed. This is consistent with observations of longer behavioral recovery times in older parabss1 flies.
D. parabss1 is a Gain-of-Function Mutation in the Voltage-Gated Sodium Channel Gene
The mutants in the BS are all similar in their behavioral and electrophysiology phenotypes. This suggested that their respective gene products might converge upon a common cell biological function or signaling pathway. Surprisingly, the 12-gene products encoded by the BS genes are disparate and do not immediately suggest any common function that might be shared. For example, the eas gene encodes ethanolamine kinase and the mutation is a two 2-base pair deletion causing a frame shift and a truncated product. This results in a defect that eliminates ethanolamine kinase activity and alters the amount of the predominant membrane lipid phosphatidylethanolamine. The sda gene encodes the transmembrane ectoenzyme aminopeptidase N that catalyzes the removal of neutral and basic amino acids from the N-termini of a variety of small peptide substrates. The sda mutation is a 2-bp insertion in the 5′ UTR that appears to reduce expression; sda phenotypes may be phenocopied by RNAi.
The parabss1 mutation is an unusual gain-of-function mutation in the voltage-gated Na+ gene, described here in more detail. Although the mutation, formerly called bss1, had been identified more than 30 years ago (Jan and Jan, 1978), molecular identification proved difficult. Recently, mapping data placed bss1 in the para Na+ channel gene and the mutation was renamed as parabss1 (Parker et al., 2011). This was a surprising result as, heretofore, all para alleles had been found to be seizure-suppressor mutations (Kuebler et al., 2001; Song and Tanouye, 2007); and parabss1 mutants do not display any of the canonical para loss-of-function phenotypes, such as a loss of action potentials and behavioral paralysis at elevated (29°C) temperatures in parats1 or paraST76 (Siddiqi and Benzer, 1976; Suzuki et al., 1971; Wu and Ganetzky, 1980). Canonical para mutations are lethal in double mutant combinations with the Na+ channel regulator mlenapts (Ganetzky, 1984). In contrast, mlenapts acts as a seizure suppressor for parabss1 (Kuebler et al., 2001). Consistent with the gain-of-function nature of the parabss1 mutation, a para+ RNAi construct is also a strong suppressor of parabss1 phenotypes (Parker et al., 2011).
The gain-of-function nature of parabss1 results in unusual gene dosage relationships that may influence interpretations of gain-of-function NaV mutations in human epilepsies, vis-à-vis loss-of-function mutations. Generally, parabss1 is not phenotypically equivalent to a deficiency deleting the para gene, and extra copies of para+ do not continuously ameliorate the effects of mutation as their dosage increases. Thus, phenotypic severity for parabss1 is
These observations contrast with para loss-of-function mutations, such as paraST76, which behave more typically. Phenotypic severity for paraST76 is
The parabss1 mutation is due to a single amino acid substitution (leu→phe) of a conserved residue at position 1699 within the hydrophobic S3b membrane-spanning segment of homology domain IV in the Na+ channel protein (Parker et al., 2011). Na+ channel α-subunits, such as encoded by para, comprise four homologous domains (I–IV), each containing six transmembrane segments (S1–S6). The ion pore is formed centrally by the collective organization of S5–S6 segments from each domain. Surrounding the ion pore are the four voltage sensors comprising the S1–S4 segments from each domain. Crucial to the action of each voltage sensor is a modular unit termed a “paddle motif,” an S3b–S4 helix–turn–helix motif that is thought to move at the protein–lipid interface, driving activation of the voltage sensors and opening and closing of the pore (Alabi et al., 2007; Bosmans et al., 2008; Catterall et al., 2008). Current thinking on Na+ channel paddle domains suggests that the voltage sensor paddles of domains I–III drive channel activation, whereas the paddle of domain IV drives channel inactivation (Bosmans et al., 2008). Consistent with this interpretation, in heterologous Xenopus ooctye expression experiments utilizing para cDNA, the functional effect of the L1699F substitution was shown to shift the voltage of fast inactivation to more positive potentials with no effect on activation voltage (Parker et al., 2011).
III. Genetic Suppression of Seizure Susceptibility in Drosophila
A. Identification of Mutations that Suppress Seizures by Reverse Genetics
This section examines a novel class of genes called “seizure suppressors.” Suppressors are modifying mutations that yield individuals phenotypically more like the wild type, that is, the mutant phenotype is “suppressed.” In some cases, the gross wild-type phenotype is completely restored; in other cases, restoration is only partial. Thus, seizure suppressors are mutations that revert or partially revert the seizure-sensitive phenotype of BS mutants, but can be genetically separated, by recombination, from the mutation that they suppress. To our knowledge, the basic approach of utilizing second-site suppressor mutations had not been previously exploited for any neurological syndrome, leading us to initiate mutant searches with the most basic of questions. (i) Are there seizure-suppressor genes, and how might they lead to new therapeutics? (ii) Can we devise screens to identify seizure suppressors? (iii) What is the entire range of potential gene products that can act as seizure suppressors? (iv) Is this range limited to nervous system-specific gene products, such as signaling molecules, or does it include non-nervous system gene products as well? There are now answers to all of these questions, such that the approach is now poised to discover an antiepileptic drug (AEDs) with good seizure-suppressing capabilities and minimal anticipated side effects. Indeed, some promising candidates may have already been identified.
Existing Drosophila neurological mutant strains were searched for ones that are seizure-resistant, that is, those that display characteristically high thresholds to evoked seizures (Table II; Kuebler et al., 2001; Song and Tanouye, 2006). Several seizure-resistant mutants are partial loss-of-function mutations of ion channel genes. Thus, the most seizure resistant of the mutants identified is shakingB2 with an evoked seizure threshold that is three times higher (shakB2 seizure threshold = 94.7 V HFS). Another seizure-resistant mutant is paraST76, a loss-of-function allele of the para Na+ channel gene that causes behavioral paralysis and a loss of action potentials at restrictive temperatures (Loughney et al., 1989; Ramaswami and Tanouye, 1989; Siddiqi and Benzer, 1976; Suzuki et al., 1971; Wu and Ganetzky, 1980). The paraST76 mutant has a seizure threshold of 65.0 6 ± 7.2, about two times that of wild type (Kuebler et al., 2001). The no action potential allele of the maleless gene (mlenapts) is also a mutation that causes seizure resistance (mlenapts seizure threshold = 72.2 ± 7.3 V HFS) (Kuebler et al., 2001). The mlenapts allele is a gain-of-function mutation in an RNA helicase-like protein (Kernan et al., 1991; Reenan et al., 2000) that causes a reduction in voltage-gated Na+ channel expression, a loss of action potentials, and behavioral paralysis at elevated temperatures (Jackson et al., 1984; Kauvar, 1982; Wu and Ganetzky, 1980; Wu et al., 1978).
Table II.
List of Seizure-Suppressor Mutants and Their Gene Products.
| Seizure-Suppressor Mutant | Gene Product |
|---|---|
| Reverse genetics | |
| paralyzed (paraST76) | Na+ channel (Kuebler et al., 2001) |
| male lethal (mlenapts) | Na+ channel regulator (Kuebler et al., 2001) |
| shakingB (shakB2) | Gap junction channel (Kuebler et al., 2001; Song and Tanouye, 2007) |
| Shaker (ShKS133) | K channel (Kuebler et al., 2001) |
| Forward genetics | |
| paralyzed (paraJS1) | Na+ channel (Song and Tanouye, 2007) |
| shakingB (shakBJS) | Gap junction channel (Song and Tanouye, 2007) |
| escargot (esgEP684 + 4 alleles) | Zn2+-finger transcription factor (Hekmat-Scafe et al., 2005) |
| meiosis-P26 (mei-P26EG16, mei-P261) | Ring finger B-box coiled-coil-NHL protein (Glasscock et al., 2005) |
| topoisomerase I (top1JS + 3 alleles) | DNA topoisomerase type I (Song et al., 2007) |
| kazal-domain protein-1 (kdp1) | Kazal-type serine protease inhibitor (Hekmat-Scafe et al., 2005) |
| kazal-domain protein-2 (kdp2) | Kazal-type serine protease inhibitor (Hekmat-Scafe et al., 2005) |
| suppressor of eas7 (su(eas7)) | Unknown (Glasscock et al., 2005) |
| suppressor of eas13 (su(eas13)) | Unknown (Glasscock et al., 2005) |
This table lists separately suppressors identified by reverse genetics and by forward genetics.
Because seizure-resistant mutants are much less susceptible to seizures than wild type, they were tested by reverse genetics to determine whether they are capable of suppressing seizures, that is if they functioned as seizure-suppressor mutations. Indeed, several were found to revert phenotypes of seizure-sensitive mutants in homozygous double mutant combinations, including paraST76, mlenapts, and shakB2 (Table III). Of these, shakB2 was chosen for a more detailed analysis as representative of the class (Song and Tanouye, 2006). The shakB gene encodes a gap junction channel, mutation in which perturbs electrical synaptic transmission (Crompton et al., 1995; Krishnan et al., 1993; Phelan et al., 1998; Thomas and Wyman, 1984). Gap junction communication is an important target for AEDs as drugs that reduce electrical transmission diminish seizures, and enhanced electrical transmission increases the frequency and severity of seizures (Carlen et al., 2000; Jahromi et al., 2002). Seizure suppression by shakB2 differs in double mutant combination with different BS mutations (Song and Tanouye, 2006; Table III). Some BS mutants show full suppression (Class III) or partial suppression (Class II) of seizure susceptibility; however, for the mutants with the most severe phenotypes, shakB2 has no effect (Class I). Loss of shakB function is responsible for this suppression by driving expression in shakB2; sda double mutants. The resulting flies regained seizure sensitivity (i.e., they lost their seizure-suppression). Thus, shakB2 may act to suppress seizures in less severe BS mutants by a mechanism similar to that proposed for drugs such as carbenoxolone that block gap junction activity (Szente et al., 2002). Observations presented here are generally consistent with such a mechanism; indeed feeding carbenoxolone to BS flies also ameliorated seizures (Song and Tanouye, 2006). These results are supportive of an electrical synaptic failure mechanism for shakB2 seizure suppression.
Table III.
The shakB2 Gap Junction Channel Mutation Acts as a Seizure Suppressor for Some Epilepsy Mutations.
| BS Mutation | Seizure Threshold (No Suppressor) (V HFS) | Seizure Threshold (+ shakB2 suppressor) (V HFS) | |
|---|---|---|---|
| Class I (no suppression) | parabss1 | 3.2 | 3.6 |
| parabss2 | 3.7 | 5.0 | |
| bas2 | 3.8 | 5.8 | |
| Class II (partial suppression) | eas | 3.4 | 15.3 |
| tko | 5.0 | 26.8 | |
| Class III (full suppression) | sda | 6.7 | 32.8 |
| jbug | 10.5 | 38.2 | |
| kdn | 20.2 | 41.6 |
Seizure susceptibility for some BS mutants are completely reverted to wild-type levels (Class III). Some mutations (Class II) show partial suppression. Some mutations show no change in seizure threshold (Class I). All values are in volts HFS (n ≥ 10 for each genotype).
B. Identification of Seizure-Suppressor Mutations by Forward Genetic Screens: Suppression by Loss-of-Function Mutations in the para Na+ Channel Gene
Forward genetic screens for new seizure-suppressor mutations are performed by mutagenesis in a seizure-sensitive genetic background. For example, Song and Tanouye (2007) utilized mutagenesis by P-element transposons in an eas genetic background. Primary screening is behavioral, selecting for exceptional bang-resistant eas flies that indicate a reversion of the eas BS paralytic phenotype (Glasscock and Tanouye, 2005; Hekmat-Scafe et al., 2005; Song and Tanouye, 2007; Song et al., 2007). Suppressor mutations (second-site suppressors) are separated from eas genetically, then mapped, cloned, and characterized (Table II). A total of nine seizure-suppressor mutations have been identified, to date by forward genetics screens, including two that validated screening procedures: a new allele of the Na+ channel gene (paraJS1) and a new allele of the gap junction channel gene (shakBJS). Several novel seizure-suppressor mutants were also identified including DNA topoisomerase I (top1JS), a Zn2+-finger transcription factor (esg), and a ring finger B-box coiled-coil-NHL protein (mei-P26EG16) (Glasscock and Tanouye, 2005; Hekmat-Scafe et al., 2005; Song and Tanouye, 2007; Song et al., 2007).
Identification of new para and shakB alleles as seizure suppressors (Song et al., 2007) is taken as validation for screening methodology, since we had previously shown by reverse genetics that extant para and mlenapts Na+ channel mutations and the shakB2 gap junction mutation are potent seizure suppressors (Kuebler et al., 2001). For paraJS1, the P-element insertion is in the 3′ UTR of para, resulting in a 45% reduction in transcription, which apparently accounts for seizure suppression. The paraJS1 mutation is a highly effective seizure suppressor; we found the behavioral phenotype of eas is suppressed by 87%, whereas the seizure threshold of eas is increased from 3.4 ± 0.5 to 26.4 ± 3.0 V, close to the wild-type threshold (Song and Tanouye, 2007). The paraJS1 mutation is a general seizure suppressor in that it suppresses sda and parabss1 mutant phenotypes, as well as eas. In a wild-type background, the paraJS1 mutation causes a weak seizure-resistant phenotype. The seizure threshold of paraJS1 flies is 51.2 ± 5.2 V HFS, about 1.5 times greater than that of wild-type flies. The paraJS1 mutation causes no other apparent phenotypes: electrophysiology, behavior, and morphology are all wild type; paraJS1 is not a temperature-sensitive paralytic mutant (Song and Tanouye, 2007). These results are consistent with our previous observations from reverse genetics that paraST76 and the mlenapts mutant that results in a dramatic reduction of para translation also function as a seizure suppressor (Kuebler et al., 2001).
C. A Mutation of DNA Topoisomerase I is a Novel and Unexpected Seizure Suppressor
A variety of different gene products have been found to contribute to seizure sensitivity in humans, rodents, and flies. This suggests that similarly, there may be a diversity of products contributing to seizure-suppression. Unexpected classes of seizure-suppressor genes are especially intriguing for study as they may yield novel insight into mechanisms underlying seizure disorder. They additionally have the potential to open the door to new ideas for therapeutics. Seizure suppressors may suggest novel targets that may lead to development of new classes of AEDs. Targets that do not otherwise compromise nervous system function may lead to drugs with minimal side effects. As example, the discovery and characterization of the DNA topoisomerase 1 allele top1JS is described in greater detail. The top1JS allele was identified in a P-element transposon screen for eas suppression (Song et al., 2007). The identification of top1JS as a seizure suppressor was surprising as DNA topoisomerases have not previously been associated with seizure, seizure control, or any other electrical excitability functions. Rather type I DNA topoisomerase is thought to resolve the torsional tension associated with DNA replication or gene transcription (Champoux, 2001).
Seizure suppression is caused by insertion of the P-element in the 5′ UTR of top1, 257 bp upstream of the translation start site resulting in a 12.5-fold reduction of transcription. The top1 gene is an essential gene of several known top1 alleles; top1JS is the only viable, all others are homozygous lethal (Lee et al., 1993; Zhang et al., 2000). The top1JS mutation is a general seizure suppressor ameliorating phenotypes of sda, eas, and bss (Song et al., 2007). As example, top1JS suppresses eas behavioral paralysis phenotypes in 63% of animals and seizure threshold is raised about 3.5-fold. One possible explanation for suppression is that top1JS causes increased neuronal apoptosis. Transcription in active neurons generates super-coiled DNA that must be continuously relaxed to sustain high levels of RNA synthesis. The binding of top1 to DNA ordinarily forms a cleavable complex leading to relaxation of the DNA supercoil (Champoux, 2001). Reduced levels of top1 activity in mutants may lead to damage of DNA and cell death. The top1JS mutant shows high levels of neuronal apoptosis and expression of the DIAP1 inhibitor of apoptosis, blocks top1JS suppression of eas seizures and this could, in part, account for seizure suppression.
IV. Chemical Suppression of Seizure Susceptibility in Drosophila
A. AEDs Reduce Seizure Sensitivity in BS Flies
Investigations of human AED effects on Drosophila BS mutant phenotypes provide several insights. They provide an additional link between the fly model and the human pathology: strengths and weaknesses of the model may be inferred from effectiveness and ineffectiveness of different AEDs. We may further infer that novel drugs developed because they are effective against Drosophila seizures may become effective treatments for human seizure disorders. The ability of known AEDs to suppress seizures in Drosophila would demonstrate the utility of this model to identify and evaluate novel anticonvulsant compounds. Finally, identifying specific AEDs that are able to suppress seizures in the BS mutants may shed light on the mechanisms causing the seizure-sensitive defect in these strains. In particular, as some AEDs show specificity to certain human epileptic syndromes, mutants may be identified from the BS mutant collection that might most closely resemble those based on drug profile. Reynolds et al. (2003) showed that phenytoin and gabapentin are effective at suppressing BS mutant behavioral phenotypes; carbamazepine, ethosuximide, and vigabactrin were not. Additional studies have shown that valproate, potassium bromide, and carbenoxolone are effective at suppressing BS phenotypes (Kuebler and Tanouye, 2002; Song and Tanouye, 2006; Song et al., 2008; Tan et al., 2004). Taken together, these combined studies indicate that many, but not all, anticonvulsants used to treat human seizure disorders are effective against Drosophila seizures.
As an example, the treatment of BS mutants with valproate is described in more detail. Valproate is a wide-spectrum AED used to treat generalized and partial seizures. It is also used increasingly to treat nonepileptic neurological syndromes such as anxiety, schizophrenia, and bipolar disorder (Greenhill and Jones, 2010; Loscher, 2002; Landmark, 2008; White et al., 2007). The broad antiepileptic efficacy of valproate is thought to reflect a combination of neuro-chemical and neurophysiological mechanisms directed against multiple molecular targets (Loscher, 2002). Valproate has been found to inhibit voltage-gated Na+ channels and block repetitive firing of action potentials, to block T-type Ca2+ channels, and to elevate GABA levels and potentiate GABA responses (Landmark, 2007; White et al., 2007). Injection of valproate into the brain or the heart suppresses seizures in BS (Howlett and Tanouye, 2011; Kuebler and Tanouye, 2002). For example, a 25-mM injection in sda mutants increased seizure threshold by about a factor of seven from 9 to 62 V HFS (Howlett and Tanouye, 2011). In contrast, for parabss1 mutants valproate effect was more modest, increasing seizure threshold by about a factor of 2.5, from 5 to 13 V HFS. The effect on eas was intermediate, increasing seizure threshold to the wild-type range, from 5 to 33 V HFS. In contrast to injected-drug, valproate-feeding methods have been less effective in suppressing BS mutant seizures and show mixed results. For example, in pksple1 homozygous mutant flies, feeding of 10 mM valproate was lethal. However, valproate significantly suppressed seizure-like behaviors in pksple1 heterozygotes (Tao et al., 2011). In parabss1 and eas mutants, valproate feeding was also ineffective at ameliorating seizure phenotypes (Song et al., 2008). For example, parabss1 flies fed valproate (10 mM) for 3 days responded especially poorly. A number of flies (11%) died after bang stimulation. For those that survived, all showed paralysis and actually took significantly longer to recover from paralysis than sucrose-fed control flies (Song et al., 2008). These results on valproate are in contrast to feeding experiments with potassium bromide, which significantly ameliorated seizure phenotypes (Song et al., 2008; Tan et al., 2004).
B. AED Development From the top1JS Seizure Suppressor: Camptothecin and Other Top1 Inhibitors
A major attraction of Drosophila-based models is the idea that mutants may be used as a platform to facilitate the development of novel therapeutics, for example, AED development inspired by seizure-suppressor mutants (Song and Tanouye, 2008, 2009). The goal would to define new drug targets, beyond Na+ channels, Ca2+ channels, and GABA-related proteins that are the main targets of current AEDs. Top1 provides an interesting test of this notion as compounds have been identified that act as potent Top1 inhibitors, and these can be compared with seizure suppression by the top1JS mutation. The involvement of top1 in seizures is surprising because DNA topoisomerases have not previously been associated with seizures or any other electrical excitability neuronal functions. Top1 is an essential nuclear enzyme that acts to relieve the torsional stress that DNA encounters during activities such as replication, transcription, and chromatin condensation (Champoux, 2001). Top1 enzyme binds to DNA and causes a single-strand break. It remains bound in an enzyme–DNA cleavage complex, facilitating DNA unwinding, and then mediating re-ligation.
Top1 inhibitors are compounds such as camptothecin (CPT), a quinoline-based alkaloid, which is a phytochemical derived from the Chinese Happy Tree (Camptotheca acuminata). CPT interferes with enzyme function by covalently binding to the top1–DNA complex, thereby blocking re-ligation (Boege et al., 1996; Pommier et al., 1998, 1999). If this occurs during replication, it is thought that there is a cell cycle checkpoint failure leading to apoptosis. In the developing brain, we would expect that Top1 functions mainly during chromosome replication and for apoptosis to be evident. In the adult brain, we might expect less replication and for Top1 function to be most important in neurons with changing transcriptional needs. The mechanism of action for CPT inhibition of Top1 is completely different from that of the top1JS mutation, which causes a 12 times transcript reduction. Nevertheless, CPT has been found to ameliorate BS seizure phenotypes in drug-feeding experiments. For example, parabss1-fed CPT recover from paralysis about two-thirds faster than control flies, and tonic–clonic-like activity is almost completely suppressed (Song et al., 2007). Drug treatment also causes a modest, even though, significant increase in seizure threshold with a great decrease in synaptic failure time. Similar results are observed with the Top1 inhibitors kaempferol and apigenin. In addition, Top1 inhibitors are tolerated much better than valproate with considerably less toxicity (Song et al., 2007).
V. Concluding Remarks. Seizure Studies in Flies: Insights From Mutant Analyses, and Comparisons With Human Seizures
Several findings from seizure investigations in flies support the utility of these studies in modeling seizure disorders in humans. Other findings that are possible in flies because of advanced genetic capabilities cannot be replicated in humans, but can provide rich insight into the genetics of seizures and seizure susceptibility. Among these observations are
All individual flies have a seizure threshold indicating a characteristic seizure susceptibility.
Seizure susceptibility can be modulated by mutation and by genetic background in characteristic and predictable ways.
Some mutations that cause seizure sensitivity in flies such as parabss1 and pksple are similar to those responsible for some human epilepsies.
Seizure-like activity in flies spreads through the central nervous system along particularly pathways that depend on functional synaptic connections and recent electrical activity.
Seizure-like activity in flies can be spatially segregated into particular regions of the central nervous system.
Electroconvulsive shock treatment in flies raises the threshold for subsequent seizure-like activity.
Drosophila seizure phenotypes can be ameliorated by the human AEDs valproate, phenytoin, gabapentin, and potassium bromide (KBr).
Na+ channel mutations are excellent seizure suppressors, consistent with the notion that many AEDs are targeting Na channels.
Several mutations that cause seizure sensitivity or seizure suppression are surprising because their gene products had not previously been implicated in seizures or any other neurological function.
Acknowledgments
This study was supported by awards from the McKnight Foundation and the NIH (NS31231) to M.A.T. We thank the members of the Tanouye laboratory for helpful discussions throughout the project.
References
- Alabi AA, Bahamonde MI, Jung HJ, Kim JI, Swartz KJ. Portability of paddle motif function and pharmacology in voltage sensors. Nature. 2007;450:370–376. doi: 10.1038/nature06266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassuk AG, Wallace RH, Buhr A, Buller AR, Afawi Z, Shimojo M, Miyata S, Chen S, Gonzalez-Alegre P, Griesbach HL, et al. A homozygous mutation in human PRICKLE1 causes an autosomal-recessive progressive myoclonus epilepsy-ataxia syndrome. Am J Hum Genet. 2008;83:572–581. doi: 10.1016/j.ajhg.2008.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boege F, Straub T, Kehr A, Boesenberg C, Christiansen K, Anderson A, Jacob F, Kohrle J. Selected novel flavones inhibit the DNA binding or the DNA religation step of eukaryotic topoisomerase I. J Biol Chem. 1996;271:2262–2270. doi: 10.1074/jbc.271.4.2262. [DOI] [PubMed] [Google Scholar]
- Bosmans F, Martin-Eauclair MF, Swartz KJ. Deconstructing voltage sensor function and pharmacology in sodium channels. Nature. 2008;456:202–209. doi: 10.1038/nature07473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlen PL, Frances S, Zhang L, Naus C, Kushnir M, Velazquez JL. The role of gap junctions in seizures. Brain Res Rev. 2000;32:235–241. doi: 10.1016/s0165-0173(99)00084-3. [DOI] [PubMed] [Google Scholar]
- Catterall WA, Dib-Hajj S, Meisler MH, Pietrobon D. Inherited neuronal ion channelopathies: new windows on complex neurological diseases. J Neurosci. 2008;28:11768–11777. doi: 10.1523/JNEUROSCI.3901-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Champoux JJ. DNA topoisomerases: structure, function and mechanism. Annu Rev Biochem. 2001;70:369–413. doi: 10.1146/annurev.biochem.70.1.369. [DOI] [PubMed] [Google Scholar]
- Crompton D, Todman M, Wilkin M, Ji S, Davies J. Essential and neural transcripts from the Drosophila shaking-B locus are differentially expressed in the embryonic mesoderm and pupal nervous system. Dev Biol. 1995;170:142–158. doi: 10.1006/dbio.1995.1203. [DOI] [PubMed] [Google Scholar]
- Fergestad T, Bostwick B, Ganetzky B. Metabolic disruption in Drosophila bang-sensitive seizure mutants. Genetics. 2006;173:1357–1364. doi: 10.1534/genetics.106.057463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganetzky B. Genetic studies of membrane excitability in Drosophila: lethal interaction between two temperature-sensitive paralytic mutations. Genetics. 1984;108:897–911. doi: 10.1093/genetics/108.4.897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganetzky B, Wu CF. Indirect suppression involving behavioral mutants with altered nerve excitability in Drosophila melanogaster. Genetics. 1982;100:597–614. doi: 10.1093/genetics/100.4.597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glasscock E, Singhania A, Tanouye MA. The mei-p26 gene encodes an RBCC-NHL protein that regulates seizure susceptibility in Drosophila. Genetics. 2005;170:1677–1689. doi: 10.1534/genetics.105.043174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glasscock E, Tanouye MA. Drosophila couch potato mutants exhibit complex neurological abnormalities including epilepsy phenotypes. Genetics. 2005;169:2137–2149. doi: 10.1534/genetics.104.028357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenhill SD, Jones RSG. Diverse antiepileptic drugs increase the ratio of background synaptic inhibition to excitation and decrease neuronal excitability in neurons of the rat entorhinal cortex in vitro. Neuroscience. 2010;167:456–474. doi: 10.1016/j.neuroscience.2010.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gubb D, Green C, Huen D, Coulson D, Johnson G, Tree D, Collier S, Roote J. The balance between isoforms of the prickle LIM domain protein is critical for planar polarity in Drosophila imaginal discs. Genes Dev. 1999;13:2315–2327. doi: 10.1101/gad.13.17.2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hekmat-Scafe DS, Dang KN, Tanouye MA. Seizure suppression by gain-of-function escargot mutations. Genetics. 2005;169:1477–1493. doi: 10.1534/genetics.104.036558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hekmat-Scafe DS, Lundy MY, Ranga R, Tanouye MA. Mutations in the K+/Cl− cotransporter gene kazachoc (kcc) increase seizure susceptibility in Drosophila. J Neurosci. 2006;26:8943–8954. doi: 10.1523/JNEUROSCI.4998-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howlett IC, Tanouye MA. Seizure-sensitivity in Drosophila is ameliorated by dorsal vessel injection of the antiepileptic drug valproate. J Neurogenet. 2011 doi: 10.3109/01677063.2013.817574. (in press) [DOI] [PubMed] [Google Scholar]
- Jackson FR, Wilson SD, Strichartz GR, Hall LM. Two types of mutants affecting voltage-sensitive sodium channels in Drosophila melanogaster. Nature. 1984;308:189–191. doi: 10.1038/308189a0. [DOI] [PubMed] [Google Scholar]
- Jacobs MP, Fischbach GD, Davis MR, Dichter MA, Dingledine R, Lowenstein DH, Morrell MJ, Noebels JL, Rogawski MA, Spencer SS, Theodore WH. Future directions for epilepsy research. Neurology. 2001;57:1536–1542. doi: 10.1212/wnl.57.9.1536. [DOI] [PubMed] [Google Scholar]
- Jahromi SS, Wentlandt K, Piran S, Carlen P. Anticonvulsant actions of gap junctional blockers in an in vitro seizure model. J Neurophysiol. 2002;88:1893–1902. doi: 10.1152/jn.2002.88.4.1893. [DOI] [PubMed] [Google Scholar]
- Jan YN, Jan LY. Genetic dissection of short-term and long-term facilitation at the Drosophila neuromuscular junction. Proc Natl Acad Sci U S A. 1978;75:515–519. doi: 10.1073/pnas.75.1.515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauvar LM. Reduced [3H]-tetrodotoxin binding in the napts paralytic mutant of Drosophila. Mol Gen Genet. 1982;187:172–173. doi: 10.1007/BF00384402. [DOI] [PubMed] [Google Scholar]
- Kernan MJ, Kuroda MI, Kreber R, Baker BS, Ganetzky B. napts, a mutation affecting sodium channel activity in Drosophila, is an allele of mle, a regulator of X chromosome transcription. Cell. 1991;66:949–959. doi: 10.1016/0092-8674(91)90440-a. [DOI] [PubMed] [Google Scholar]
- Krishnan SN, Frei E, Swain G, Wyman RJ. Passover, a gene required for synaptic connectivity in the giant fiber system of Drosophila. Cell. 1993;73:967–977. doi: 10.1016/0092-8674(93)90274-t. [DOI] [PubMed] [Google Scholar]
- Kuebler D, Tanouye MA. Modifications of seizure susceptibility in Drosophila. J Neurophysiol. 2000;83:998–1009. doi: 10.1152/jn.2000.83.2.998. [DOI] [PubMed] [Google Scholar]
- Kuebler D, Tanouye MA. The anticonvulsant sodium valproate reduces seizure-susceptibility in mutant Drosophila. Brain Res. 2002;958:36–42. doi: 10.1016/s0006-8993(02)03431-5. [DOI] [PubMed] [Google Scholar]
- Kuebler D, Zhang HG, Ren X, Tanouye MA. Genetic suppression of seizure susceptibility in Drosophila. J Neurophysiol. 2001;86:1211–1225. doi: 10.1152/jn.2001.86.3.1211. [DOI] [PubMed] [Google Scholar]
- Landmark CJ. Targets for antiepileptic drugs in the synapse. Med Sci Monit. 2007;13:RA1–RA7. [PubMed] [Google Scholar]
- Landmark CJ. Antiepileptic drugs in non-epilepsy disorders: relations between mechanisms of action and clinical efficacy. CNS Drugs. 2008;22:27–47. doi: 10.2165/00023210-200822010-00003. [DOI] [PubMed] [Google Scholar]
- Lee J, Wu CF. Electroconvulsive seizure behavior in Drosophila: analysis of the physiological repertoire underlying a stereotyped action pattern in bang-sensitive mutants. J Neurosci. 2002;22:11065–11079. doi: 10.1523/JNEUROSCI.22-24-11065.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MP, Brown SD, Chen A, Hsieh TS. DNA topoisomerase I is essential in Drosophila melanogaster. Proc Natl Acad Sci U S A. 1993;90:6656–6660. doi: 10.1073/pnas.90.14.6656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loscher W. Basic pharmacology of valproate: a review after 35 years of clinical use for the treatment of epilepsy. CNS Drugs. 2002;16:669–694. doi: 10.2165/00023210-200216100-00003. [DOI] [PubMed] [Google Scholar]
- Loughney K, Kreber R, Ganetzky B. Molecular analysis of the para locus, a sodium channel gene in Drosophila. Cell. 1989;58:1143–1154. doi: 10.1016/0092-8674(89)90512-6. [DOI] [PubMed] [Google Scholar]
- Mahoney K, Moore SJ, Buckley D, Alam M, Parfrey P, Penney S, Merner N, Hodgkinson K, Young TL. Variable neurologic phenotype in a GEFS+ family with a novel mutation in SCN1A. Seizure. 2009;18:492–497. doi: 10.1016/j.seizure.2009.04.009. [DOI] [PubMed] [Google Scholar]
- Meisler MH, Kearney JA. Sodium channel mutations in epilepsy and other neurological disorders. J Clin Invest. 2005;115:2010–2017. doi: 10.1172/JCI25466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulley JC, Scheffer IE, Petrou S, Dibbens LM, Berkovic SF, Harkin LA. SCN1A mutations and epilepsy. Hum Mutat. 2005;25:535–542. doi: 10.1002/humu.20178. [DOI] [PubMed] [Google Scholar]
- Noebels JL. The biology of epilepsy genes. Annu Rev Neurosci. 2003;26:599–625. doi: 10.1146/annurev.neuro.26.010302.081210. [DOI] [PubMed] [Google Scholar]
- Parker L, Padilla M, Du Y, Dong K, Tanouye MA. Drosophila as a model for epilepsy: bss is a gain-of-function mutation in the Para sodium channel gene that leads to seizures. Genetics. 2011;187:523–534. doi: 10.1534/genetics.110.123299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlidis P, Ramaswami M, Tanouye MA. The Drosophila easily shocked gene: a mutation in a phospholipid synthetic pathway causes seizure, neuronal failure, and paralysis. Cell. 1994;79:23–33. doi: 10.1016/0092-8674(94)90397-2. [DOI] [PubMed] [Google Scholar]
- Pavlidis P, Tanouye MA. Seizures and failures in the giant fiber pathway of Drosophila bang-sensitive paralytic mutants. J Neurosci. 1995;15:5810–5819. doi: 10.1523/JNEUROSCI.15-08-05810.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phelan P, Stebbings LA, Baines RA, Bacon JP, Davies JA, Ford C. Drosophila shaking-B protein forms gap junctions in paired Xenopus oocytes. Nature. 1998;391:181–184. doi: 10.1038/34426. [DOI] [PubMed] [Google Scholar]
- Pommier Y, Pourquier P, Fan Y, Strumberg D. Mechanism of action of eukaryotic DNA topoisomerase I and drugs targeted to the enzyme. Biochim Biophys Acta. 1998;1400:83–105. doi: 10.1016/s0167-4781(98)00129-8. [DOI] [PubMed] [Google Scholar]
- Pommier Y, Pourquier P, Urasaki Y, Wu J, Laco GS. Topoisomerase I inhibitors: selectivity and cellular resistance. Drug Resist Update. 1999;2:307–318. doi: 10.1054/drup.1999.0102. [DOI] [PubMed] [Google Scholar]
- Ramaswami M, Tanouye MA. Two sodium channel genes in Drosophila: implications for channel diversity. Proc Natl Acad Sci U S A. 1989;86:2079–2082. doi: 10.1073/pnas.86.6.2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reenan RA, Hanrahan CJ, Ganetzky B. The mle(napts) RNA helicase mutation in Drosophila results in a splicing catastrophe of the para Na+ channel transcript in a region of RNA editing. Neuron. 2000;25:139–149. doi: 10.1016/s0896-6273(00)80878-8. [DOI] [PubMed] [Google Scholar]
- Reid CA, Berkovic SF, Petrou S. Mechanisms of human inherited epilepsies. Prog Neurobiol. 2009;87:41–57. doi: 10.1016/j.pneurobio.2008.09.016. [DOI] [PubMed] [Google Scholar]
- Reynolds ER, Stauffer EA, Feeney L, Rojahn E, Jacobs B, McKeever C. Treatment with the antiepileptic drugs phenytoin and gabapentin ameliorates seizure and paralysis of Drosophila bang-sensitive mutants. J Neurobiol. 2003;58:503–513. doi: 10.1002/neu.10297. [DOI] [PubMed] [Google Scholar]
- Royden CS, Pirrotta V, Jan LY. The tko locus, site of a behavioral mutation in D. melanogaster, codes for a protein homologous to prokaryotic ribosomal protein S12. Cell. 1987;51:165–173. doi: 10.1016/0092-8674(87)90144-9. [DOI] [PubMed] [Google Scholar]
- Shneker BF, Fountain NB. Epilepsy. Dis Mon. 2003;49:426–478. doi: 10.1016/s0011-5029(03)00065-8. [DOI] [PubMed] [Google Scholar]
- Siddiqi O, Benzer S. Neurophysiological defects in temperature-sensitive paralytic mutants of Drosophila melanogaster. Proc Natl Acad Sci U S A. 1976;73:3253–3257. doi: 10.1073/pnas.73.9.3253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song J, Hu J, Tanouye MA. Seizure suppression by top1 mutations in Drosophila. J Neurosci. 2007;27:2927–2937. doi: 10.1523/JNEUROSCI.3944-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song J, Parker L, Hormozi L, Tanouye MA. DNA topoisomerase I inhibitors ameliorate seizure-like behaviors and paralysis in a Drosophila model of epilepsy. Neuroscience. 2008;156:722–728. doi: 10.1016/j.neuroscience.2008.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song J, Tanouye MA. Seizure suppression by shakB2, a gap junction mutation in Drosophila. J Neurophysiol. 2006;95:627–635. doi: 10.1152/jn.01059.2004. [DOI] [PubMed] [Google Scholar]
- Song J, Tanouye MA. A role for para sodium channel gene 3′ UTR in the modification of Drosophila seizure susceptibility. Dev Neurobiol. 2007;67:1944–1956. doi: 10.1002/dneu.20519. [DOI] [PubMed] [Google Scholar]
- Song J, Tanouye MA. From bench to drug: human seizure modeling using Drosophila. Prog Neurobiol. 2008;84:182–191. doi: 10.1016/j.pneurobio.2007.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song J, Tanouye MA. The genetics and molecular biology of seizure-susceptibility in Drosophila. In: Baraban S, editor. Animal Models of Epilepsy: Methods and Innovation. Springer-Verlag; New York, LLC: 2009. pp. 27–44. [Google Scholar]
- Suzuki D, Grigliatti T, Williamson R. Temperature-sensitive mutations in Drosophila melanogaster, VII. A mutation (parats) causing reversible adult paralysis. Proc Natl Acad Sci U S A. 1971;68:890–893. doi: 10.1073/pnas.68.5.890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szente M, Gajda Z, Said, Said Ali K, Hermesz E. Involvement of electrical coupling in the in vivo ictal epileptiform activity induced by 4-aminopyridine in the neocortex. Neuroscience. 2002;115:1067–1078. doi: 10.1016/s0306-4522(02)00533-x. [DOI] [PubMed] [Google Scholar]
- Tan JS, Lin F, Tanouye MA. Potassium bromide, an anticonvulsant, is effective at alleviating seizures in the Drosophila bang-sensitive mutant bang senseless. Brain Res. 2004;1020:45–52. doi: 10.1016/j.brainres.2004.05.111. [DOI] [PubMed] [Google Scholar]
- Tanouye MA, Wyman RJ. Motor outputs of giant nerve fiber in Drosophila. J Neurophysiol. 1980;44:405–421. doi: 10.1152/jn.1980.44.2.405. [DOI] [PubMed] [Google Scholar]
- Tao H, Manak JR, Sowers L, Mei X, Kiyonari H, Abe T, et al. Mutations in Prickle orthologs cause seizures in flies, mice, and humans. Am J Hum Genet. 2011;88:138–149. doi: 10.1016/j.ajhg.2010.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas JB, Wyman RJ. Mutations altering synaptic connectivity between identified neurons in Drosophila melanogaster. J Neurosci. 1984;4:530–538. doi: 10.1523/JNEUROSCI.04-02-00530.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White HS, Smith MD, Wilcox KS. Mechanisms of action of antiepileptic drugs. Int Rev Neurobiol. 2007;81:85–110. doi: 10.1016/S0074-7742(06)81006-8. [DOI] [PubMed] [Google Scholar]
- Wu CF, Ganetzky B. Genetic alteration of nerve membrane excitability in temperature-sensitive paralytic mutants of Drosophila melanogaster. Nature. 1980;286:814–816. doi: 10.1038/286814a0. [DOI] [PubMed] [Google Scholar]
- Wu CF, Ganetzky B, Jan LY, Jan YN. A Drosophila mutant with a temperature-sensitive block in nerve conduction. Proc Natl Acad Sci U S A. 1978;75:4047–4051. doi: 10.1073/pnas.75.8.4047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang CX, Chen AD, Gettel NJ, Hsieh TS. Essential functions of DNA Topoisomerase I in Drosophila melanogaster. Dev Biol. 2000;222:27–40. doi: 10.1006/dbio.2000.9704. [DOI] [PubMed] [Google Scholar]
- Zhang H, Tan J, Reynolds E, Kuebler D, Faulhaber S, Tanouye MA. The Drosophila slamdance gene: a mutation in an aminopeptidase can cause seizure, paralysis and neuronal failure. Genetics. 2002;162:1283–1299. doi: 10.1093/genetics/162.3.1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YQ, Roote J, Brogna S, Davis AW, Barbash DA, Nash D, Ashburner M. Stress sensitive B encodes an adenine nucleotide translocase in Drosophila melanogaster. Genetics. 1999;153:891–903. doi: 10.1093/genetics/153.2.891. [DOI] [PMC free article] [PubMed] [Google Scholar]